Compliance Just Got Easier: Stay ahead of regulatory changes with instant notifications on updates that matter.
['Air Programs']
['Air Quality']
05/13/2025

Copyright 2026 J. J. Keller & Associate, Inc. For re-use options please contact copyright@jjkeller.com or call 800-558-5011.
Method 201 - Determination of PM10 Emissions (Exhaust Gas Recycle Procedure).
Method 201A - Determination of PM10 and PM2.5 Emissions From Stationary Sources (Constant Sampling Rate Procedure)
Method 202 - Dry Impinger Method for Determining Condensable Particulate Emissions From Stationary Sources
Method 203A - Visual Determination of Opacity of Emissions from Stationary Sources for Time-Averaged Regulations.
Method 203B - Visual Determination of Opacity of Emissions from Stationary Sources for Time-Exception Regulations.
Method 203C - Visual Determination of Opacity of Emissions from Stationary Sources for Instantaneous Regulations.
Method 204 - Criteria for and Verification of a Permanent or Temporary Total Enclosure.
Method 204A - Volatile Organic Compounds Content in Liquid Input Stream.
Method 204B - Volatile Organic Compounds Emissions in Captured Stream.
Method 204C - Volatile Organic Compounds Emissions in Captured Stream (Dilution Technique).
Method 204D - Volatile Organic Compounds Emissions in Uncaptured Stream from Temporary Total Enclosure.
Method 204E - Volatile Organic Compounds Emissions in Uncaptured Stream from Building Enclosure.
Method 204F - Volatile Organic Compounds Content in Liquid Input Stream (Distillation Approach).
Method 205 - Verification of Gas Dilution Systems for Field Instrument Calibrations
Method 207 - Pre-Survey Procedure for Corn Wet-Milling Facility Emission Sources
1.0 Presented herein are recommended test methods for measuring air pollutantemanating from an emission source. They are provided for States to use in their plans to meet the requirements of subpart K - Source Surveillance.
2.0 The State may also choose to adopt other methods to meet the requirements of subpart K of this part, subject to the normal plan review process.
3.0 The State may also meet the requirements of subpart K of this part by adopting, again subject to the normal plan review process, any of the relevant methods in appendix A to 40 CFR part 60.
4.0 Quality Assurance Procedures. The performance testing shall include a test method performance audit (PA) during the performance test. The PAs consist of blind audit samples supplied by an accredited audit sample provider and analyzed during the performance test in order to provide a measure of test data bias. Gaseous audit samples are designed to audit the performance of the sampling system as well as the analytical system and must be collected by the sampling system during the compliance test just as the compliance samples are collected. If a liquid or solid audit sample is designed to audit the sampling system, it must also be collected by the sampling system during the compliance test. If multiple sampling systems or sampling trains are used during the compliance test for any of the test methods, the tester is only required to use one of the sampling systems per method to collect the audit sample. The audit sample must be analyzed by the same analyst using the same analytical reagents and analytical system and at the same time as the compliance samples. Retests are required when there is a failure to produce acceptable results for an audit sample. However, if the audit results do not affect the compliance or noncompliance status of the affected facility, the compliance authority may waive the reanalysis requirement, further audits, or retests and accept the results of the compliance test. Acceptance of the test results shall constitute a waiver of the reanalysis requirement, further audits, or retests. The compliance authority may also use the audit sample failure and the compliance test results as evidence to determine the compliance or noncompliance status of the affected facility. A blind audit sample is a sample whose value is known only to the sample provider and is not revealed to the tested facility until after it reports the measured value of the audit sample. For pollutants that exist in the gas phase at ambient temperature, the audit sample shall consist of an appropriate concentration of the pollutant in air or nitrogen that will be introduced into the sampling system of the test method at or near the same entry point as a sample from the emission source. If no gas phase audit samples are available, an acceptable alternative is a sample of the pollutant in the same matrix that would be produced when the sample is recovered from the sampling system as required by the test method. For samples that exist only in a liquid or solid form at ambient temperature, the audit sample shall consist of an appropriate concentration of the pollutant in the same matrix that would be produced when the sample is recovered from the sampling system as required by the test method. An accredited audit sample provider (AASP) is an organization that has been accredited to prepare audit samples by an independent, third party accrediting body.
a. The source owner, operator, or representative of the tested facility shall obtain an audit sample, if commercially available, from an AASP for each test method used for regulatory compliance purposes. No audit samples are required for the following test methods: Methods 3A and 3C of appendix A-3 of part 60 of this chapter, Methods 6C, 7E, 9, and 10 of appendix A-4 of part 60, Methods 18 and 19 of appendix A-6 of part 60, Methods 20, 22, and 25A of appendix A-7 of part 60, Methods 30A and 30B of appendix A-8 of part 60, and Methods 303, 318, 320, and 321 of appendix A of part 63 of this chapter. If multiple sources at a single facility are tested during a compliance test event, only one audit sample is required for each method used during a compliance test. The compliance authority responsible for the compliance test may waive the requirement to include an audit sample if they believe that an audit sample is not necessary. "Commercially available" means that two or more independent AASPs have blind audit samples available for purchase. If the source owner, operator, or representative cannot find an audit sample for a specific method, the owner, operator, or representative shall consult the EPA Web site at the following URL, http://www.epa.gov/ttn/emc, to confirm whether there is a source that can supply an audit sample for that method. If the EPA Web site does not list an available audit sample at least 60 days prior to the beginning of the compliance test, the source owner, operator, or representative shall not be required to include an audit sample as part of the quality assurance program for the compliance test. When ordering an audit sample, the source owner, operator, or representative shall give the sample provider an estimate for the concentration of each pollutant that is emitted by the source or the estimated concentration of each pollutant based on the permitted level and the name, address, and phone number of the compliance authority. The source owner, operator, or representative shall report the results for the audit sample along with a summary of the emissions test results for the audited pollutant to the compliance authority and shall report the results of the audit sample to the AASP. The source owner, operator, or representative shall make both reports at the same time and in the same manner or shall report to the compliance authority first and then report to the AASP. If the method being audited is a method that allows the samples to be analyzed in the field, and the tester plans to analyze the samples in the field, the tester may analyze the audit samples prior to collecting the emission samples provided a representative of the compliance authority is present at the testing site. The tester may request and the compliance authority may grant a waiver to the requirement that a representative of the compliance authority must be present at the testing site during the field analysis of an audit sample. The source owner, operator, or representative may report the results of the audit sample to the compliance authority and then report the results of the audit sample to the AASP prior to collecting any emission samples. The test protocol and final test report shall document whether an audit sample was ordered and utilized and the pass/fail results as applicable.
b. An AASP shall have and shall prepare, analyze, and report the true value of audit samples in accordance with a written technical criteria document that describes how audit samples will be prepared and distributed in a manner that will ensure the integrity of the audit sample program. An acceptable technical criteria document shall contain standard operating procedures for all of the following operations:
1. Preparing the sample;
2. Confirming the true concentration of the sample;
3. Defining the acceptance limits for the results from a well qualified tester. This procedure must use well established statistical methods to analyze historical results from well qualified testers. The acceptance limits shall be set so that there is 95 percent confidence that 90 percent of well qualified labs will produce future results that are within the acceptance limit range;
4. Providing the opportunity for the compliance authority to comment on the selected concentration level for an audit sample;
5. Distributing the sample to the user in a manner that guarantees that the true value of the sample is unknown to the user;
6. Recording the measured concentration reported by the user and determining if the measured value is within acceptable limits;
7. Report the results from each audit sample in a timely manner to the compliance authority and to the source owner, operator, or representative by the AASP. The AASP shall make both reports at the same time and in the same manner or shall report to the compliance authority first and then report to the source owner, operator, or representative. The results shall include the name of the facility tested, the date on which the compliance test was conducted, the name of the company performing the sample collection, the name of the company that analyzed the compliance samples including the audit sample, the measured result for the audit sample, and whether the testing company passed or failed the audit. The AASP shall report the true value of the audit sample to the compliance authority. The AASP may report the true value to the source owner, operator, or representative if the AASP's operating plan ensures that no laboratory will receive the same audit sample twice.
8. Evaluating the acceptance limits of samples at least once every two years to determine in consultation with the voluntary consensus standard body if they should be changed;
9. Maintaining a database, accessible to the compliance authorities, of results from the audit that shall include the name of the facility tested, the date on which the compliance test was conducted, the name of the company performing the sample collection, the name of the company that analyzed the compliance samples including the audit sample, the measured result for the audit sample, the true value of the audit sample, the acceptance range for the measured value, and whether the testing company passed or failed the audit.
c. The accrediting body shall have a written technical criteria document that describes how it will ensure that the AASP is operating in accordance with the AASP technical criteria document that describes how audit samples are to be prepared and distributed. This document shall contain standard operating procedures for all of the following operations:
1. Checking audit samples to confirm their true value as reported by the AASP;
2. Performing technical systems audits of the AASP's facilities and operating procedures at least once every 2 years.
3. Providing standards for use by the voluntary consensus standard body to approve the accrediting body that will accredit the audit sample providers.
d. The technical criteria documents for the accredited sample providers and the accrediting body shall be developed through a public process guided by a voluntary consensus standards body (VCSB). The VCSB shall operate in accordance with the procedures and requirements in the Office of Management and Budget Circular A-119. A copy of Circular A-119 is available upon request by writing the Office of Information and Regulatory Affairs, Office of Management and Budget, 725 17th Street, NW., Washington, DC 20503, by calling (202) 395-6880 or by downloading online at http://standards.gov/standards_gov/a119.cfm. The VCSB shall approve all accrediting bodies. The Administrator will review all technical criteria documents. If the technical criteria documents do not meet the minimum technical requirements in this Appendix M, paragraphs b. through d., the technical criteria documents are not acceptable and the proposed audit sample program is not capable of producing audit samples of sufficient quality to be used in a compliance test. All acceptable technical criteria documents shall be posted on the EPA Web site at the following URL, http://www.epa.gov/ttn/emc.
Method 201 - Determination of PM10 Emissions
(exhaust gas recycle procedure)
1. Applicability and Principle
1.1 Applicability. This method applies to the in-stack measurement of particulate matter (PM) emissions equal to or less than an aerodynamic diameter of nominally 10 µm (PM10) from stationary sources. The EPA recognizes that condensible emissions not collected by an in-stack method are also PM10, and that emissions that contribute to ambient PM10 levels are the sum of condensible emissions and emissions measured by an in-stack PM10 method, such as this method or Method 201A. Therefore, for establishing source contributions to ambient levels of PM10, such as for emission inventory purposes, EPA suggests that source PM10 measurement include both in-stack PM10 and condensible emissions. Condensible missions may be measured by an impinger analysis in combination with this method.
1.2 Principle. A gas sample is isokinetically extracted from the source. An in-stack cyclone is used to separate PM greater than PM10, and an in-stack glass fiber filter is used to collect the PM10. To maintain isokinetic flow rate conditions at the tip of the probe and a constant flow rate through the cyclone, a clean, dried portion of the sample gas at stack temperature is recycled into the nozzle. The particulate mass is determined gravimetrically after removal of uncombined water.
2. Apparatus
Note:
Method 5 as cited in this method refers to the method in 40 CFR part 60, appendix A.
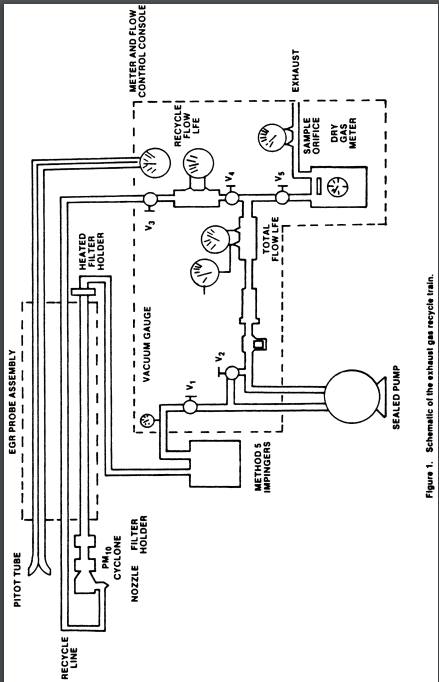
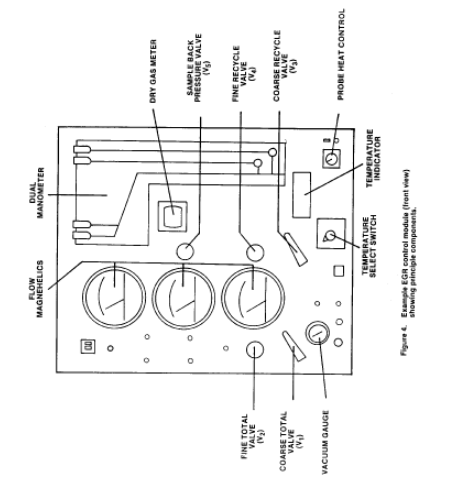
2.1 Sampling Train. A schematic of the exhaust of the exhaust gas recycle (EGR) train is shown in Figure 1 of this method.
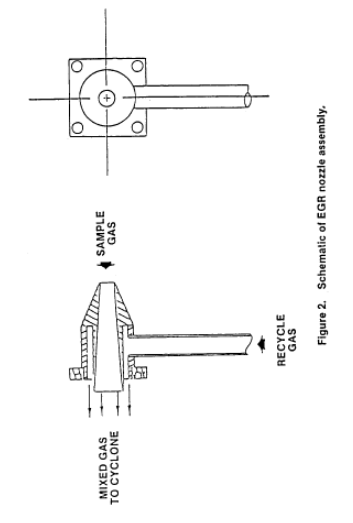
2.1.1 Nozzle with Recycle Attachment. Stainless steel (316 or equivalent) with a sharp tapered leading edge, and recycle attachment welded directly on the side of the nozzle (see schematic in Figure 2 of this method). The angle of the taper shall be on the outside. Use only straight sampling nozzles. "Gooseneck" or other nozzle extensions designed to turn the sample gas flow 90°, as in Method 5 are not acceptable. Locate a thermocouple in the recycle attachment to measure the temperature of the recycle gas as shown in Figure 3 of this method. The recycle attachment shall be made of stainless steel and shall be connected to the probe and nozzle with stainless steel fittings. Two nozzle sizes, e.g., 0.125 and 0.160 in., should be available to allow isokinetic sampling to be conducted over a range of flow rates. Calibrate each nozzle as described in Method 5, Section 5.1.
2.1.2 PM10 Sizer. Cyclone, meeting the specifications in Section 5.7 of this method.
2.1.3 Filter Holder. 63mm, stainless steel. An Andersen filter, part number SE274, has been found to be acceptable for the in-stack filter.
Note:
Mention of trade names or specific products does not constitute endorsement by the Environmental Protection Agency.
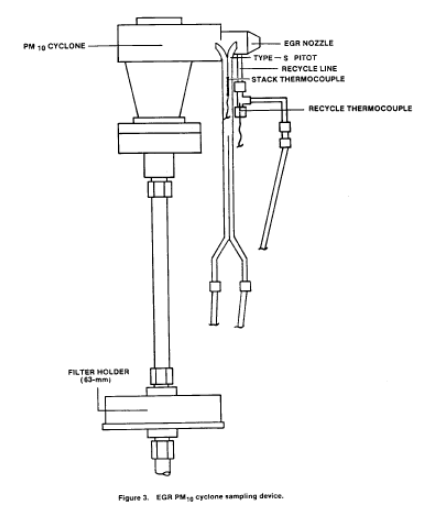
2.1.4 Pitot Tube. Same as in Method 5, Section 2.1.3. Attach the pitot to the pitot lines with stainless steel fittings and to the cyclone in a configuration similar to that shown in Figure 3 of this method. The pitot lines shall be made of heat resistant material and attached to the probe with stainless steel fittings.
2.1.5 EGR Probe. Stainless steel, 15.9-mm ( 5/8-in.) ID tubing with a probe liner, stainless steel 9.53-mm ( 3/8-in.) ID stainless steel recycle tubing, two 6.35-mm ( 1/4-in.) ID stainless steel tubing for the pitot tube extensions, three thermocouple leads, and one power lead, all contained by stainless steel tubing with a diameter of approximately 51 mm (2.0 in.). Design considerations should include minimum weight construction materials sufficient for probe structural strength. Wrap the sample and recycle tubes with a heating tape to heat the sample and recycle gases to stack temperature.
2.1.6 Condenser. Same as in Method 5, Section 2.1.7.
2.1.7 Umbilical Connector. Flexible tubing with thermocouple and power leads of sufficient length to connect probe to meter and flow control console.
2.1.8 Vacuum Pump. Leak-tight, oil-less, noncontaminating, with an absolute filter, "HEPA" type, at the pump exit. A Gast Model 0522-V103 G18DX pump has been found to be satisfactory.
2.1.9 Meter and Flow Control Console. System consisting of a dry gas meter and calibrated orifice for measuring sample flow rate and capable of measuring volume to ±2 percent, calibrated laminar flow elements (LFE's) or equivalent for measuring total and sample flow rates, probe heater control, and manometers and magnehelic gauges (as shown in Figures 4 and 5 of this method), or equivalent. Temperatures needed for calculations include stack, recycle, probe, dry gas meter, filter, and total flow. Flow measurements include velocity head (Δp), orifice differential pressure (ΔH), total flow, recycle flow, and total back-pressure through the system.
2.1.10 Barometer. Same as in Method 5, Section 2.1.9.
2.1.11 Rubber Tubing. 6.35-mm (1/4-in.) ID flexible rubber tubing.
2.2 Sample Recovery.
2.2.1 Nozzle, Cyclone, and Filter Holder Brushes. Nylon bristle brushes property sized and shaped for cleaning the nozzle, cyclone, filter holder, and probe or probe liner, with stainless steel wire shafts and handles.
2.2.2 Wash Bottles, Glass Sample Storage Containers, Petri Dishes, Graduated Cylinder and Balance, Plastic Storage Containers, and Funnels. Same as Method 5, Sections 2.2.2 through 2.2.6 and 2.2.8, respectively.
2.3 Analysis. Same as in Method 5, Section 2.3.
3. Reagents
The reagents used in sampling, sample recovery, and analysis are the same as that specified in Method 5, Sections 3.1, 3.2, and 3.3, respectively.
4. Procedure
4.1 Sampling. The complexity of this method is such that, in order to obtain reliable results, testers should be trained and experienced with the test procedures.
4.1.1 Pretest Preparation. Same as in Method 5, Section 4.1.1.
4.1.2 Preliminary Determinations. Same as Method 5, Section 4.1.2, except use the directions on nozzle size selection in this section. Use of the EGR method may require a minimum sampling port diameter of 0.2 m (6 in.). Also, the required maximum number of sample traverse points at any location shall be 12.
4.1.2.1 The cyclone and filter holder must be in-stack or at stack temperature during sampling. The blockage effects of the EGR sampling assembly will be minimal if the cross-sectional area of the sampling assembly is 3 percent or less of the cross-sectional area of the duct and a pitot coefficient of 0.84 may be assigned to the pitot. If the cross-sectional area of the assembly is greater than 3 percent of the cross-sectional area of the duct, then either determine the pitot coefficient at sampling conditions or use a standard pitot with a known coefficient in a configuration with the EGR sampling assembly such that flow disturbances are minimized.
4.1.2.2 Construct a setup of pressure drops for various Δp's and temperatures. A computer is useful for these calculations. An example of the output of the EGR setup program is shown in Figure 6 of this method, and directions on its use are in section 4.1.5.2 of this method. Computer programs, written in IBM BASIC computer language, to do these types of setup and reduction calculations for the EGR procedure, are available through the National Technical Information Services (NTIS), Accession number PB90-500000, 5285 Port Royal Road, Springfield, VA 22161.
4.1.2.3 The EGR setup program allows the tester to select the nozzle size based on anticipated average stack conditions and prints a setup sheet for field use. The amount of recycle through the nozzle should be between 10 and 80 percent. Inputs for the EGR setup program are stack temperature (minimum, maximum, and average), stack velocity (minimum, maximum, and average), atmospheric pressure, stack static pressure, meter box temperature, stack moisture, percent 02, and percent CO2 in the stack gas, pitot coefficient (Cp), orifice Δ H2, flow rate measurement calibration values [slope (m) and y-intercept (b) of the calibration curve], and the number of nozzles available and their diameters.
4.1.2.4 A less rigorous calculation for the setup sheet can be done manually using the equations on the example worksheets in Figures 7, 8, and 9 of this method, or by a Hewlett-Packard HP41 calculator using the program provided in appendix D of the EGR operators manual, entitled Applications Guide for Source PM10Exhaust Gas Recycle Sampling System. This calculation uses an approximation of the total flow rate and agrees within 1 percent of the exact solution for pressure drops at stack temperatures from 38 to 260°C (100 to 500°F) and stack moisture up to 50 percent. Also, the example worksheets use a constant stack temperature in the calculation, ingoring the complicated temperature dependence from all three pressure drop equations. Errors for this at stack temperatures ±28°C (±50°F) of the temperature used in the setup calculations are within 5 percent for flow rate and within 5 percent for cyclone cut size.
4.1.2.5 The pressure upstream of the LFE's is assumed to be constant at 0.6 in. Hg in the EGR setup calculations.
4.1.2.6 The setup sheet constructed using this procedure shall be similar to Figure 6 of this method. Inputs needed for the calculation are the same as for the setup computer except that stack velocities are not needed.
4.1.3 Preparation of Collection Train. Same as in Method 5, Section 4.1.3, except use the following directions to set up the train.
4.1.3.1 Assemble the EGR sampling device, and attach it to probe as shown in Figure 3 of this method. If stack temperatures exceed 260°C (500°F), then assemble the EGR cyclone without the O-ring and reduce the vacuum requirement to 130 mm Hg (5.0 in. Hg) in the leak-check procedure in Section 4.1.4.3.2 of this method.
4.1.3.2 Connect the proble directly to the filter holder and condenser as in Method 5. Connect the condenser and probe to the meter and flow control console with the umbilical connector. Plug in the pump and attach pump lines to the meter and flow control console.
4.1.4 Leak-Check Procedure. The leak-check for the EGR Method consists of two parts: the sample-side and the recycle-side. The sample-side leak-check is required at the beginning of the run with the cyclone attached, and after the run with the cyclone removed. The cyclone is removed before the post-test leak-check to prevent any disturbance of the collected sample prior to analysis. The recycle-side leak-check tests the leak tight integrity of the recycle components and is required prior to the first test run and after each shipment.
4.1.4.1 Pretest Leak-Check. A pretest leak-check of the entire sample-side, including the cyclone and nozzle, is required. Use the leak-check procedure in Section 4.1.4.3 of this method to conduct a pretest leak-check.
4.1.4.2 Leak-Checks During Sample Run. Same as in Method 5, Section 4.1.4.1.
4.1.4.3 Post-Test Leak-Check. A leak-check is required at the conclusion of each sampling run. Remove the cyclone before the leak-check to prevent the vacuum created by the cooling of the probe from disturbing the collected sample and use the following procedure to conduct a post-test leak-check.
4.1.4.3.1 The sample-side leak-check is performed as follows: After removing the cyclone, seal the probe with a leak-tight stopper. Before starting pump, close the coarse total valve and both recycle valves, and open completely the sample back pressure valve and the fine total valve. After turning the pump on, partially open the coarse total valve slowly to prevent a surge in the manometer. Adjust the vacuum to at least 381 mm Hg (15.0 in. Hg) with the fine total valve. If the desired vacuum is exceeded, either leak-check at this higher vacuum or end the leak-check as shown below and start over.
Caution: Do not decrease the vacuum with any of the valves. This may cause a rupture of the filter.
Note:
A lower vacuum may be used, provided that it is not exceeded during the test.
4.1.4.3.2 Leak rates in excess of 0.00057 m 3/min (0.020 ft 3/min) are unacceptable. If the leak rate is too high, void the sampling run.
4.1.4.3.3 To complete the leak-check, slowly remove the stopper from the nozzle until the vacuum is near zero, then immediately turn off the pump. This procedure sequence prevents a pressure surge in the manometer fluid and rupture of the filter.
4.1.4.3.4 The recycle-side leak-check is performed as follows: Close the coarse and fine total valves and sample back pressure valve. Plug the sample inlet at the meter box. Turn on the power and the pump, close the recycle valves, and open the total flow valves. Adjust the total flow fine adjust valve until a vacuum of 25 inches of mercury is achieved. If the desired vacuum is exceeded, either leak-check at this higher vacuum, or end the leak-check and start over. Minimum acceptable leak rates are the same as for the sample-side. If the leak rate is too high, void the sampling run.
4.1.5 EGR Train Operation. Same as in Method 5, Section 4.1.5, except omit references to nomographs and recommendations about changing the filter assembly during a run.
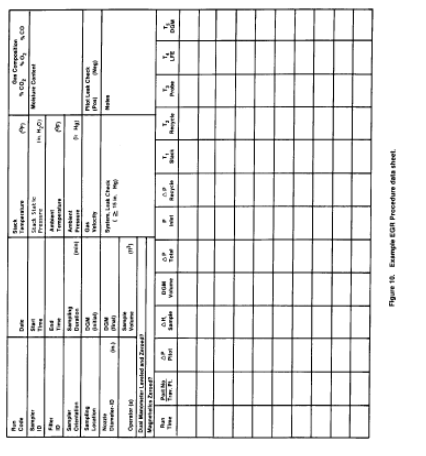
4.1.5.1 Record the data required on a data sheet such as the one shown in Figure 10 of this method. Make periodic checks of the manometer level and zero to ensure correct ΔH and Δp values. An acceptable procedure for checking the zero is to equalize the pressure at both ends of the manometer by pulling off the tubing, allowing the fluid to equilibrate and, if necessary, to re-zero. Maintain the probe temperature to within 11°C (20°F) of stack temperature.
4.1.5.2 The procedure for using the example EGR setup sheet is as follows: Obtain a stack velocity reading from the pitot manometer (Δp), and find this value on the ordinate axis of the setup sheet. Find the stack temperature on the abscissa. Where these two values intersect are the differential pressures necessary to achieve isokineticity and 10 µm cut size (interpolation may be necessary).
4.1.5.3 The top three numbers are differential pressures (in. H2 O), and the bottom number is the percent recycle at these flow settings. Adjust the total flow rate valves, coarse and fine, to the sample value (ΔH) on the setup sheet, and the recycle flow rate valves, coarse and fine, to the recycle flow on the setup sheet.
4.1.5.4 For startup of the EGR sample train, the following procedure is recommended. Preheat the cyclone in the stack for 30 minutes. Close both the sample and recycle coarse valves. Open the fine total, fine recycle, and sample back pressure valves halfway. Ensure that the nozzle is properly aligned with the sample stream. After noting the Δp and stack temperature, select the appropriate ΔH and recycle from the EGR setup sheet. Start the pump and timing device simultaneously. Immediately open both the coarse total and the coarse recycle valves slowly to obtain the approximate desired values. Adjust both the fine total and the fine recycle valves to achieve more precisely the desired values. In the EGR flow system, adjustment of either valve will result in a change in both total and recycle flow rates, and a slight iteration between the total and recycle valves may be necessary. Because the sample back pressure valve controls the total flow rate through the system, it may be necessary to adjust this valve in order to obtain the correct flow rate.
Note:
Isokinetic sampling and proper operation of the cyclone are not achieved unless the correct ΔH and recycle flow rates are maintained.
4.1.5.5 During the test run, monitor the probe and filter temperatures periodically, and make adjustments as necessary to maintain the desired temperatures. If the sample loading is high, the filter may begin to blind or the cyclone may clog. The filter or the cyclone may be replaced during the sample run. Before changing the filter or cyclone, conduct a leak-check (Section 4.1.4.2 of this method). The total particulate mass shall be the sum of all cyclone and the filter catch during the run. Monitor stack temperature and Δp periodically, and make the necessary adjustments in sampling and recycle flow rates to maintain isokinetic sampling and the proper flow rate through the cyclone. At the end of the run, turn off the pump, close the coarse total valve, and record the final dry gas meter reading. Remove the probe from the stack, and conduct a post-test leak-check as outlined in Section 4.1.4.3 of this method.
4.2 Sample Recovery. Allow the probe to cool. When the probe can be safely handled, wipe off all external PM adhering to the outside of the nozzle, cyclone, and nozzle attachment, and place a cap over the nozzle to prevent losing or gaining PM. Do not cap the nozzle tip tightly while the sampling train is cooling, as this action would create a vacuum in the filter holder. Disconnect the probe from the umbilical connector, and take the probe to the cleanup site. Sample recovery should be conducted in a dry indoor area or, if outside, in an area protected from wind and free of dust. Cap the ends of the impingers and carry them to the cleanup site. Inspect the components of the train prior to and during disassembly to note any abnormal conditions. Disconnect the pitot from the cyclone. Remove the cyclone from the probe. Recover the sample as follows:
4.2.1 Container Number 1 (Filter). The recovery shall be the same as that for Container Number 1 in Method 5, Section 4.2.
4.2.2 Container Number 2 (Cyclone or Large PM Catch). The cyclone must be disassembled and the nozzle removed in order to recover the large PM catch. Quantitatively recover the PM from the interior surfaces of the nozzle and the cyclone, excluding the "turn around" cup and the interior surfaces of the exit tube. The recovery shall be the same as that for Container Number 2 in Method 5, Section 4.2.
4.2.3 Container Number 3 (PM10). Quantitatively recover the PM from all of the surfaces from cyclone exit to the front half of the in-stack filter holder, including the "turn around" cup and the interior of the exit tube. The recovery shall be the same as that for Container Number 2 in Method 5, Section 4.2.
4.2.4 Container Number 4 (Silica Gel). Same as that for Container Number 3 in Method 5, Section 4.2.
4.2.5 Impinger Water. Same as in Method 5, Section 4.2, under "Impinger Water."
4.3 Analysis. Same as in Method 5, Section 4.3, except handle EGR Container Numbers 1 and 2 like Container Number 1 in Method 5, EGR Container Numbers 3, 4, and 5 like Container Number 3 in Method 5, and EGR Container Number 6 like Container Number 3 in Method 5. Use Figure 11 of this method to record the weights of PM collected.
4.4 Quality Control Procedures. Same as in Method 5, Section 4.4.
4.5 PM10 Emission Calculation and Acceptability of Results. Use the EGR reduction program or the procedures in section 6 of this method to calculate PM10 emissions and the criteria in section 6.7 of this method to determine the acceptability of the results.
5. Calibration
Maintain an accurate laboratory log of all calibrations.
5.1 Probe Nozzle. Same as in Method 5, Section 5.1.
5.2 Pitot Tube. Same as in Method 5, Section 5.2.
5.3 Meter and Flow Control Console.
5.3.1 Dry Gas Meter. Same as in Method 5, Section 5.3.
5.3.2 LFE Gauges. Calibrate the recycle, total, and inlet total LFE gauges with a manometer. Read and record flow rates at 10, 50, and 90 percent of full scale on the total and recycle pressure gauges. Read and record flow rates at 10, 20, and 30 percent of full scale on the inlet total LFE pressure gauge. Record the total and recycle readings to the nearest 0.3 mm (0.01 in.). Record the inlet total LFE readings to the nearest 3 mm (0.1 in.). Make three separate measurements at each setting and calculate the average. The maximum difference between the average pressure reading and the average manometer reading shall not exceed 1 mm (0.05 in.). If the differences exceed the limit specified, adjust or replace the pressure gauge. After each field use, check the calibration of the pressure gauges.
5.3.3 Total LFE. Same as the metering system in Method 5, Section 5.3.
5.3.4 Recycle LFE. Same as the metering system in Method 5, Section 5.3, except completely close both the coarse and fine recycle valves.
5.4 Probe Heater. Connect the probe to the meter and flow control console with the umbilical connector. Insert a thermocouple into the probe sample line approximately half the length of the probe sample line. Calibrate the probe heater at 66°C (150°F), 121°C (250°F), and 177°C (350°F). Turn on the power, and set the probe heater to the specified temperature. Allow the heater to equilibrate, and record the thermocouple temperature and the meter and flow control console temperature to the nearest 0.5°C (1°F). The two temperatures should agree within 5.5°C (10°F). If this agreement is not met, adjust or replace the probe heater controller.
5.5 Temperature Gauges. Connect all thermocouples, and let the meter and flow control console equilibrate to ambient temperature. All thermocouples shall agree to within 1.1°C (2.0°F) with a standard mercury-in-glass thermometer. Replace defective thermocouples.
5.6 Barometer. Calibrate against a standard mercury-in-glass barometer.
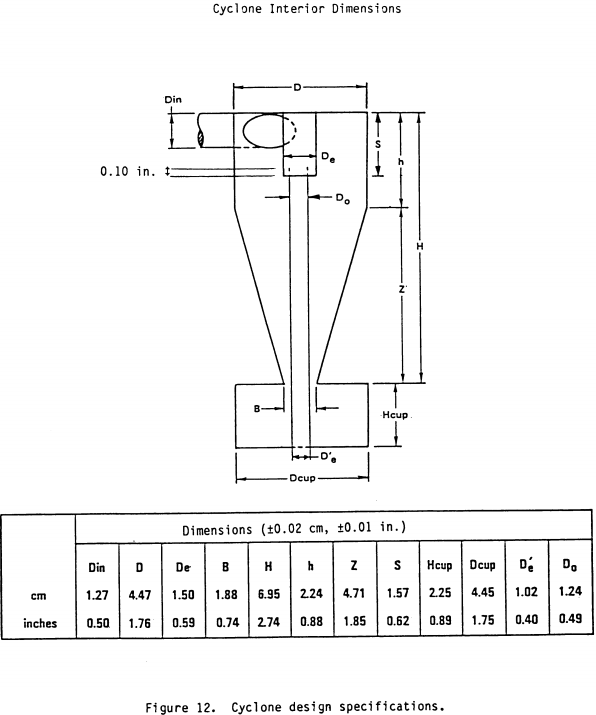
5.7 Probe Cyclone and Nozzle Combinations. The probe cyclone and nozzle combinations need not be calibrated if the cyclone meets the design specifications in Figure 12 of this method and the nozzle meets the design specifications in appendix B of the Application Guide for the Source PM310Exhaust Gas Recycle Sampling System, EPA/600/3-88-058. This document may be obtained from Roy Huntley at (919) 541-1060. If the nozzles do not meet the design specifications, then test the cyclone and nozzle combination for conformity with the performance specifications (PS's) in Table 1 of this method. The purpose of the PS tests is to determine if the cyclone's sharpness of cut meets minimum performance criteria. If the cyclone does not meet design specifications, then, in addition to the cyclone and nozzle combination conforming to the PS's, calibrate the cyclone and determine the relationship between flow rate, gas viscosity, and gas density. Use the procedures in Section 5.7.5 of this method to conduct PS tests and the procedures in Section 5.8 of this method to calibrate the cyclone. Conduct the PS tests in a wind tunnel described in Section 5.7.1 of this method and using a particle generation system described in Section 5.7.2 of this method. Use five particle sizes and three wind velocities as listed in Table 2 of this method. Perform a minimum of three replicate measurements of collection efficiency for each of the 15 conditions listed, for a minimum of 45 measurements.
5.7.1 Wind Tunnel. Perform calibration and PS tests in a wind tunnel (or equivalent test apparatus) capable of establishing and maintaining the required gas stream velocities within 10 percent.
5.7.2 Particle Generation System. The particle generation system shall be capable of producing solid monodispersed dye particles with the mass median aerodynamic diameters specified in Table 2 of this method. The particle size distribution verification should be performed on an integrated sample obtained during the sampling period of each test. An acceptable alternative is to verify the size distribution of samples obtained before and after each test, with both samples required to meet the diameter and monodispersity requirements for an acceptable test run.
5.7.2.1 Establish the size of the solid dye particles delivered to the test section of the wind tunnel using the operating parameters of the particle generation system, and verify the size during the tests by microscopic examination of samples of the particles collected on a membrane filter. The particle size, as established by the operating parameters of the generation system, shall be within the tolerance specified in Table 2 of this method. The precision of the particle size verification technique shall be at least ±0.5 µm, and the particle size determined by the verification technique shall not differ by more than 10 percent from that established by the operating parameters of the particle generation system.
5.7.2.2 Certify the monodispersity of the particles for each test either by microscopic inspection of collected particles on filters or by other suitable monitoring techniques such as an optical particle counter followed by a multichannel pulse height analyzer. If the proportion of multiplets and satellites in an aerosol exceeds 10 percent by mass, the particle generation system is unacceptable for purposes of this test. Multiplets are particles that are agglomerated, and satellites are particles that are smaller than the specified size range.
5.7.3 Schematic Drawings. Schematic drawings of the wind tunnel and blower system and other information showing complete procedural details of the test atmosphere generation, verification, and delivery techniques shall be furnished with calibration data to the reviewing agency.
5.7.4 Flow Rate Measurement. Determine the cyclone flow rates with a dry gas meter and a stopwatch, or a calibrated orifice system capable of measuring flow rates to within 2 percent.
5.7.5 Performance Specification Procedure. Establish the test particle generator operation and verify the particle size microscopically. If mondispersity is to be verified by measurements at the beginning and the end of the run rather than by an integrated sample, these measurements may be made at this time.
5.7.5.1 The cyclone cut size (D50) is defined as the aerodynamic diameter of a particle having a 50 percent probability of penetration. Determine the required cyclone flow rate at which D50 is 10 µm. A suggested procedure is to vary the cyclone flow rate while keeping a constant particle size of 10 µm. Measure the PM collected in the cyclone (mc), exit tube (mt), and filter (mf). Compute the cyclone efficiency (Ec) as follows:
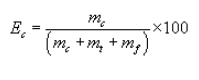
5.7.5.2 Perform three replicates and calculate the average cyclone efficiency as follows:
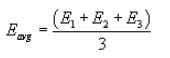
where E1, E2, and E3 are replicate measurements of Ec.
5.7.5.3 Calculate the standard deviation (σ) for the replicate measurements of Ec as follows:
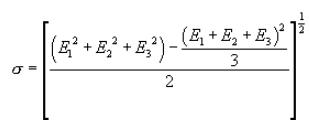
if σ exceeds 0.10, repeat the replicate runs.
5.7.5.4 Using the cyclone flow rate that produces D50 for 10 µm, measure the overall efficiency of the cyclone and nozzle, Eo, at the particle sizes and nominal gas velocities in Table 2 of this method using this following procedure.
5.7.5.5 Set the air velocity in the wind tunnel to one of the nominal gas velocities from Table 2 of this method. Establish isokinetic sampling conditions and the correct flow rate through the sampler (cyclone and nozzle) using recycle capacity so that the D50 is 10 µm. Sample long enough to obtain ±5 percent precision on the total collected mass as determined by the precision and the sensitivity of the measuring technique. Determine separately the nozzle catch (mn), cyclone catch (mc), cyclone exit tube catch (mt), and collection filter catch (mf).
5.7.5.6 Calculate the overall efficiency (Eo) as follows:
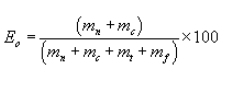
5.7.5.7 Do three replicates for each combination of gas velocities and particle sizes in Table 2 of this method. Calculate Eo for each particle size following the procedures described in this section for determining efficiency. Calculate the standard deviation (σ) for the replicate measurements. If σ exceeds 0.10, repeat the replicate runs.
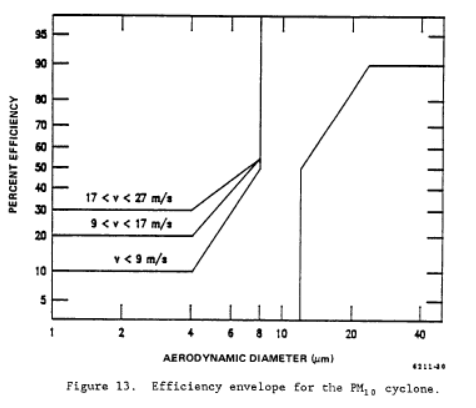
5.7.6 Criteria for Acceptance. For each of the three gas stream velocities, plot the average Eo as a function of particle size on Figure 13 of this method. Draw a smooth curve for each velocity through all particle sizes. The curve shall be within the banded region for all sizes, and the average Ec for a D50 for 10 µm shall be 50 ±0.5 percent.
5.8 Cyclone Calibration Procedure. The purpose of this section is to develop the relationship between flow rate, gas viscosity, gas density, and D50. This procedure only needs to be done on those cyclones that do not meet the design specifications in Figure 12 of this method.
5.8.1 Calculate cyclone flow rate. Determine the flow rates and D50's for three different particle sizes between 5 µm and 15 µm, one of which shall be 10 µm. All sizes must be within 0.5 µm. For each size, use a different temperature within 60°C (108°F) of the temperature at which the cyclone is to be used and conduct triplicate runs. A suggested procedure is to keep the particle size constant and vary the flow rate. Some of the values obtained in the PS tests in Section 5.7.5 may be used.
5.8.1.1 On log-log graph paper, plot the Reynolds number (Re) on the abscissa, and the square root of the Stokes 50 number [(STK50) 1/2] on the ordinate for each temperature. Use the following equations:
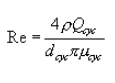
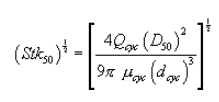
where:
Qcyc = Cyclone flow rate cm 3/sec.
ρ = Gas density, g/cm 3.
dcyc = Diameter of cyclone inlet, cm.
µcyc = Viscosity of gas through the cyclone, poise.
D50 = Cyclone cut size, cm.
5.8.1.2 Use a linear regression analysis to determine the slope (m), and the y-intercept (b). Use the following formula to determine Q, the cyclone flow rate required for a cut size of 10 µm.
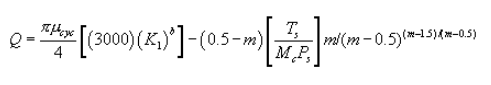
where:
Q = Cyclone flow rate for a cut size of 10 µm, cm 3/sec.
Ts = Stack gas temperature,°K,
d = Diameter of nozzle, cm.
K1 = 4.077 × 10ˆ’3.
5.8.2. Directions for Using Q. Refer to Section 5 of the EGR operators manual for directions in using this expression for Q in the setup calculations.
6. Calculations
6.1 The EGR data reduction calculations are performed by the EGR reduction computer program, which is written in IBM BASIC computer language and is available through NTIS, Accession number PB90-500000, 5285 Port Royal Road, Springfield, Virginia 22161. Examples of program inputs and outputs are shown in Figure 14 of this method.
6.1.1 Calculations can also be done manually, as specified in Method 5, Sections 6.3 through 6.7, and 6.9 through 6.12, with the addition of the following:
6.1.2 Nomenclature.
Bc = Moisture fraction of mixed cyclone gas, by volume, dimensionless.
C1 = Viscosity constant, 51.12 micropoise for°K (51.05 micropoise for° R).
C2 = Viscosity constant, 0.372 micropoise/°K (0.207 micropoise/° R).
C3 = Viscosity constant, 1.05 × 10ˆ’4 micropoise/°K 2 (3.24 × 10ˆ’5 micropoise/° R 2).
C4 = Viscosity constant, 53.147 micropoise/fraction O2.
C5 = Viscosity constant, 74.143 micropoise/fraction H2 O.
D50 = Diameter of particles having a 50 percent probability of penetration, µm.
f02 = Stack gas fraction O2 by volume, dry basis.
K1 = 0.3858°K/mm Hg (17.64° R/in. Hg).
Mc = Wet molecular weight of mixed gas through the PM10 cyclone, g/g-mole (lb/lb-mole).
Md = Dry molecular weight of stack gas, g/g-mole (lb/lb-mole).
Pbar = Barometer pressure at sampling site, mm Hg (in. Hg).
Pin1 = Gauge pressure at inlet to total LFE, mm H2 O (in. H2 O).
P3 = Absolute stack pressure, mm Hg (in. Hg).
Q2 = Total cyclone flow rate at wet cyclone conditions, m 3/min (ft 3/min).
Qs(std) = Total cyclone flow rate at standard conditons, dscm/min (dscf/min).
Tm = Average temperature of dry gas meter,°K (°R).
Ts = Average stack gas temperature,°K (°R).
Vw(std) = Volume of water vapor in gas sample (standard conditions), scm (scf).
XT = Total LFE linear calibration constant, m 3/[(min)(mm H2 O]) { ft 3/[(min)(in. H2 O)]}.
YT = Total LFE linear calibration constant, dscm/min (dscf/min).
Δ PT = Pressure differential across total LFE, mm H2 O, (in. H2 O).
θ = Total sampling time, min.
µcyc = Viscosity of mixed cyclone gas, micropoise.
µLFE = Viscosity of gas laminar flow elements, micropoise.
µstd = Viscosity of standard air, 180.1 micropoise.
6.2 PM10 Particulate Weight. Determine the weight of PM10 by summing the weights obtained from Container Numbers 1 and 3, less the acetone blank.
6.3 Total Particulate Weight. Determine the particulate catch for PM greater than PM10 from the weight obtained from Container Number 2 less the acetone blank, and add it to the PM10 particulate weight.
6.4 PM10 Fraction. Determine the PM10 fraction of the total particulate weight by dividing the PM10 particulate weight by the total particulate weight.
6.5 Total Cyclone Flow Rate. The average flow rate at standard conditions is determined from the average pressure drop across the total LFE and is calculated as follows:
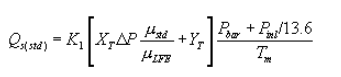
The flow rate, at actual cyclone conditions, is calculated as follows:
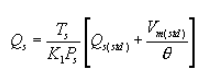
The flow rate, at actual cyclone conditions, is calculated as follows:
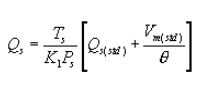
6.6 Aerodynamic Cut Size. Use the following procedure to determine the aerodynamic cut size (D50).
6.6.1 Determine the water fraction of the mixed gas through the cyclone by using the equation below.
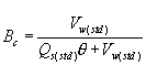
6.6.2 Calculate the cyclone gas viscosity as follows:
µcyc = C1 + C2 Ts + C3 Ts2 + C4 f02 ˆ’ C5 Bc
6.6.3 Calculate the molecular weight on a wet basis of the cyclone gas as follows:
Mc = Md(1 ˆ’ Bc) + 18.0(Bc)
6.6.4 If the cyclone meets the design specification in Figure 12 of this method, calculate the actual D50 of the cyclone for the run as follows:
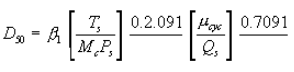
where β1 = 0.1562.
6.6.5 If the cyclone does not meet the design specifications in Figure 12 of this method, then use the following equation to calculate D50.
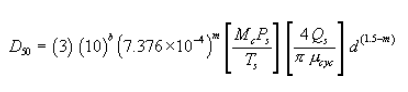
where:
m = Slope of the calibration curve obtained in Section 5.8.2.
b = y-intercept of the calibration curve obtained in Section 5.8.2.
6.7 Acceptable Results. Acceptability of anisokinetic variation is the same as Method 5, Section 6.12.
6.7.1 If 9.0 µm ‰¤D50 ‰¤11 µm and 90 ‰¤I ‰¤110, the results are acceptable. If D50 is greater than 11 µm, the Administrator may accept the results. If D50 is less than 9.0 µm, reject the results and repeat the test.
7. Bibliography
1. Same as Bibliography in Method 5.
2. McCain, J.D., J.W. Ragland, and A.D. Williamson. Recommended Methodology for the Determination of Particles Size Distributions in Ducted Sources, Final Report. Prepared for the California Air Resources Board by Southern Research Institute. May 1986.
3. Farthing, W.E., S.S. Dawes, A.D. Williamson, J.D. McCain, R.S. Martin, and J.W. Ragland. Development of Sampling Methods for Source PM-10 Emissions. Southern Research Institute for the Environmental Protection Agency. April 1989.
4. Application Guide for the Source PM10Exhaust Gas Recycle Sampling System, EPA/600/3-88-058.
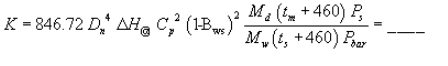
EXAMPLE EMISSION GAS RECYCLE SETUP SHEET
VERSION 3.1 MAY 1986
TEST I.D.: SAMPLE SETUP
RUN DATE: 11/24/86
LOCATION: SOURCE SIM
OPERATOR(S): RH JB
NOZZLE DIAMETER (IN): .25
STACK CONDITIONS:
AVERAGE TEMPERATURE (F): 200.0
AVERAGE VELOCITY (FT/SEC): 15.0
AMBIENT PRESSURE (IN HG): 29.92
STACK PRESSURE (IN H20): .10
GAS COMPOSITION:
| H20 = 10.0% | MD = 28.84 |
| O2 = 20.9% | MW = 27.75 |
| CO2 = .0% | (LB/LB MOLE) |
TARGET PRESSURE DROPS
TEMPERATURE (F)
| DP(PTO) | 150 | 161 | 172 | 183 | 194 | 206 | 217 | 228 |
| 0.026 | SAMPLE | .49 | .49 | .48 | .47 | .46 | .45 | .45 |
| TOTAL | 1.90 | 1.90 | 1.91 | 1.92 | 1.92 | 1.92 | 1.93 | |
| RECYCLE | 2.89 | 2.92 | 2.94 | 2.97 | 3.00 | 3.02 | 3.05 | |
| % RCL | 61% | 61% | 62% | 62% | 63% | 63% | 63% | |
| .031 | .58 | .56 | .55 | .55 | .55 | .54 | .53 | .52 |
| 1.88 | 1.89 | 1.89 | 1.90 | 1.91 | 1.91 | 1.91 | 1.92 | |
| 2.71 | 2.74 | 2.77 | 2.80 | 2.82 | 2.85 | 2.88 | 2.90 | |
| 57% | 57% | 58% | 58% | 59% | 59% | 60% | 60% | |
| .035 | .67 | .65 | .64 | .63 | .62 | .61 | .670 | .59 |
| 1.88 | 1.88 | 1.89 | 1.89 | 1.90 | 1.90 | 1.91 | 1.91 | |
| 2.57 | 2.60 | 2.63 | 2.66 | 2.69 | 2.72 | 2.74 | 2.74 | |
| 54% | 55% | 55% | 56% | 56% | 57% | 57% | 57% | |
| .039 | .75 | .74 | .72 | .71 | .70 | .69 | .67 | .66 |
| 1.87 | 1.88 | 1.88 | 1.89 | 1.89 | 1.90 | 1.90 | 1.91 | |
| 2.44 | 2.47 | 2.50 | 2.53 | 2.56 | 2.59 | 2.62 | 2.65 | |
| 51% | 52% | 52% | 53% | 53% | 54% | 54% | 55% | |
| Figure 6. Example EGR setup sheet. | ||||||||
| Barometric pressure, Pbar, in. Hg | = | ___ | |
| Stack static pressure, Pg, in. H2 O | = | ___ | |
| Average stack temperature, ts,°F | = | ___ | |
| Meter temperature, tm,°F | = | ___ | |
| Gas analysis: | |||
| %CO2 | = | ___ | |
| %O2 | = | ___ | |
| %N2 + %CO | = | ___ | |
| Fraction moisture content, Bws | = | ___ | |
| Calibration data: | |||
| Nozzle diameter, Dn in | = | ___ | |
| Pitot coefficient, Cp | = | ___ | |
| ΔH2, in. H2O | = | ___ | |
| Molecular weight of stack gas, dry basis: | |||
| Md = 0.44 | |||
| (%CO2) + 0.32 | = | lb/lb mole | |
| (%O2) + 0.28 | |||
| (%N2 + %CO) | |||
| Molecular weight of stack gas, wet basis: | |||
| Mw = Md (1-Bws) + 18Bws | = | ___ | lb/lb mole |
| Absolute stack pressure: | |||
| Ps = Pbar + (Pg/13.6) | = | ___ | in. Hg |

Desired meter orifice pressure (ΔH) for velocity head of stack gas (Δp):

Figure 7. Example worksheet 1, meter orifice pressure head calculation.
| Barometric pressure, Pbar, in. Hg | = | ___ | |
| Absolute stack pressure, Ps, in. Hg | = | ___ | |
| Average stack temperature, Ts,°R | = | ___ | |
| Meter temperature, Tm,°R | = | ___ | |
| Molecular weight of stack gas, wet basis, Md lb/lb mole | = | ___ | |
| Pressure upstream of LFE, in. Hg | = | 0.6 | |
| Gas analysis: | |||
| %O2 | = | ___ | |
| Fraction moisture content, Bws | = | ___ | |
| Calibration data: | |||
| Nozzle diameter, Dn, in | = | ___ | |
| Pitot coefficient, Cp | = | ___ | |
| Total LFE calibration constant, Xt | = | ___ | |
| Total LFE calibration constant, Tt | = | ___ | |
| Absolute pressure upstream of LFE: | |||
| PLFE = Pbar + 0.6 | = | ___ | in. Hg |
| Viscosity of gas in total LFE: | |||
| µLFE = 152.418 + 0.2552 Tm + 3.2355 × 10ˆ’5 Tm2 + 0.53147 (%O2) | = | ___ | |
| Viscosity of dry stack gas: | |||
| µd = 152.418 + 0.2552 Ts + 3.2355 × 10ˆ’5 Ts2 + 0.53147 (%O2) | = | ___ |
Constants:
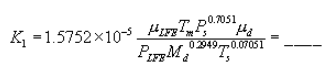
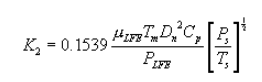
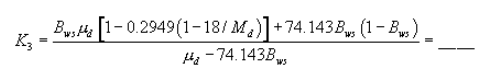
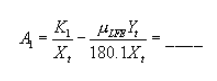
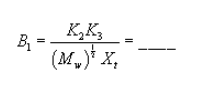
Total LFE pressure head:
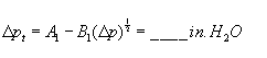
Figure 8. Example worksheet 1, meter orifice pressure head calculation.
| Barometric pressure, Pbar, in. Hg | = | ___ | |
| Absolute stack pressure, Ps, in. Hg | = | ___ | |
| Average stack temperature, Ts,°R | = | ___ | |
| Meter temperature, Tm,°R | = | ___ | |
| Molecular weight of stack gas, dry basis, Md lb/lb mole | = | ___ | |
| Viscosity of LFE gasµLFE,poise | = | ___ | |
| Absolute pressure upstream of LFE, PPLEin. Hg | = | ___ | |
| Calibration data: | |||
| Nozzle diameter, Dn, in | = | ___ | |
| Pitot coefficient, Cp | = | ___ | |
| Recycle LFE calibration constant, Xt | = | ___ | |
| Recycle LFE calibration constant, Yt | = | ___ |
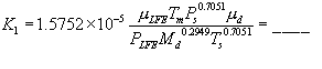
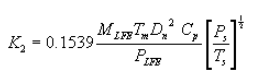
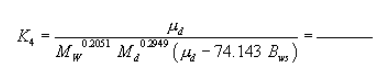
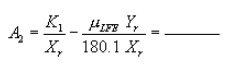
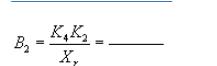
Pressure head for recycle LFE:
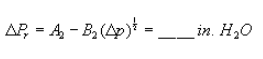
Figure 9. Example worksheet 3, recycle LFE pressure head.
Plant
Date
Run no.
Filter no.
Amount liquid lost during transport
Acetone blank volume, ml
Acetone wash volume, ml (2) - - - (3)
Acetone blank conc., mg/mg (Equation 5-4, Method 5)
Acetone wash blank, mg (Equation 5-5, Method 5)
| Container number | Weight of particulate matter, mg | ||
|---|---|---|---|
| Final weight | Tare weight | Weight gain | |
| 1 | |||
| 3 | |||
| Total | |||
| Less acetone blank | |||
| Weight of PM10 | |||
| 2 | |||
| Less acetone blank | |||
| Total particulate weight | |||
Figure 11. EGR method analysis sheet.
| Parameter | Units | Specification |
|---|---|---|
| 1. Collection efficiency | Percent | Such that collection efficiency falls within envelope specified by Section 5.7.6 and Figure 13. |
| 2. Cyclone cut size (D50) | µm | 10 ±1 µm aerodynamic diameter. |
| Particle size (µm) a | Target gas velocities (m/sec) | ||
|---|---|---|---|
| 7 ±1.0 | 15 ±1.5 | 25 ±2.5 | |
| 5 ±0.5 | |||
| 7 ±0.5 | |||
| 10 ±0.5 | |||
| 14 ±1.0 | |||
| 20 ±1.0 | |||
| (a) Mass median aerodynamic diameter. | |||
Emission Gas Recycle, Data Reduction, Version 3.4 MAY 1986
Test ID. Code: Chapel Hill 2.
Test Location: Baghouse Outlet.
Test Site: Chapel Hill.
Test Date: 10/20/86.
Operators(s): JB RH MH.
Entered Run Data
| Temperatures: | |
| T(STK) | 251.0 F |
| T(RCL) | 259.0 F |
| T(LFE) | 81.0 F |
| T(DGM) | 76.0 F |
| System Pressures: | |
| DH(ORI) | 1.18 INWG |
| DP(TOT) | 1.91 INWG |
| P(INL) | 12.15 INWG |
| DP(RCL) | 2.21 INWG |
| DP(PTO) | 0.06 INWG |
| Miscellanea: | |
| P(BAR) | 29.99 INWG |
| DP(STK) | 0.10 INWG |
| V(DGM) | 13.744 FT3 |
| TIME | 60.00 MIN |
| % CO2 | 8.00 |
| % O2 | 20.00 |
| NOZ (IN) | 0.2500 |
| Water Content: | |
| Estimate | 0.0% |
| or | |
| Condenser | 7.0 ML |
| Column | 0.0 GM |
| Raw Masses: | |
| Cyclone 1 | 21.7 MG |
| Filter | 11.7 MG |
| Impinger Residue | 0.0 MG |
| Blank Values: | |
| CYC Rinse | 0.0 MG |
| Filter Holder Rinse | 0.0 MG |
| Filter Blank | 0.0 MG |
| Impinger Rinse | 0.0 MG |
| Calibration Values: | |
| CP(PITOT) | 0.840 |
| DH@(ORI) | 10.980 |
| M(TOT LFE) | 0.2298 |
| B(TOT LFE) | ˆ’.0058 |
| M(RCL LFE) | 0.0948 |
| B(RCL LFE) | ˆ’.0007 |
| DGM GAMMA | 0.9940 |
Reduced Data
| Stack Velocity (FT/SEC) | 15.95 |
| Stack Gas Moisture (%) | 2.4 |
| Sample Flow Rate (ACFM) | 0.3104 |
| Total Flow Rate (ACFM) | 0.5819 |
| Recycle Flow Rate (ACFM) | 0.2760 |
| Percent Recycle | 46.7 |
| Isokinetic Ratio (%) | 95.1 |
| (Particulate) | (MG/DNCM) | (GR/ACF) | (GR/DCF) | (LB/DSCF) (X 1E6) | ||
|---|---|---|---|---|---|---|
| (UM) | (% <) | |||||
| Cyclone 1 | 10.15 | 35.8 | 56.6 | 0.01794 | 0.02470 | 3.53701 |
| Backup Filter | 30.5 | 0.00968 | 0.01332 | 1.907 | ||
| Particulate Total | 87.2 | 0.02762 | 0.03802 | 5.444 | ||
| Note: Figure 14. Example inputs and outputs of the EGR reduction program. | ||||||
METHOD 201A - DETERMINATION OF PM10 AND PM2.5 EMISSIONS FROM STATIONARY SOURCES (Constant Sampling Rate Procedure)
1.0 Scope and Applicability
1.1 Scope. The U.S. Environmental Protection Agency (U.S. EPA or "we") developed this method to describe the procedures that the stack tester ("you") must follow to measure filterable particulate matter (PM) emissions equal to or less than a nominal aerodynamic diameter of 10 micrometers (PM10) and 2.5 micrometers (PM2.5). This method can be used to measure coarse particles (i.e., the difference between the measured PM10 concentration and the measured PM2.5 concentration).
1.2 Applicability. This method addresses the equipment, preparation, and analysis necessary to measure filterable PM. You can use this method to measure filterable PM from stationary sources only. Filterable PM is collected in stack with this method (i.e., the method measures materials that are solid or liquid at stack conditions). If the gas filtration temperature exceeds 29.4 °C (85 °F), then you may use the procedures in this method to measure only filterable PM (material that does not pass through a filter or a cyclone/filter combination). If the gas filtration temperature exceeds 29.4 °C (85 °F), and you must measure both the filterable and condensable (material that condenses after passing through a filter) components of total primary (direct) PM emissions to the atmosphere, then you must combine the procedures in this method with the procedures in Method 202 of appendix M to this part for measuring condensable PM. However, if the gas filtration temperature never exceeds 29.4 °C (85 °F), then use of Method 202 of appendix M to this part is not required to measure total primary PM.
1.3 Responsibility. You are responsible for obtaining the equipment and supplies you will need to use this method. You must also develop your own procedures for following this method and any additional procedures to ensure accurate sampling and analytical measurements.
1.4 Additional Methods. To obtain results, you must have a thorough knowledge of the following test methods found in appendices A-1 through A-3 of 40 CFR part 60:
(a) Method 1 - Sample and velocity traverses for stationary sources.
(b) Method 2 - Determination of stack gas velocity and volumetric flow rate (Type S pitot tube).
(c) Method 3 - Gas analysis for the determination of dry molecular weight.
(d) Method 4 - Determination of moisture content in stack gases.
(e) Method 5 - Determination of particulate matter emissions from stationary sources.
1.5 Limitations. You cannot use this method to measure emissions in which water droplets are present because the size separation of the water droplets may not be representative of the dry particle size released into the air. To measure filterable PM10 and PM2.5 in emissions where water droplets are known to exist, we recommend that you use Method 5 of appendix A-3to part 60. Because of the temperature limit of the O-rings used in this sampling train, you must follow the procedures in Section 8.6.1 to test emissions from stack gas temperatures exceeding 205°C (400°F).
1.6 Conditions. You can use this method to obtain particle sizing at 10 micrometers and or 2.5 micrometers if you sample within 80 and 120 percent of isokinetic flow. You can also use this method to obtain total filterable particulate if you sample within 90 to 110 percent of isokinetic flow, the number of sampling points is the same as required by Method 5 of appendix A-3 to part 60 or Method 17 of appendix A-6 to part 60, and the filter temperature is within an acceptable range for these methods. For Method 5, the acceptable range for the filter temperature is generally 120 °C (248 °F) unless a higher or lower temperature is specified. The acceptable range varies depending on the source, control technology and applicable rule or permit condition. To satisfy Method 5 criteria, you may need to remove the in-stack filter and use an out-of-stack filter and recover the PM in the probe between the PM2.5 particle sizer and the filter. In addition, to satisfy Method 5 and Method 17 criteria, you may need to sample from more than 12 traverse points. Be aware that this method determines in-stack PM10 and PM2.5 filterable emissions by sampling from a required maximum of 12 sample points, at a constant flow rate through the train (the constant flow is necessary to maintain the size cuts of the cyclones), and with a filter that is at the stack temperature. In contrast, Method 5 or Method 17 trains are operated isokinetically with varying flow rates through the train. Method 5 and Method 17 require sampling from as many as 24 sample points. Method 5 uses an out-of-stack filter that is maintained at a constant temperature of 120 °C (248 °F). Further, to use this method in place of Method 5 or Method 17, you must extend the sampling time so that you collect the minimum mass necessary for weighing each portion of this sampling train. Also, if you are using this method as an alternative to a test method specified in a regulatory requirement (e.g., a requirement to conduct a compliance or performance test), then you must receive approval from the authority that established the regulatory requirement before you conduct the test.
2.0 Summary of Method
2.1 Summary. To measure PM10 and PM2.5, extract a sample of gas at a predetermined constant flow rate through an in-stack sizing device. The particle-sizing device separates particles with nominal aerodynamic diameters of 10 micrometers and 2.5 micrometers. To minimize variations in the isokinetic sampling conditions, you must establish well-defined limits. After a sample is obtained, remove uncombined water from the particulate, then use gravimetric analysis to determine the particulate mass for each size fraction. The original method, as promulgated in 1990, has been changed by adding a PM2.5 cyclone downstream of the PM10 cyclone. Both cyclones were developed and evaluated as part of a conventional five-stage cascade cyclone train. The addition of a PM2.5 cyclone between the PM10 cyclone and the stack temperature filter in the sampling train supplements the measurement of PM10 with the measurement of PM2.5. Without the addition of the PM2.5 cyclone, the filterable particulate portion of the sampling train may be used to measure total and PM10 emissions. Likewise, with the exclusion of the PM10 cyclone, the filterable particulate portion of the sampling train may be used to measure total and PM2.5 emissions. Figure 1 of Section 17 presents the schematic of the sampling train configured with this change.
3.0 Definitions
3.1 Condensable particulate matter (CPM) means material that is vapor phase at stack conditions, but condenses and/or reacts upon cooling and dilution in the ambient air to form solid or liquid PM immediately after discharge from the stack. Note that all CPM is assumed to be in the PM2.5 size fraction.
3.2 Constant weight means a difference of no more than 0.5 mg or one percent of total weight less tare weight, whichever is greater, between two consecutive weighings, with no less than six hours of desiccation time between weighings.
3.3 Filterable particulate matter (PM) means particles that are emitted directly by a source as a solid or liquid at stack or release conditions and captured on the filter of a stack test train.
3.4 Primary particulate matter (PM) (also known as direct PM) means particles that enter the atmosphere as a direct emission from a stack or an open source. Primary PM has two components: Filterable PM and condensable PM. These two PM components have no upper particle size limit.
3.5 Primary PM2.5 (also known as direct PM2.5, total PM2.5, PM2.5, or combined filterable PM2.5 and condensable PM) means PM with an aerodynamic diameter less than or equal to 2.5 micrometers. These solid particles are emitted directly from an air emissions source or activity, or are the gaseous or vaporous emissions from an air emissions source or activity that condense to form PM at ambient temperatures. Direct PM2.5 emissions include elemental carbon, directly emitted organic carbon, directly emitted sulfate, directly emitted nitrate, and other inorganic particles (including but not limited to crustal material, metals, and sea salt).
3.6 Primary PM10 (also known as direct PM10, total PM10, PM10, or the combination of filterable PM10 and condensable PM) means PM with an aerodynamic diameter equal to or less than 10 micrometers.
4.0 Interferences
You cannot use this method to measure emissions where water droplets are present because the size separation of the water droplets may not be representative of the dry particle size released into the air. Stacks with entrained moisture droplets may have water droplets larger than the cut sizes for the cyclones. These water droplets normally contain particles and dissolved solids that become PM10 and PM2.5 following evaporation of the water.
5.0 Safety
5.1 Disclaimer. Because the performance of this method may require the use of hazardous materials, operations, and equipment, you should develop a health and safety plan to ensure the safety of your employees who are on site conducting the particulate emission test. Your plan should conform with all applicable Occupational Safety and Health Administration, Mine Safety and Health Administration, and Department of Transportation regulatory requirements. Because of the unique situations at some facilities and because some facilities may have more stringent requirements than is required by State or federal laws, you may have to develop procedures to conform to the plant health and safety requirements.
6.0 Equipment and Supplies
Figure 2 of Section 17 shows details of the combined cyclone heads used in this method. The sampling train is the same as Method 17 of appendix A-6 to part 60 with the exception of the PM10 and PM2.5 sizing devices. The following sections describe the sampling train's primary design features in detail.
6.1 Filterable Particulate Sampling Train Components.
6.1.1 Nozzle. You must use stainless steel (316 or equivalent) or fluoropolymer-coated stainless steel nozzles with a sharp tapered leading edge. We recommend one of the 12 nozzles listed in Figure 3 of Section 17 because they meet design specifications when PM10 cyclones are used as part of the sampling train. We also recommend that you have a large number of nozzles in small diameter increments available to increase the likelihood of using a single nozzle for the entire traverse. We recommend one of the nozzles listed in Figure 4A or 4B of Section 17 because they meet design specifications when PM2.5 cyclones are used without PM10 cyclones as part of the sampling train.
6.1.2 PM10 and PM2.5 Sizing Device.
6.1.2.1 Use stainless steel (316 or equivalent) or fluoropolymer-coated PM10 and PM2.5 sizing devices. You may use sizing devices constructed of high-temperature specialty metals such as Inconel, Hastelloy, or Haynes 230. (See also Section 8.6.1.) The sizing devices must be cyclones that meet the design specifications shown in Figures 3, 4A, 4B, 5, and 6 of Section 17. Use a caliper to verify that the dimensions of the PM10 and PM2.5 sizing devices are within ±0.02 cm of the design specifications. Example suppliers of PM10 and PM2.5 sizing devices include the following:
(a) Environmental Supply Company, Inc., 2142 E. Geer Street, Durham, North Carolina 27704. Telephone No.: (919) 956-9688; Fax: (919) 682-0333.
(b) Apex Instruments, 204 Technology Park Lane, Fuquay-Varina, North Carolina 27526. Telephone No.: (919) 557-7300 (phone); Fax: (919) 557-7110.
6.1.2.2 You may use alternative particle sizing devices if they meet the requirements in Development and Laboratory Evaluation of a Five-Stage Cyclone System, EPA-600/7-78-008 (http://cfpub.epa.gov/ols).
6.1.3 Filter Holder. Use a filter holder that is stainless steel (316 or equivalent). A heated glass filter holder may be substituted for the steel filter holder when filtration is performed out-of-stack. Commercial-size filter holders are available depending upon project requirements, including commercial stainless steel filter holders to support 25-, 47-, 63-, 76-, 90-, 101-, and 110-mm diameter filters. Commercial size filter holders contain a fluoropolymer O-ring, a stainless steel screen that supports the particulate filter, and a final fluoropolymer O-ring. Screw the assembly together and attach to the outlet of cyclone IV. The filter must not be compressed between the fluoropolymer O-ring and the filter housing.
6.1.4 Pitot Tube. You must use a pitot tube made of heat resistant tubing. Attach the pitot tube to the probe with stainless steel fittings. Follow the specifications for the pitot tube and its orientation to the inlet nozzle given in Section 6.1.1.3 of Method 5 of appendix A-3 to part 60.
6.1.5 Probe Extension and Liner. The probe extension must be glass- or fluoropolymer-lined. Follow the specifications in Section 6.1.1.2 of Method 5 of appendix A-3 to part 60. If the gas filtration temperature never exceeds 30°C (85°F), then the probe may be constructed of stainless steel without a probe liner and the extension is not recovered as part of the PM.
6.1.6 Differential Pressure Gauge, Condensers, Metering Systems, Barometer, and Gas Density Determination Equipment. Follow the requirements in Sections 6.1.1.4 through 6.1.3 of Method 5 of appendix A-3 to part 60, as applicable.
6.2 Sample Recovery Equipment.
6.2.1 Filterable Particulate Recovery. Use the following equipment to quantitatively determine the amount of filterable PM recovered from the sampling train.
(a) Cyclone and filter holder brushes.
(b) Wash bottles. Two wash bottles are recommended. Any container material is acceptable, but wash bottles used for sample and blank recovery must not contribute more than 0.1 mg of residual mass to the CPM measurements.
(c) Leak-proof sample containers. Containers used for sample and blank recovery must not contribute more than 0.05 mg of residual mass to the CPM measurements.
(d) Petri dishes. For filter samples; glass, polystyrene, or polyethylene, unless otherwise specified by the Administrator.
(e) Graduated cylinders. To measure condensed water to within 1 ml or 0.5 g. Graduated cylinders must have subdivisions not greater than 2 ml.
(f) Plastic storage containers. Air-tight containers to store silica gel.
6.2.2 Analysis Equipment.
(a) Funnel. Glass or polyethylene, to aid in sample recovery.
(b) Rubber policeman. To aid in transfer of silica gel to container; not necessary if silica gel is weighed in the field.
(c) Analytical balance. Analytical balance capable of weighing at least 0.0001 g (0.1 mg).
(d) Balance. To determine the weight of the moisture in the sampling train components, use an analytical balance accurate to ±0.5 g.
(e) Fluoropolymer beaker liners.
7.0 Reagents, Standards, and Sampling Media
7.1 Sample Collection. To collect a sample, you will need a filter and silica gel. You must also have water and crushed ice. These items must meet the following specifications.
7.1.1 Filter. Use a nonreactive, nondisintegrating glass fiber, quartz, or polymer filter that does not a have an organic binder. The filter must also have an efficiency of at least 99.95 percent (less than 0.05 percent penetration) on 0.3 micrometer dioctyl phthalate particles. You may use test data from the supplier's quality control program to document the PM filter efficiency.
7.1.2 Silica Gel. Use an indicating-type silica gel of 6 to 16 mesh. You must obtain approval from the regulatory authority that established the requirement to use this test method to use other types of desiccants (equivalent or better) before you use them. Allow the silica gel to dry for two hours at 175°C (350°F) if it is being reused. You do not have to dry new silica gel if the indicator shows the silica is active for moisture collection.
7.1.3 Crushed Ice. Obtain from the best readily available source.
7.1.4 Water. Use deionized, ultra-filtered water that contains 1.0 part per million by weight (1 milligram/liter) residual mass or less to recover and extract samples.
7.2 Sample Recovery and Analytical Reagents. You will need acetone and anhydrous calcium sulfate for the sample recovery and analysis. Unless otherwise indicated, all reagents must conform to the specifications established by the Committee on Analytical Reagents of the American Chemical Society. If such specifications are not available, then use the best available grade. Additional information on each of these items is in the following paragraphs.
7.2.1 Acetone. Use acetone that is stored in a glass bottle. Do not use acetone from a metal container because it will likely produce a high residue in the laboratory and field reagent blanks. You must use acetone with blank values less than 1 part per million by weight residue. Analyze acetone blanks prior to field use to confirm low blank values. In no case shall a blank value of greater than 0.0001 percent (1 part per million by weight) of the weight of acetone used in sample recovery be subtracted from the sample weight (i.e., the maximum blank correction is 0.1 mg per 100 g of acetone used to recover samples).
7.2.2 Particulate Sample Desiccant. Use indicating-type anhydrous calcium sulfate to desiccate samples prior to weighing.
8.0 Sample Collection, Preservation, Storage, and Transport
8.1 Qualifications. This is a complex test method. To obtain reliable results, you should be trained and experienced with in-stack filtration systems (such as cyclones, impactors, and thimbles) and impinger and moisture train systems.
8.2 Preparations. Follow the pretest preparation instructions in Section 8.1 of Method 5 of appendix A-3 to part 60.
8.3 Site Setup. You must complete the following to properly set up for this test:
(a) Determine the sampling site location and traverse points.
(b) Calculate probe/cyclone blockage.
(c) Verify the absence of cyclonic flow.
(d) Complete a preliminary velocity profile and select a nozzle(s) and sampling rate.
8.3.1 Sampling Site Location and Traverse Point Determination. Follow the standard procedures in Method 1 of appendix A-1 to part 60 to select the appropriate sampling site. Choose a location that maximizes the distance from upstream and downstream flow disturbances.
(a) Traverse points. The required maximum number of total traverse points at any location is 12, as shown in Figure 7 of Section 17. You must prevent the disturbance and capture of any solids accumulated on the inner wall surfaces by maintaining a 1-inch distance from the stack wall (0.5 inch for sampling locations less than 36.4 inches in diameter with the pitot tube and 32.4 inches without the pitot tube). During sampling, when the PM2.5 cyclone is used without the PM10, traverse points closest to the stack walls may not be reached because the inlet to a PM2.5 cyclone is located approximately 2.75 inches from the end of the cyclone. For these cases, you may collect samples using the procedures in Section 11.3.2.2 of Method 1 of appendix A-3 to part 60. You must use the traverse point closest to the unreachable sampling points as replacement for the unreachable points. You must extend the sampling time at the replacement sampling point to include the duration of the unreachable traverse points.
(b) Round or rectangular duct or stack. If a duct or stack is round with two ports located 90° apart, use six sampling points on each diameter. Use a 3x4 sampling point layout for rectangular ducts or stacks. Consult with the Administrator to receive approval for other layouts before you use them.
(c) Sampling ports. You must determine if the sampling ports can accommodate the in-stack cyclones used in this method. You may need larger diameter sampling ports than those used by Method 5 of appendix A-3 to part 60 or Method 17 of appendix A-6 to part 60 for total filterable particulate sampling. When you use nozzles smaller than 0.16 inch in diameter and either a PM10 or a combined PM10 and PM2.5 sampling apparatus, the sampling port diameter may need to be six inches in diameter to accommodate the entire apparatus because the conventional 4-inch diameter port may be too small due to the combined dimension of the PM10 cyclone and the nozzle extending from the cyclone, which will likely exceed the internal diameter of the port. A 4-inch port should be adequate for the single PM2.5 sampling apparatus. However, do not use the conventional 4-inch diameter port in any circumstances in which the combined dimension of the cyclone and the nozzle extending from the cyclone exceeds the internal diameter of the port. (Note: If the port nipple is short, you may be able to "hook" the sampling head through a smaller port into the duct or stack.)
8.3.2 Probe/Cyclone Blockage Calculations. Follow the procedures in the next two sections, as appropriate.
8.3.2.1 Ducts with diameters greater than 36.4 inches. Based on commercially available cyclone assemblies for this procedure, ducts with diameters greater than 36.4 inches have blockage effects less than three percent, as illustrated in Figure 8 of Section 17. You must minimize the blockage effects of the combination of the in-stack nozzle/cyclones, pitot tube, and filter assembly that you use by keeping the cross-sectional area of the assembly at three percent or less of the cross-sectional area of the duct.
8.3.2.2 Ducts with diameters between 25.7 and 36.4 inches. Ducts with diameters between 25.7 and 36.4 inches have blockage effects ranging from three to six percent, as illustrated in Figure 8 of Section 17. Therefore, when you conduct tests on these small ducts, you must adjust the observed velocity pressures for the estimated blockage factor whenever the combined sampling apparatus blocks more than three percent of the stack or duct (see Sections 8.7.2.2 and 8.7.2.3 on the probe blockage factor and the final adjusted velocity pressure, respectively). (Note: Valid sampling with the combined PM2.5/PM10 cyclones cannot be performed with this method if the average stack blockage from the sampling assembly is greater than six percent, i.e., the stack diameter is less than 26.5 inches.)
8.3.3 Cyclonic Flow. Do not use the combined cyclone sampling head at sampling locations subject to cyclonic flow. Also, you must follow procedures in Method 1 of appendix A-1 to part 60 to determine the presence or absence of cyclonic flow and then perform the following calculations:
(a) As per Section 11.4 of Method 1 of appendix A-1 to part 60, find and record the angle that has a null velocity pressure for each traverse point using an S-type pitot tube.
(b) Average the absolute values of the angles that have a null velocity pressure. Do not use the sampling location if the average absolute value exceeds 20°. (Note: You can minimize the effects of cyclonic flow conditions by moving the sampling location, placing gas flow straighteners upstream of the sampling location, or applying a modified sampling approach as described in EPA Guideline Document GD-008, Particulate Emissions Sampling in Cyclonic Flow. You may need to obtain an alternate method approval from the regulatory authority that established the requirement to use this test method prior to using a modified sampling approach.)
8.3.4 Preliminary Velocity Profile. Conduct a preliminary velocity traverse by following Method 2 of appendix A-1 to part 60 velocity traverse procedures. The purpose of the preliminary velocity profile is to determine all of the following:
(a) The gas sampling rate for the combined probe/cyclone sampling head in order to meet the required particle size cut.
(b) The appropriate nozzle to maintain the required gas sampling rate for the velocity pressure range and isokinetic range. If the isokinetic range cannot be met (e.g., batch processes, extreme process flow or temperature variation), void the sample or use methods subject to the approval of the Administrator to correct the data. The acceptable variation from isokinetic sampling is 80 to 120 percent and no more than 100 ± 21 percent (2 out of 12 or 5 out of 24) sampling points outside of this criteria.
(c) The necessary sampling duration to obtain sufficient particulate catch weights.
8.3.4.1 Preliminary traverse. You must use an S-type pitot tube with a conventional thermocouple to conduct the traverse. Conduct the preliminary traverse as close as possible to the anticipated testing time on sources that are subject to hour-by-hour gas flow rate variations of approximately ± 20 percent and/or gas temperature variations of approximately ± 28°C (± 50°F). (Note: You should be aware that these variations can cause errors in the cyclone cut diameters and the isokinetic sampling velocities.)
8.3.4.2 Velocity pressure range. Insert the S-type pitot tube at each traverse point and record the range of velocity pressures measured on data form in Method 2 of appendix A-1 to part 60. You will use this later to select the appropriate nozzle.
8.3.4.3 Initial gas stream viscosity and molecular weight. Determine the average gas temperature, average gas oxygen content, average carbon dioxide content, and estimated moisture content. You will use this information to calculate the initial gas stream viscosity (Equation 3) and molecular weight (Equations 1 and 2). (Note: You must follow the instructions outlined in Method 4 of appendix A-3 to part 60 or Alternative Moisture Measurement Method Midget Impingers (ALT-008) to estimate the moisture content. You may use a wet bulb-dry bulb measurement or hand-held hygrometer measurement to estimate the moisture content of sources with gas temperatures less than 71°C (160°F).)
8.3.4.4 Approximate PM concentration in the gas stream. Determine the approximate PM concentration for the PM2.5 and the PM2.5 to PM10 components of the gas stream through qualitative measurements or estimates from precious stack particulate emissions tests. Having an idea of the particulate concentration in the gas stream is not essential but will help you determine the appropriate sampling time to acquire sufficient PM weight for better accuracy at the source emission level. The collectible PM weight requirements depend primarily on the types of filter media and weighing capabilities that are available and needed to characterize the emissions. Estimate the collectible PM concentrations in the greater than 10 micrometer, less than or equal to 10 micrometers and greater than 2.5 micrometers, and less than or equal to 2.5 micrometer size ranges. Typical PM concentrations are listed in Table 1 of Section 17. Additionally, relevant sections of AP-42, Compilation of Air Pollutant Emission Factors, may contain particle size distributions for processes characterized in those sections, and appendix B2 of AP-42 contains generalized particle size distributions for nine industrial process categories (e.g., stationary internal combustion engines firing gasoline or diesel fuel, calcining of aggregate or unprocessed ores). The generalized particle size distributions can be used if source-specific particle size distributions are unavailable. Appendix B2 of AP-42 also contains typical collection efficiencies of various particulate control devices and example calculations showing how to estimate uncontrolled total particulate emissions, uncontrolled size-specific emissions, and controlled size-specific particulate emissions. (http://www.epa.gov/ttnchie1/ap42.)
8.4 Pre-test Calculations. You must perform pre-test calculations to help select the appropriate gas sampling rate through cyclone I (PM10) and cyclone IV (PM2.5). Choosing the appropriate sampling rate will allow you to maintain the appropriate particle cut diameters based upon preliminary gas stream measurements, as specified in Table 2 of Section 17.
8.4.1 Gas Sampling Rate. The gas sampling rate is defined by the performance curves for both cyclones, as illustrated in Figure 10 of Section 17. You must use the calculations in Section 8.5 to achieve the appropriate cut size specification for each cyclone. The optimum gas sampling rate is the overlap zone defined as the range below the cyclone IV 2.25 micrometer curve down to the cyclone I 11.0 micrometer curve (area between the two dark, solid lines in Figure 10 of Section 17).
8.4.2 Choosing the Appropriate Sampling Rate. You must select a gas sampling rate in the middle of the overlap zone (discussed in Section 8.4.1), as illustrated in Figure 10 of Section 17, to maximize the acceptable tolerance for slight variations in flow characteristics at the sampling location. The overlap zone is also a weak function of the gas composition. (Note: The acceptable range is limited, especially for gas streams with temperatures less than approximately 100°F. At lower temperatures, it may be necessary to perform the PM10 and PM2.5 separately in order to meet the necessary particle size criteria shown in Table 2 of Section 17.)
8.5 Test Calculations. You must perform all of the calculations in Table 3 of Section 17 and the calculations described in Sections 8.5.1 through 8.5.5.
8.5.1 Assumed Reynolds Number. You must select an assumed Reynolds number (Nre) using Equation 10 and an estimated sampling rate or from prior experience under the stack conditions determined using Methods 1 through 4 to part 60. You will perform initial test calculations based on an assumed Nre for the test to be performed. You must verify the assumed Nre by substituting the sampling rate (Qs) calculated in Equation 7 into Equation 10. Then use Table 5 of Section 17 to determine if the Nre used in Equation 5 was correct.
8.5.2 Final Sampling Rate. Recalculate the final Qs if the assumed Nre used in your initial calculation is not correct. Use Equation 7 to recalculate the optimum Qs.
8.5.3 Meter Box ΔH. Use Equation 11 to calculate the meter box orifice pressure drop (ΔH) after you calculate the optimum sampling rate and confirm the Nre. (Note: The stack gas temperature may vary during the test, which could affect the sampling rate. If the stack gas temperature varies, you must make slight adjustments in the meter box ΔH to maintain the correct constant cut diameters. Therefore, use Equation 11 to recalculate the ΔH values for 50°F above and below the stack temperature measured during the preliminary traverse (see Section 8.3.4.1), and document this information in Table 4 of Section 17.)
8.5.4 Choosing a Sampling Nozzle. Select one or more nozzle sizes to provide for near isokinetic sampling rate (see Section 1.6). This will also minimize an isokinetic sampling error for the particles at each point. First calculate the mean stack gas velocity (vs) using Equation 13. See Section 8.7.2 for information on correcting for blockage and use of different pitot tube coefficients. Then use Equation 14 to calculate the diameter (D) of a nozzle that provides for isokinetic sampling at the mean vs at flow Qs. From the available nozzles one size smaller and one size larger than this diameter, D, select the most appropriate nozzle. Perform the following steps for the selected nozzle.
8.5.4.1 Minimum/maximum nozzle/stack velocity ratio. Use Equation 15 to determine the velocity of gas in the nozzle. Use Equation 16 to calculate the minimum nozzle/stack velocity ratio (Rmin). Use Equation 17 to calculate the maximum nozzle/stack velocity ratio (Rmax).
8.5.4.2 Minimum gas velocity. Use Equation 18 to calculate the minimum gas velocity (vmin) if Rmin is an imaginary number (negative value under the square root function) or if Rmin is less than 0.5. Use Equation 19 to calculate vmin if Rmin is ‰¥0.5.
8.5.4.3 Maximum stack velocity. Use Equation 20 to calculate the maximum stack velocity (vmax) if Rmax is less than 1.5. Use Equation 21 to calculate the stack velocity if Rmax is ‰¥1.5.
8.5.4.4 Conversion of gas velocities to velocity pressure. Use Equation 22 to convert vmin to minimum velocity pressure, Δpmin. Use Equation 23 to convert vmax to maximum velocity pressure, Δpmax.
8.5.4.5 Comparison to observed velocity pressures. Compare minimum and maximum velocity pressures with the observed velocity pressures at all traverse points during the preliminary test (see Section 8.3.4.2).
8.5.5 Optimum Sampling Nozzle. The nozzle you selected is appropriate if all the observed velocity pressures during the preliminary test fall within the range of the Δpmin and Δpmax. Make sure the following requirements are met then follow the procedures in Sections 8.5.5.1 and 8.5.5.2.
(a) Choose an optimum nozzle that provides for isokinetic sampling conditions as close to 100 percent as possible. This is prudent because even if there are slight variations in the gas flow rate, gas temperature, or gas composition during the actual test, you have the maximum assurance of satisfying the isokinetic criteria. Generally, one of the two candidate nozzles selected will be closer to optimum (see Section 8.5.4).
(b) When testing is for PM2.5 only, you are allowed a 16 percent failure rate, rounded to the nearest whole number, of sampling points that are outside the range of the Δpmin and Δpmax. If the coarse fraction for PM10 determination is included, you are allowed only an eight percent failure rate of the sampling points, rounded to the nearest whole number, outside the Δpmin and Δpmax.
8.5.5.1 Precheck. Visually check the selected nozzle for dents before use.
8.5.5.2 Attach the pre-selected nozzle. Screw the pre-selected nozzle onto the main body of cyclone I using fluoropolymer tape. Use a union and cascade adaptor to connect the cyclone IV inlet to the outlet of cyclone I (see Figure 2 of Section 17).
8.6 Sampling Train Preparation. A schematic of the sampling train used in this method is shown in Figure 1 of Section 17. First, assemble the train and complete the leak check on the combined cyclone sampling head and pitot tube. Use the following procedures to prepare the sampling train. (Note: Do not contaminate the sampling train during preparation and assembly. Keep all openings, where contamination can occur, covered until just prior to assembly or until sampling is about to begin.)
8.6.1 Sampling Head and Pitot Tube. Assemble the combined cyclone train. The O-rings used in the train have a temperature limit of approximately 205°C (400°F). Use cyclones with stainless steel sealing rings for stack temperatures above 205°C (400°F) up to 260°C (500°F). You must also keep the nozzle covered to protect it from nicks and scratches. This method may not be suitable for sources with stack gas temperatures exceeding 260°C (500°F) because the threads of the cyclone components may gall or seize, thus preventing the recovery of the collected PM and rendering the cyclone unusable for subsequent use. You may use stainless steel cyclone assemblies constructed with bolt-together rather than screw-together assemblies at temperatures up to 538°C (1,000°F). You must use "break-away" or expendable stainless steel bolts that can be over-torqued and broken if necessary to release cyclone closures, thus allowing you to recover PM without damaging the cyclone flanges or contaminating the samples. You may need to use specialty metals to achieve reliable particulate mass measurements above 538°C (1,000°F). The method can be used at temperatures up to 1,371°C (2,500°F) using specially constructed high-temperature stainless steel alloys (Hastelloy or Haynes 230) with bolt-together closures using break-away bolts.
8.6.2 Filterable Particulate Filter Holder and Pitot Tube. Attach the pre-selected filter holder to the end of the combined cyclone sampling head (see Figure 2 of Section 17). Attach the S-type pitot tube to the combined cyclones after the sampling head is fully attached to the end of the probe. (Note: The pitot tube tip must be mounted slightly beyond the combined head cyclone sampling assembly and at least one inch off the gas flow path into the cyclone nozzle. This is similar to the pitot tube placement in Method 17 of appendix A-6 to part 60.) Securely fasten the sensing lines to the outside of the probe to ensure proper alignment of the pitot tube. Provide unions on the sensing lines so that you can connect and disconnect the S-type pitot tube tips from the combined cyclone sampling head before and after each run. Calibrate the pitot tube on the sampling head according to the most current ASTM International D3796 because the cyclone body is a potential source flow disturbance and will change the pitot coefficient value from the baseline (isolated tube) value.
8.6.3 Filter. You must number and tare the filters before use. To tare the filters, desiccate each filter at 20 ±5.6°C (68 ±10°F) and ambient pressure for at least 24 hours and weigh at intervals of at least six hours to a constant weight. (See Section 3.0 for a definition of constant weight.) Record results to the nearest 0.1 mg. During each weighing, the filter must not be exposed to the laboratory atmosphere for longer than two minutes and a relative humidity above 50 percent. Alternatively, the filters may be oven-dried at 104°C (220°F) for two to three hours, desiccated for two hours, and weighed. Use tweezers or clean disposable surgical gloves to place a labeled (identified) and pre-weighed filter in the filter holder. You must center the filter and properly place the gasket so that the sample gas stream will not circumvent the filter. The filter must not be compressed between the gasket and the filter housing. Check the filter for tears after the assembly is completed. Then screw or clamp the filter housing together to prevent the seal from leaking.
8.6.4 Moisture Trap. If you are measuring only filterable particulate (or you are sure that the gas filtration temperature will be maintained below 30°C (85°F)), then an empty modified Greenburg Smith impinger followed by an impinger containing silica gel is required. Alternatives described in Method 5 of appendix A-3 to part 60 may also be used to collect moisture that passes through the ambient filter. If you are measuring condensable PM in combination with this method, then follow the procedures in Method 202 of appendix M of this part for moisture collection.
8.6.5 Leak Check. Use the procedures outlined in Section 8.4 of Method 5 of appendix A-3 to part 60 to leak check the entire sampling system. Specifically perform the following procedures:
8.6.5.1 Sampling train. You must pretest the entire sampling train for leaks. The pretest leak check must have a leak rate of not more than 0.02 actual cubic feet per minute or four percent of the average sample flow during the test run, whichever is less. Additionally, you must conduct the leak check at a vacuum equal to or greater than the vacuum anticipated during the test run. Enter the leak check results on the analytical data sheet (see Section 11.1) for the specific test. (Note: Do not conduct a leak check during port changes.)
8.6.5.2 Pitot tube assembly. After you leak check the sample train, perform a leak check of the pitot tube assembly. Follow the procedures outlined in Section 8.4.1 of Method 5 of appendix A-3 to part 60.
8.6.6 Sampling Head. You must preheat the combined sampling head to the stack temperature of the gas stream at the test location (±28 °C, ±50 °F). This will heat the sampling head and prevent moisture from condensing from the sample gas stream.
8.6.6.1 Warmup. You must complete a passive warmup (of 30-40 min) within the stack before the run begins to avoid internal condensation.
8.6.6.2 Shortened warmup. You can shorten the warmup time by thermostated heating outside the stack (such as by a heat gun). Then place the heated sampling head inside the stack and allow the temperature to equilibrate.
8.7 Sampling Train Operation. Operate the sampling train the same as described in Section 4.1.5 of Method 5 of appendix A-3 to part 60, but use the procedures in this section for isokinetic sampling and flow rate adjustment. Maintain the flow rate calculated in Section 8.4.1 throughout the run, provided the stack temperature is within 28°C (50°F) of the temperature used to calculate ΔH. If stack temperatures vary by more than 28°C (50°F), use the appropriate ΔH value calculated in Section 8.5.3. Determine the minimum number of traverse points as in Figure 7 of Section 17. Determine the minimum total projected sampling time based on achieving the data quality objectives or emission limit of the affected facility. We recommend that you round the number of minutes sampled at each point to the nearest 15 seconds. Perform the following procedures:
8.7.1 Sample Point Dwell Time. You must calculate the flow rate-weighted dwell time (that is, sampling time) for each sampling point to ensure that the overall run provides a velocity-weighted average that is representative of the entire gas stream. Vary the dwell time at each traverse point proportionately with the point velocity. Calculate the dwell time at each of the traverse points using Equation 24. You must use the data from the preliminary traverse to determine the average velocity pressure (Δpavg). You must use the velocity pressure measured during the sampling run to determine the velocity pressure at each point (Δpn). Here, Ntp equals the total number of traverse points. Each traverse point must have a dwell time of at least two minutes.
8.7.2 Adjusted Velocity Pressure. When selecting your sampling points using your preliminary velocity traverse data, your preliminary velocity pressures must be adjusted to take into account the increase in velocity due to blockage. Also, you must adjust your preliminary velocity data for differences in pitot tube coefficients. Use the following instructions to adjust the preliminary velocity pressure.
8.7.2.1 Different pitot tube coefficient. You must use Equation 25 to correct the recorded preliminary velocity pressures if the pitot tube mounted on the combined cyclone sampling head has a different pitot tube coefficient than the pitot tube used during the preliminary velocity traverse (see Section 8.3.4).
8.7.2.2 Probe blockage factor. You must use Equation 26 to calculate an average probe blockage correction factor (bf) if the diameter of your stack or duct is between 25.7 and 36.4 inches for the combined PM2.5/PM10 sampling head and pitot and between 18.8 and 26.5 inches for the PM2.5 cyclone and pitot. A probe blockage factor is calculated because of the flow blockage caused by the relatively large cross-sectional area of the cyclone sampling head, as discussed in Section 8.3.2.2 and illustrated in Figures 8 and 9 of Section 17. You must determine the cross-sectional area of the cyclone head you use and determine its stack blockage factor. (Note: Commercially-available sampling heads (including the PM10 cyclone, PM2.5 cyclone, pitot and filter holder) have a projected area of approximately 31.2 square inches when oriented into the gas stream.) As the probe is moved from the outermost to the innermost point, the amount of blockage that actually occurs ranges from approximately 13 square inches to the full 31.2 square inches plus the blockage caused by the probe extension. The average cross-sectional area blocked is 22 square inches.
8.7.2.3 Final adjusted velocity pressure. Calculate the final adjusted velocity pressure (Δps2) using Equation 27. (Note: Figures 8 and 9 of Section 17 illustrate that the blockage effect of the combined PM10, PM2.5 cyclone sampling head, and pitot tube increases rapidly below stack diameters of 26.5 inches. Therefore, the combined PM10, PM2.5 filter sampling head and pitot tube is not applicable for stacks with a diameter less than 26.5 inches because the blockage is greater than six percent. For stacks with a diameter less than 26.5 inches, PM2.5 particulate measurements may be possible using only a PM2.5 cyclone, pitot tube, and in-stack filter. If the blockage exceeds three percent but is less than six percent, you must follow the procedures outlined in Method 1A of appendix A-1 to part 60 to conduct tests. You must conduct the velocity traverse downstream of the sampling location or immediately before the test run.
8.7.3 Sample Collection. Collect samples the same as described in Section 4.1.5 of Method 5 of appendix A-3 to part 60, except use the procedures in this section for isokinetic sampling and flow rate adjustment. Maintain the flow rate calculated in Section 8.5 throughout the run, provided the stack temperature is within 28°C (50°F) of the temperature used to calculate ΔH. If stack temperatures vary by more than 28°C (50°F), use the appropriate ΔH value calculated in Section 8.5.3. Calculate the dwell time at each traverse point as in Equation 24. In addition to these procedures, you must also use running starts and stops if the static pressure at the sampling location is less than minus 5 inches water column. This prevents back pressure from rupturing the sample filter. If you use a running start, adjust the flow rate to the calculated value after you perform the leak check (see Section 8.4).
8.7.3.1 Level and zero manometers. Periodically check the level and zero point of the manometers during the traverse. Vibrations and temperature changes may cause them to drift.
8.7.3.2 Portholes. Clean the portholes prior to the test run. This will minimize the chance of collecting deposited material in the nozzle.
8.7.3.3 Sampling procedures. Verify that the combined cyclone sampling head temperature is at stack temperature. You must maintain the temperature of the cyclone sampling head within ±10°C (±18°F) of the stack temperature. (Note: For many stacks, portions of the cyclones and filter will be external to the stack during part of the sampling traverse. Therefore, you must heat and/or insulate portions of the cyclones and filter that are not within the stack in order to maintain the sampling head temperature at the stack temperature. Maintaining the temperature will ensure proper particle sizing and prevent condensation on the walls of the cyclones.) To begin sampling, remove the protective cover from the nozzle. Position the probe at the first sampling point with the nozzle pointing directly into the gas stream. Immediately start the pump and adjust the flow to calculated isokinetic conditions. Ensure the probe/pitot tube assembly is leveled. (Note: When the probe is in position, block off the openings around the probe and porthole to prevent unrepresentative dilution of the gas stream. Take care to minimize contamination from material used to block the flow or insulate the sampling head during collection at the first sampling point.)
(a) Traverse the stack cross-section, as required by Method 1 of appendix A-1 to part 60, with the exception that you are only required to perform a 12-point traverse. Do not bump the cyclone nozzle into the stack walls when sampling near the walls or when removing or inserting the probe through the portholes. This will minimize the chance of extracting deposited materials.
(b) Record the data required on the field test data sheet for each run. Record the initial dry gas meter reading. Then take dry gas meter readings at the following times: the beginning and end of each sample time increment; when changes in flow rates are made; and when sampling is halted. Compare the velocity pressure measurements (Equations 22 and 23) with the velocity pressure measured during the preliminary traverse. Keep the meter box ΔH at the value calculated in Section 8.5.3 for the stack temperature that is observed during the test. Record all point-by-point data and other source test parameters on the field test data sheet. Do not leak check the sampling system during port changes.
(c) Maintain flow until the sampling head is completely removed from the sampling port. You must restart the sampling flow prior to inserting the sampling head into the sampling port during port changes.
(d) Maintain the flow through the sampling system at the last sampling point. At the conclusion of the test, remove the pitot tube and combined cyclone sampling head from the stack while the train is still operating (running stop). Make sure that you do not scrape the pitot tube or the combined cyclone sampling head against the port or stack walls. Then stop the pump and record the final dry gas meter reading and other test parameters on the field test data sheet. (Note: After you stop the pump, make sure you keep the combined cyclone head level to avoid tipping dust from the cyclone cups into the filter and/or down-comer lines.)
8.7.4 Process Data. You must document data and information on the process unit tested, the particulate control system used to control emissions, any non-particulate control system that may affect particulate emissions, the sampling train conditions, and weather conditions. Record the site barometric pressure and stack pressure on the field test data sheet. Discontinue the test if the operating conditions may cause non-representative particulate emissions.
8.7.4.1 Particulate control system data. Use the process and control system data to determine whether representative operating conditions were maintained throughout the testing period.
8.7.4.2 Sampling train data. Use the sampling train data to confirm that the measured particulate emissions are accurate and complete.
8.7.5 Sample Recovery. First remove the sampling head (combined cyclone/filter assembly) from the train probe. After the sample head is removed, perform a post-test leak check of the probe and sample train. Then recover the components from the cyclone/filter. Refer to the following sections for more detailed information.
8.7.5.1 Remove sampling head. After cooling and when the probe can be safely handled, wipe off all external surfaces near the cyclone nozzle and cap the inlet to the cyclone to prevent PM from entering the assembly. Remove the combined cyclone/filter sampling head from the probe. Cap the outlet of the filter housing to prevent PM from entering the assembly.
8.7.5.2 Leak check probe/sample train assembly (post-test). Leak check the remainder of the probe and sample train assembly (including meter box) after removing the combined cyclone head/filter. You must conduct the leak rate at a vacuum equal to or greater than the maximum vacuum achieved during the test run. Enter the results of the leak check onto the field test data sheet. If the leak rate of the sampling train (without the combined cyclone sampling head) exceeds 0.02 actual cubic feet per minute or four percent of the average sampling rate during the test run (whichever is less), the run is invalid and must be repeated.
8.7.5.3 Weigh or measure the volume of the liquid collected in the water collection impingers and silica trap. Measure the liquid in the first impingers to within 1 ml using a clean graduated cylinder or by weighing it to within 0.5 g using a balance. Record the volume of the liquid or weight of the liquid present to be used to calculate the moisture content of the effluent gas.
8.7.5.4 Weigh the silica impinger. If a balance is available in the field, weigh the silica impinger to within 0.5 g. Note the color of the indicating silica gel in the last impinger to determine whether it has been completely spent and make a notation of its condition. If you are measuring CPM in combination with this method, the weight of the silica gel can be determined before or after the post-test nitrogen purge is complete (See Section 8.5.3 of Method 202 of appendix M to this part).
8.7.5.5 Recovery of PM. Recovery involves the quantitative transfer of particles in the following size range: greater than 10 micrometers; less than or equal to 10 micrometers but greater than 2.5 micrometers; and less than or equal to 2.5 micrometers. You must use a nylon or fluoropolymer brush and an acetone rinse to recover particles from the combined cyclone/filter sampling head. Use the following procedures for each container:
(a) Container #1, Less than or equal to PM2.5 micrometer filterable particulate. Use tweezers and/or clean disposable surgical gloves to remove the filter from the filter holder. Place the filter in the Petri dish that you labeled with the test identification and Container #1. Using a dry brush and/or a sharp-edged blade, carefully transfer any PM and/or filter fibers that adhere to the filter holder gasket or filter support screen to the Petri dish. Seal the container. This container holds particles less than or equal to 2.5 micrometers that are caught on the in-stack filter. (Note: If the test is conducted for PM10 only, then Container #1 would be for less than or equal to PM10 micrometer filterable particulate.)
(b) Container #2, Greater than PM10micrometer filterable particulate. Quantitatively recover the PM from the cyclone I cup and brush cleaning and acetone rinses of the cyclone cup, internal surface of the nozzle, and cyclone I internal surfaces, including the outside surface of the downcomer line. Seal the container and mark the liquid level on the outside of the container you labeled with test identification and Container #2. You must keep any dust found on the outside of cyclone I and cyclone nozzle external surfaces out of the sample. This container holds PM greater than 10 micrometers.
(c) Container #3, Filterable particulate less than or equal to 10 micrometer and greater than 2.5 micrometers. Place the solids from cyclone cup IV and the acetone (and brush cleaning) rinses of the cyclone I turnaround cup (above inner downcomer line), inside of the downcomer line, and interior surfaces of cyclone IV into Container #3. Seal the container and mark the liquid level on the outside of the container you labeled with test identification and Container #3. This container holds PM less than or equal to 10 micrometers but greater than 2.5 micrometers.
(d) Container #4, Less than or equal to PM2.5micrometers acetone rinses of the exit tube of cyclone IV and front half of the filter holder. Place the acetone rinses (and brush cleaning) of the exit tube of cyclone IV and the front half of the filter holder in container #4. Seal the container and mark the liquid level on the outside of the container you labeled with test identification and Container #4. This container holds PM that is less than or equal to 2.5 micrometers.
(e) Container #5, Cold impinger water. If the water from the cold impinger used for moisture collection has been weighed in the field, it can be discarded. Otherwise, quantitatively transfer liquid from the cold impinger that follows the ambient filter into a clean sample bottle (glass or plastic). Mark the liquid level on the bottle you labeled with test identification and Container #5. This container holds the remainder of the liquid water from the emission gases. If you collected condensable PM using Method 202 of appendix M to this part in conjunction with using this method, you must follow the procedures in Method 202 of appendix M to this part to recover impingers and silica used to collect moisture.
(f) Container #6, Silica gel absorbent. Transfer the silica gel to its original container labeled with test identification and Container #6 and seal. A funnel may make it easier to pour the silica gel without spilling. A rubber policeman may be used as an aid in removing the silica gel from the impinger. It is not necessary to remove the small amount of silica gel dust particles that may adhere to the impinger wall and are difficult to remove. Since the gain in weight is to be used for moisture calculations, do not use any water or other liquids to transfer the silica gel. If the silica gel has been weighed in the field to measure water content, it can be discarded. Otherwise, the contents of Container #6 are weighed during sample analysis.
(g) Container #7, Acetone field reagent blank. Take approximately 200 ml of the acetone directly from the wash bottle you used and place it in Container #7 labeled "Acetone Field Reagent Blank."
8.7.6 Transport Procedures. Containers must remain in an upright position at all times during shipping. You do not have to ship the containers under dry or blue ice.
9.0 Quality Control
9.1 Daily Quality Checks. You must perform daily quality checks of field log books and data entries and calculations using data quality indicators from this method and your site-specific test plan. You must review and evaluate recorded and transferred raw data, calculations, and documentation of testing procedures. You must initial or sign log book pages and data entry forms that were reviewed.
9.2 Calculation Verification. Verify the calculations by independent, manual checks. You must flag any suspect data and identify the nature of the problem and potential effect on data quality. After you complete the test, prepare a data summary and compile all the calculations and raw data sheets.
9.3 Conditions. You must document data and information on the process unit tested, the particulate control system used to control emissions, any non-particulate control system that may affect particulate emissions, the sampling train conditions, and weather conditions. Discontinue the test if the operating conditions may cause non-representative particulate emissions.
9.4 Field Analytical Balance Calibration Check. Perform calibration check procedures on field analytical balances each day that they are used. You must use National Institute of Standards and Technology (NIST)-traceable weights at a mass approximately equal to the weight of the sample plus container you will weigh.
10.0 Calibration and Standardization
Maintain a log of all filterable particulate sampling and analysis calibrations. Include copies of the relevant portions of the calibration and field logs in the final test report.
10.1 Gas Flow Velocities. You must use an S-type pitot tube that meets the required EPA specifications (EPA Publication 600/4-77-0217b) during these velocity measurements. (Note: If, as specified in Section 8.7.2.3, testing is performed in stacks less than 26.5 inches in diameter, testers may use a standard pitot tube according to the requirements in Method 1 or 2 of appendix A-3 to part 60 of this chapter.) You must also complete the following:
(a) Visually inspect the S-type pitot tube before sampling.
(b) Leak check both legs of the pitot tube before and after sampling.
(c) Maintain proper orientation of the S-type pitot tube while making measurements.
10.1.1 S-type Pitot Tube Orientation. The S-type pitot tube is properly oriented when the yaw and the pitch axis are 90 degrees to the air flow.
10.1.2 Average Velocity Pressure Record. Instead of recording either high or low values, record the average velocity pressure at each point during flow measurements.
10.1.3 Pitot Tube Coefficient. Determine the pitot tube coefficient based on physical measurement techniques described in Method 2 of appendix A-1 to part 60. (Note: You must calibrate the pitot tube on the sampling head because of potential interferences from the cyclone body. Refer to Section 8.7.2 for additional information.)
10.2 Thermocouple Calibration. You must calibrate the thermocouples using the procedures described in Section 10.3.1 of Method 2 of appendix A-1 to part 60 or Alternative Method 2 Thermocouple Calibration (ALT-011). Calibrate each temperature sensor at a minimum of three points over the anticipated range of use against a NIST-traceable thermometer. Alternatively, a reference thermocouple and potentiometer calibrated against NIST standards can be used.
10.3 Nozzles. You may use stainless steel (316 or equivalent), high-temperature steel alloy, or fluoropolymer-coated nozzles for isokinetic sampling. Make sure that all nozzles are thoroughly cleaned, visually inspected, and calibrated according to the procedure outlined in Section 10.1 of Method 5 of appendix A-3 to part 60.
10.4 Dry Gas Meter Calibration. Calibrate your dry gas meter following the calibration procedures in Section 16.1 of Method 5 of appendix A-3 to part 60. Also, make sure you fully calibrate the dry gas meter to determine the volume correction factor prior to field use. Post-test calibration checks must be performed as soon as possible after the equipment has been returned to the shop. Your pre-test and post-test calibrations must agree within ±5 percent.
11.0 Analytical Procedures
11.1 Analytical Data Sheet. Record all data on the analytical data sheet. Obtain the data sheet from Figure 5-6 of Method 5 of appendix A-3 to part 60. Alternatively, data may be recorded electronically using software applications such as the Electronic Reporting Tool located at http://www.epa.gov/ttn/chief/ert/ert_tool.html.
11.2 Dry Weight of PM. Determine the dry weight of particulate following procedures outlined in this section.
11.2.1 Container #1, Less than or Equal to PM2.5 Micrometer Filterable Particulate. Transfer the filter and any loose particulate from the sample container to a tared weighing dish or pan that is inert to solvent or mineral acids. Desiccate for 24 hours in a dessicator containing anhydrous calcium sulfate. Weigh to a constant weight and report the results to the nearest 0.1 mg. (See Section 3.0 for a definition of Constant weight.) If constant weight requirements cannot be met, the filter must be treated as described in Section 11.2.1 of Method 202 of appendix M to this part. Note: The nozzle and front half wash and filter collected at or below 30°C (85°F) may not be heated and must be maintained at or below 30°C (85°F).
11.2.2 Container #2, Greater than PM10 Micrometer Filterable Particulate Acetone Rinse. Separately treat this container like Container #4.
11.2.3 Container #3, Filterable Particulate Less than or Equal to 10 Micrometer and Greater than 2.5 Micrometers Acetone Rinse. Separately treat this container like Container #4.
11.2.4 Container #4, Less than or Equal to PM2.5 Micrometers Acetone Rinse of the Exit Tube of Cyclone IV and Front Half of the Filter Holder. Note the level of liquid in the container and confirm on the analysis sheet whether leakage occurred during transport. If a noticeable amount of leakage has occurred, either void the sample or use methods (subject to the approval of the Administrator) to correct the final results. Quantitatively transfer the contents to a tared 250 ml beaker or tared fluoropolymer beaker liner, and evaporate to dryness at room temperature and pressure in a laboratory hood. Desiccate for 24 hours and weigh to a constant weight. Report the results to the nearest 0.1 mg.
11.2.5 Container #5, Cold Impinger Water. If the amount of water has not been determined in the field, note the level of liquid in the container and confirm on the analysis sheet whether leakage occurred during transport. If a noticeable amount of leakage has occurred, either void the sample or use methods (subject to the approval of the Administrator) to correct the final results. Measure the liquid in this container either volumetrically to ±1 ml or gravimetrically to ±0.5 g.
11.2.6 Container #6, Silica Gel Absorbent. Weigh the spent silica gel (or silica gel plus impinger) to the nearest 0.5 g using a balance. This step may be conducted in the field.
11.2.7 Container #7, Acetone Field Reagent Blank. Use 150 ml of acetone from the blank container used for this analysis. Transfer 150 ml of the acetone to a clean 250-ml beaker or tared fluoropolymer beaker liner. Evaporate the acetone to dryness at room temperature and pressure in a laboratory hood. Following evaporation, desiccate the residue for 24 hours in a desiccator containing anhydrous calcium sulfate. Weigh and report the results to the nearest 0.1 mg.
12.0 Calculations and Data Analysis
12.1 Nomenclature. Report results in International System of Units (SI units) unless the regulatory authority that established the requirement to use this test method specifies reporting in English units. The following nomenclature is used.
A = Area of stack or duct at sampling location, square inches.
An = Area of nozzle, square feet.
bf = Average blockage factor calculated in Equation 26, dimensionless.
Bws = Moisture content of gas stream, fraction (e.g., 10 percent H2O is Bws = 0.10).
C = Cunningham correction factor for particle diameter, Dp, and calculated using the actual stack gas temperature, dimensionless.
%CO2 = Carbon Dioxide content of gas stream, percent by volume.
Ca = Acetone blank concentration, mg/mg.
CfPM10 = Conc. of filterable PM10, gr/DSCF.
CfPM2.5 = Conc. of filterable PM2.5, gr/DSCF.
Cp = Pitot coefficient for the combined cyclone pitot, dimensionless.
Cp€² = Coefficient for the pitot used in the preliminary traverse, dimensionless.
Cr = Re-estimated Cunningham correction factor for particle diameter equivalent to the actual cut size diameter and calculated using the actual stack gas temperature, dimensionless.
Ctf = Conc. of total filterable PM, gr/DSCF.
C1 = ˆ’150.3162 (micropoise)
C2 = 18.0614 (micropoise/K 0.5) = 13.4622 (micropoise/R 0.5)
C3 = 1.19183 × 10 6 (micropoise/K 2) = 3.86153 × 10 6 (micropoise/R 2)
C4 = 0.591123 (micropoise)
C5 = 91.9723 (micropoise)
C6 = 4.91705 × 10ˆ’5 (micropoise/K 2) = 1.51761 × 10ˆ’5 (micropoise/R 2)
D = Inner diameter of sampling nozzle mounted on Cyclone I, inches.
Dp = Physical particle size, micrometers.
D50 = Particle cut diameter, micrometers.
D50-1 = Re-calculated particle cut diameters based on re-estimated Cr, micrometers.
D50LL = Cut diameter for cyclone I corresponding to the 2.25 micrometer cut diameter for cyclone IV, micrometers.
D50N = D50 value for cyclone IV calculated during the Nth iterative step, micrometers.
D50(N + 1) = D50 value for cyclone IV calculated during the N + 1 iterative step, micrometers.
D50T = Cyclone I cut diameter corresponding to the middle of the overlap zone shown in Figure 10 of Section 17, micrometers.
I = Percent isokinetic sampling, dimensionless.
Kp = 85.49, ((ft/sec)/(pounds/mole -°R)).
ma = Mass of residue of acetone after evaporation, mg.
Md = Molecular weight of dry gas, pounds/pound mole.
mg = Milligram.
mg/L = Milligram per liter.
Mw = Molecular weight of wet gas, pounds/pound mole.
M1 = Milligrams of PM collected on the filter, less than or equal to 2.5 micrometers.
M2 = Milligrams of PM recovered from Container #2 (acetone blank corrected), greater than 10 micrometers.
M3 = Milligrams of PM recovered from Container #3 (acetone blank corrected), less than or equal to 10 and greater than 2.5 micrometers.
M4 = Milligrams of PM recovered from Container #4 (acetone blank corrected), less than or equal to 2.5 micrometers.
Ntp = Number of iterative steps or total traverse points.
Nre = Reynolds number, dimensionless.
%O2,wet = Oxygen content of gas stream, % by volume of wet gas.
(Note: The oxygen percentage used in Equation 3 is on a wet gas basis. That means that since oxygen is typically measured on a dry gas basis, the measured percent O2 must be multiplied by the quantity (1-Bws) to convert to the actual volume fraction. Therefore, %O2,wet = (1-Bws) * %O2, dry)
Pbar = Barometric pressure, inches Hg.
Ps = Absolute stack gas pressure, inches Hg.
Qs = Sampling rate for cyclone I to achieve specified D50.
QsST = Dry gas sampling rate through the sampling assembly, DSCFM.
QI = Sampling rate for cyclone I to achieve specified D50.
Rmax = Nozzle/stack velocity ratio parameter, dimensionless.
Rmin = Nozzle/stack velocity ratio parameter, dimensionless.
Tm = Meter box and orifice gas temperature,°R.
tn = Sampling time at point n, min.
tr = Total projected run time, min.
Ts = Absolute stack gas temperature,°R.
t1 = Sampling time at point 1, min.
vmax = Maximum gas velocity calculated from Equations 18 or 19, ft/sec.
vmin = Minimum gas velocity calculated from Equations 16 or 17, ft/sec.
vn = Sample gas velocity in the nozzle, ft/sec.
vs = Velocity of stack gas, ft/sec.
Va = Volume of acetone blank, ml.
Vaw = Volume of acetone used in sample recovery wash, ml.
Vc = Quantity of water captured in impingers and silica gel, ml.
Vm = Dry gas meter volume sampled, ACF.
Vms = Dry gas meter volume sampled, corrected to standard conditions, DSCF.
Vws = Volume of water vapor, SCF.
Vic = Volume of impinger contents sample, ml.
Wa = Weight of blank residue in acetone used to recover samples, mg.
W2,3,4 = Weight of PM recovered from Containers #2, #3, and #4, mg.
Z = Ratio between estimated cyclone IV D50 values, dimensionless.
ΔH = Meter box orifice pressure drop, inches W.C.
ΔH@ = Pressure drop across orifice at flow rate of 0.75 SCFM at standard conditions, inches W.C.
(Note: Specific to each orifice and meter box.)
[(Δp) 0.5]avg = Average of square roots of the velocity pressures measured during the preliminary traverse, inches W.C.
Δpm = Observed velocity pressure using S-type pitot tube in preliminary traverse, inches W.C.
Δpavg = Average velocity pressure, inches W.C.
Δpmax = Maximum velocity pressure, inches W.C.
Δpmin = Minimum velocity pressure, inches W.C.
Δpn = Velocity pressure measured at point n during the test run, inches W.C.
Δps = Velocity pressure calculated in Equation 25, inches W.C.
Δps1 = Velocity pressure adjusted for combined cyclone pitot tube, inches W.C.
Δps2 = Velocity pressure corrected for blockage, inches W.C.
Δp1 = Velocity pressure measured at point 1, inches W.C.
γ = Dry gas meter gamma value, dimensionless.
µ = Gas viscosity, micropoise.
θ = Total run time, min.
ρa = Density of acetone, mg/ml (see label on bottle).
12.0 = Constant calculated as 60 percent of 20.5 square inch cross-sectional area of combined cyclone head, square inches.
12.2 Calculations. Perform all of the calculations found in Table 6 of Section 17. Table 6 of Section 17 also provides instructions and references for the calculations.
12.3 Analyses. Analyze D50 of cyclone IV and the concentrations of the PM in the various size ranges.
12.3.1 D50 of Cyclone IV. To determine the actual D50 for cyclone IV, recalculate the Cunningham correction factor and the Reynolds number for the best estimate of cyclone IV D50. The following sections describe additional information on how to recalculate the Cunningham correction factor and determine which Reynolds number to use.
12.3.1.1 Cunningham correction factor. Recalculate the initial estimate of the Cunningham correction factor using the actual test data. Insert the actual test run data and D50 of 2.5 micrometers into Equation 4. This will give you a new Cunningham correction factor based on actual data.
12.3.1.2 Initial D50for cyclone IV. Determine the initial estimate for cyclone IV D50 using the test condition Reynolds number calculated with Equation 10 as indicated in Table 3 of Section 17. Refer to the following instructions.
(a) If the Reynolds number is less than 3,162, calculate the D50 for cyclone IV with Equation 34, using actual test data.
(b) If the Reynolds number is greater than or equal to 3,162, calculate the D50 for cyclone IV with Equation 35 using actual test data.
(c) Insert the "new" D50 value calculated by either Equation 34 or 35 into Equation 36 to re-establish the Cunningham Correction Factor (Cr). (Note: Use the test condition calculated Reynolds number to determine the most appropriate equation (Equation 34 or 35).)
12.3.1.3 Re-establish cyclone IV D50. Use the re-established Cunningham correction factor (calculated in the previous step) and the calculated Reynolds number to determine D50-1.
(a) Use Equation 37 to calculate the re-established cyclone IV D50-1 if the Reynolds number is less than 3,162.
(b) Use Equation 38 to calculate the re-established cyclone IV D50-1 if the Reynolds number is greater than or equal to 3,162.
12.3.1.4 Establish "Z" values. The "Z" value is the result of an analysis that you must perform to determine if the Cr is acceptable. Compare the calculated cyclone IV D50 (either Equation 34 or 35) to the re-established cyclone IV D50-1 (either Equation 36 or 37) values based upon the test condition calculated Reynolds number (Equation 39). Follow these procedures.
(a) Use Equation 39 to calculate the "Z" values. If the "Z" value is between 0.99 and 1.01, the D50-1 value is the best estimate of the cyclone IV D50 cut diameter for your test run.
(b) If the "Z" value is greater than 1.01 or less than 0.99, re-establish a Cr based on the D50-1 value determined in either Equations 36 or 37, depending upon the test condition Reynolds number.
(c) Use the second revised Cr to re-calculate the cyclone IV D50.
(d) Repeat this iterative process as many times as necessary using the prescribed equations until you achieve the criteria documented in Equation 40.
12.3.2 Particulate Concentration. Use the particulate catch weights in the combined cyclone sampling train to calculate the concentration of PM in the various size ranges. You must correct the concentrations for the acetone blank.
12.3.2.1 Acetone blank concentration. Use Equation 42 to calculate the acetone blank concentration (Ca).
12.3.2.2 Acetone blank residue weight. Use Equation 44 to calculate the acetone blank weight (Wa (2,3,4)). Subtract the weight of the acetone blank from the particulate weight catch in each size fraction.
12.3.2.3 Particulate weight catch per size fraction. Correct each of the PM weights per size fraction by subtracting the acetone blank weight (i.e., M2,3,4-Wa). (Note: Do not subtract a blank value of greater than 0.1 mg per 100 ml of the acetone used from the sample recovery.) Use the following procedures.
(a) Use Equation 45 to calculate the PM recovered from Containers #1, #2, #3, and #4. This is the total collectible PM (Ctf).
(b) Use Equation 46 to determine the quantitative recovery of PM10 (CfPM10) from Containers #1, #3, and #4.
(c) Use Equation 47 to determine the quantitative recovery of PM2.5 (CfPM2.5) recovered from Containers #1 and #4.
12.4 Reporting. You must prepare a test report following the guidance in EPA Guidance Document 043, Preparation and Review of Test Reports (December 1998).
12.5 Equations. Use the following equations to complete the calculations required in this test method.
Molecular Weight of Dry Gas. Calculate the molecular weight of the dry gas using Equation 1.
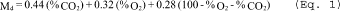
Molecular Weight of Wet Gas. Calculate the molecular weight of the stack gas on a wet basis using Equation 2.
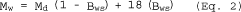
Gas Stream Viscosity. Calculate the gas stream viscosity using Equation 3. This equation uses constants for gas temperatures in°R.
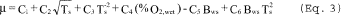
Cunningham Correction Factor. The Cunningham correction factor is calculated for a 2.25 micrometer diameter particle.
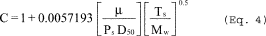
Lower Limit Cut Diameter for Cyclone I for NreLess than 3,162. The Cunningham correction factor is calculated for a 2.25 micrometer diameter particle.
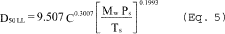
Cut Diameter for Cyclone I for the Middle of the Overlap Zone.
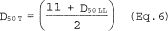
Sampling Rate Using Both PM10and PM2.5Cyclones.
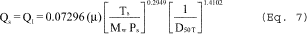
Sampling Rate Using Only PM2.5 Cyclone.
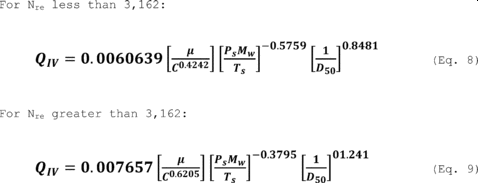
Reynolds Number.
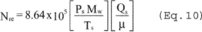
Meter Box Orifice Pressure Drop.
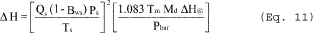
Lower Limit Cut Diameter for Cyclone I for NreGreater than or Equal to 3,162. The Cunningham correction factor is calculated for a 2.25 micrometer diameter particle.
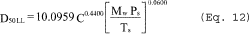
Velocity of Stack Gas. Correct the mean preliminary velocity pressure for Cp and blockage using Equations 25, 26, and 27.
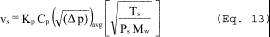
Calculated Nozzle Diameter for Acceptable Sampling Rate.
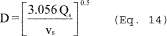
Velocity of Gas in Nozzle.
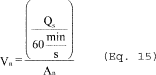
Minimum Nozzle/Stack Velocity Ratio Parameter.
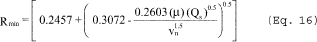
Maximum Nozzle/Stack Velocity Ratio Parameter.
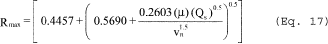
Minimum Gas Velocity for RminLess than 0.5.

Minimum Gas Velocity for RminGreater than or Equal to 0.5.
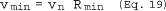
Maximum Gas Velocity for RmaxLess than to 1.5.
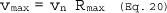
Maximum Gas Velocity for RmaxGreater than or Equal to 1.5.
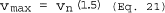
Minimum Velocity Pressure.
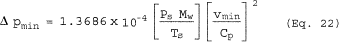
Maximum Velocity Pressure.
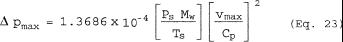
Sampling Dwell Time at Each Point. Ntp is the total number of traverse points. You must use the preliminary velocity traverse data.
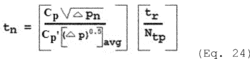
Adjusted Velocity Pressure.
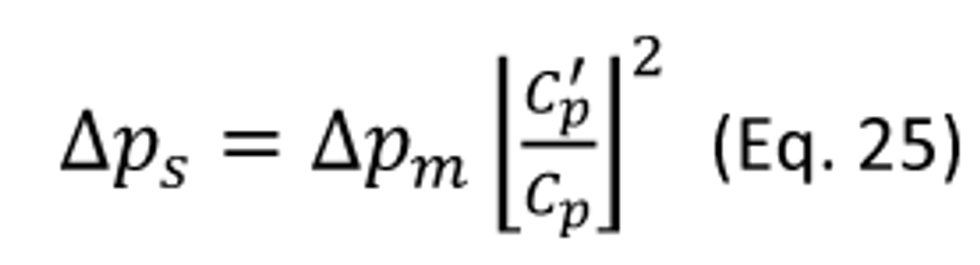
Average Probe Blockage Factor.

Velocity Pressure.
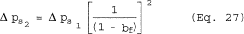
Dry Gas Volume Sampled at Standard Conditions.
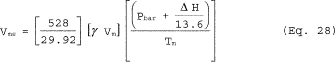
Sample Flow Rate at Standard Conditions.
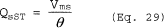
Volume of Water Vapor.
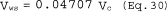
Moisture Content of Gas Stream.
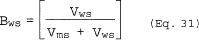
Sampling Rate.
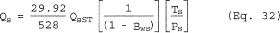
(Note: The viscosity and Reynolds Number must be recalculated using the actual stack temperature, moisture, and oxygen content.)
Actual Particle Cut Diameter for Cyclone I. This is based on actual temperatures and pressures measured during the test run.
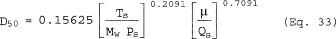
Particle Cut Diameter for NreLess than 3,162 for Cyclone IV. C must be recalculated using the actual test data and a D50 for 2.5 micrometer diameter particle size.
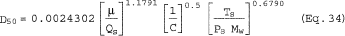
Particle Cut Diameter for NreGreater than or Equal to 3,162 for Cyclone IV. C must be recalculated using the actual test run data and a D50 for 2.5 micrometer diameter particle size.
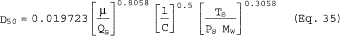
Re-estimated Cunningham Correction Factor. You must use the actual test run Reynolds Number (Nre) value and select the appropriate D50 from Equation 33 or 34 (or Equation 37 or 38 if reiterating).
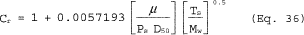
Re-calculated Particle Cut Diameter for NreLess than 3,162.
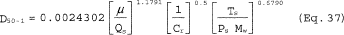
Re-calculated Particle Cut Diameter for N Greater than or Equal to 3,162.
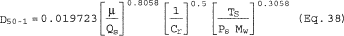
Ratio (Z) Between D50and D50-1Values.

Acceptance Criteria for Z Values. The number of iterative steps is represented by N.
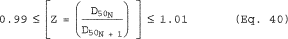
Percent Isokinetic Sampling.
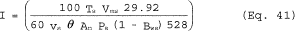
Acetone Blank Concentration.
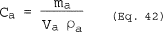
Acetone Blank Correction Weight.
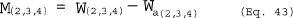
Acetone Blank Weight.
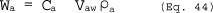
Concentration of Total Filterable PM.
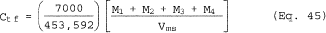
Concentration of Filterable PM10.
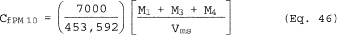
Concentration of Filterable PM2.5.
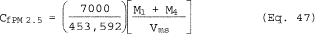
13.0 Method Performance
13.1 Field evaluation of PM10 and total PM showed that the precision of constant sampling rate method was the same magnitude as Method 17 of appendix A-6 to part 60 (approximately five percent). Precision in PM10 and total PM between multiple trains showed standard deviations of four to five percent and total mass compared to 4.7 percent observed for Method 17 in simultaneous test runs at a Portland cement clinker cooler exhaust. The accuracy of the constant sampling rate PM10 method for total mass, referenced to Method 17, was ˆ’2 ±4.4 percent (Farthing, 1988a).
13.2 Laboratory evaluation and guidance for PM10 cyclones were designed to limit error due to spatial variations to 10 percent. The maximum allowable error due to an isokinetic sampling was limited to ±20 percent for 10 micrometer particles in laboratory tests (Farthing, 1988b).
13.3 A field evaluation of the revised Method 201A by EPA showed that the detection limit was 2.54 mg for total filterable PM, 1.44 mg for filterable PM10, and 1.35 mg for PM2.5. The precision resulting from 10 quadruplicate tests (40 test runs) conducted for the field evaluation was 6.7 percent relative standard deviation. The field evaluation also showed that the blank expected from Method 201A was less than 0.9 mg (EPA, 2010).
14.0 Alternative Procedures
Alternative methods for estimating the moisture content (ALT-008) and thermocouple calibration (ALT-011) can be found at http://www.epa.gov/ttn/emc/approalt.html.
15.0 Waste Management
[Reserved]
16.0 References
(1) Dawes, S.S., and W.E. Farthing. 1990. "Application Guide for Measurement of PM2.5 at Stationary Sources," U.S. Environmental Protection Agency, Atmospheric Research and Exposure Assessment Laboratory, Research Triangle Park, NC, 27511, EPA-600/3-90/057 (NTIS No.: PB 90-247198).
(2) Farthing, et al. 1988a. "PM10 Source Measurement Methodology: Field Studies," EPA 600/3-88/055, NTIS PB89-194278/AS, U.S. Environmental Protection Agency, Research Triangle Park, NC 27711.
(3) Farthing, W.E., and S.S. Dawes. 1988b. "Application Guide for Source PM10 Measurement with Constant Sampling Rate," EPA/600/3-88-057, U.S. Environmental Protection Agency, Research Triangle Park, NC 27711.
(4) Richards, J.R. 1996. "Test protocol: PCA PM10/PM2.5 Emission Factor Chemical Characterization Testing," PCA R&D Serial No. 2081, Portland Cement Association.
(5) U.S. Environmental Protection Agency, Federal Reference Methods 1 through 5 and Method 17, 40 CFR part 60,r">Appendix A-1 through A-3 and A-6.
(6) U.S. Environmental Protection Agency. 2010. "Field Evaluation of an Improved Method for Sampling and Analysis of Filterable and Condensable Particulate Matter." Office of Air Quality Planning and Standards, Sector Policy and Program Division Monitoring Policy Group. Research Triangle Park, NC 27711.
17.0 Tables, Diagrams, Flowcharts, and Validation Data
You must use the following tables, diagrams, flowcharts, and data to complete this test method successfully.
| Particle size range | Concentration and % by weight |
|---|---|
| Total collectible particulate | 0.015 gr/DSCF. |
| Less than or equal to 10 and greater than 2.5 micrometers | 40% of total collectible PM. |
| ‰¤2.5 micrometers | 20% of total collectible PM. |
| Cyclone |
Min. cut
diameter (micrometer) |
Max. cut
diameter (micrometer) |
|---|---|---|
| PM10 Cyclone (Cyclone I from five stage cyclone) | 9 | 11 |
| PM2.5 Cyclone (Cyclone IV from five stage cyclone) | 2.25 | 2.75 |
| If you are using . . . | To calculate . . . | Then use . . . |
|---|---|---|
| Preliminary data | Dry gas molecular weight, Md | Equation 1. |
| Dry gas molecular weight (Md) and preliminary moisture content of the gas stream | wet gas molecular weight, MW | Equation 2. a |
| Stack gas temperature, and oxygen and moisture content of the gas stream | gas viscosity, µ | Equation 3. |
| Gas viscosity, µ | Cunningham correction factor b, C | Equation 4. |
|
Reynolds Number
c (Nre)
Nre less than 3,162 | Preliminary lower limit cut diameter for cyclone I, D50LL | Equation 5. |
| D50LL from Equation 5 | Cut diameter for cyclone I for middle of the overlap zone, D50T | Equation 6. |
| D50T from Equation 6 | Final sampling rate for cyclone I, QI(Qs) | Equation 7. |
| D50 for PM2.5 cyclone and Nre less than 3,162 | Final sampling rate for cyclone IV, QIV | Equation 8. |
| D50 for PM2.5 cyclone and Nre greater than or equal to 3,162 | Final sampling rate for cyclone IV, QIV | Equation 9. |
| QI(Qs) from Equation 7 | Verify the assumed Reynolds number, Nre | Equation 10. |
| a Use Method 4 to determine the moisture content of the stack gas. Use a wet bulb-dry bulb measurement device or hand-held hygrometer to estimate moisture content of sources with gas temperature less than 160°F.
b For the lower cut diameter of cyclone IV, 2.25 micrometer. c Verify the assumed Reynolds number, using the procedure in Section 8.5.1, before proceeding to Equation 11. | ||
| Stack Temperature (°R) | Ts - 50° | Ts | Ts + 50° |
|---|---|---|---|
| ΔH, (inches W.C.) | a | a | a |
| a These values are to be filled in by the stack tester. | |||
| If the Nre is . . . | Then . . . | And . . . |
|---|---|---|
| Less than 3,162 | Calculate ΔH for the meter box | Assume original D50LL is correct |
| Greater than or equal to 3,162 | Recalculate D50LL using Equation 12 | Substitute the "new" D50LL into Equation 6 to recalculate D50T. |
| Calculations | Instructions and References |
|---|---|
| Average dry gas meter temperature | See field test data sheet. |
| Average orifice pressure drop | See field test data sheet. |
| Dry gas volume (Vms) | Use Equation 28 to correct the sample volume measured by the dry gas meter to standard conditions (20°C, 760 mm Hg or 68°F, 29.92 inches Hg). |
| Dry gas sampling rate (QsST) | Must be calculated using Equation 29. |
| Volume of water condensed (Vws) | Use Equation 30 to determine the water condensed in the impingers and silica gel combination. Determine the total moisture catch by measuring the change in volume or weight in the impingers and weighing the silica gel. |
| Moisture content of gas stream (Bws) | Calculate this using Equation 31. |
| Sampling rate (Qs) | Calculate this using Equation 32. |
| Test condition Reynolds number a | Use Equation 10 to calculate the actual Reynolds number during test conditions. |
| Actual D50 of cyclone I | Calculate this using Equation 33. This calculation is based on the average temperatures and pressures measured during the test run. |
| Stack gas velocity (vs) | Calculate this using Equation 13. |
| Percent isokinetic rate (%I) | Calculate this using Equation 41. |
| a Calculate the Reynolds number at the cyclone IV inlet during the test based on: (1) The sampling rate for the combined cyclone head, (2) the actual gas viscosity for the test, and (3) the dry and wet gas stream molecular weights. | |
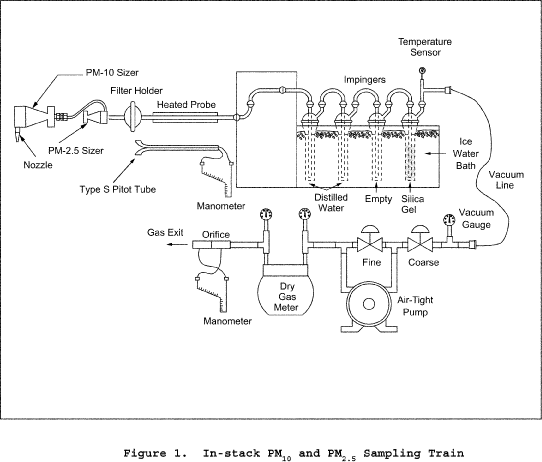
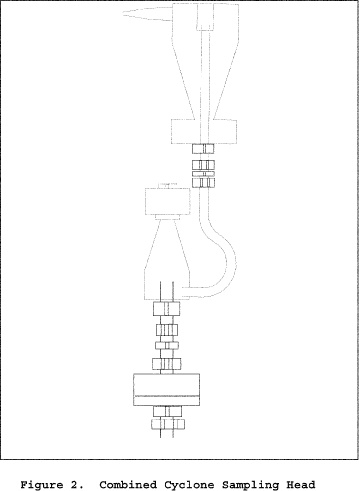
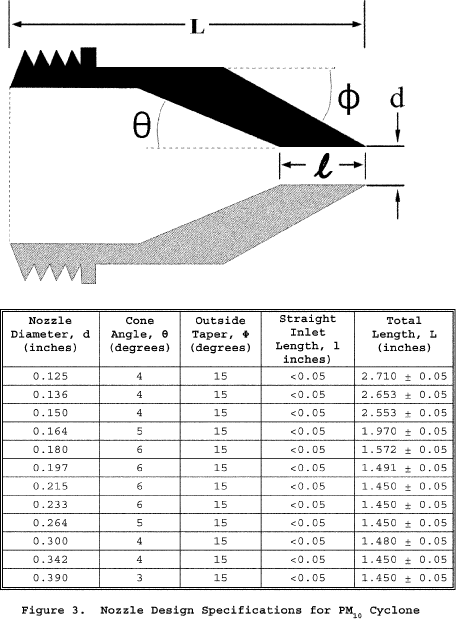
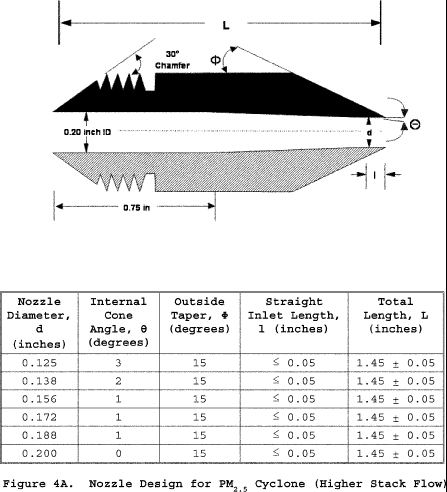
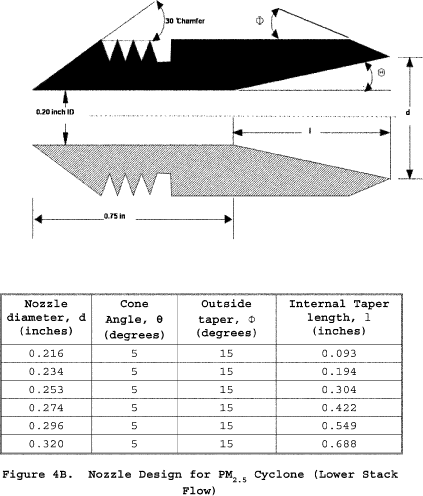
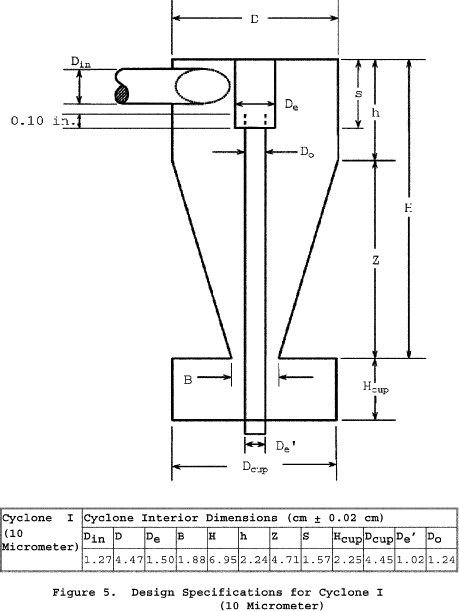
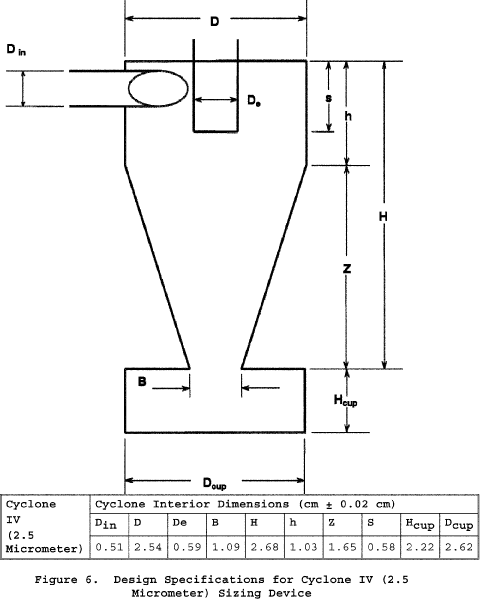
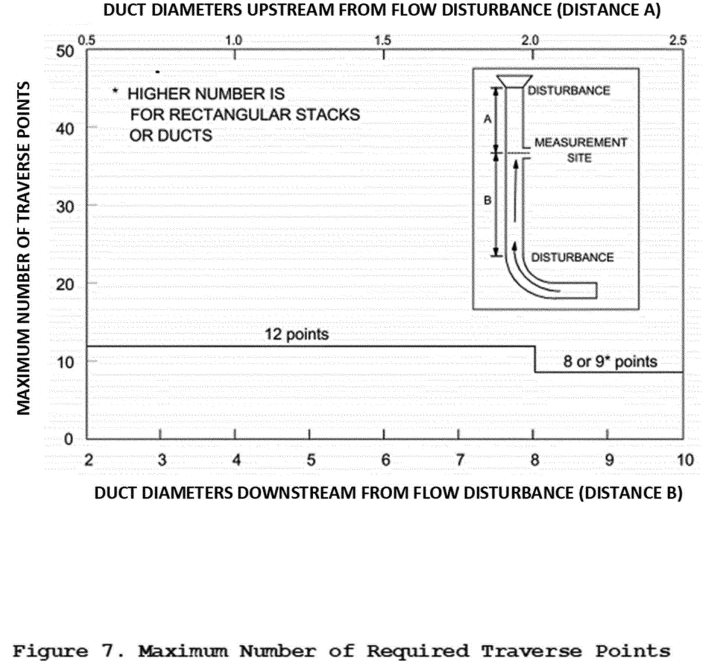
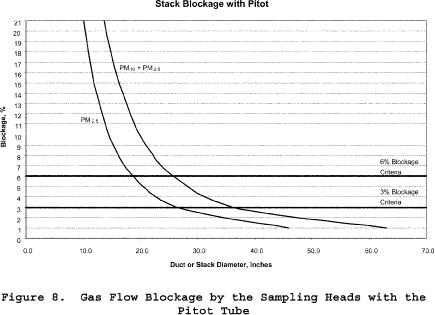
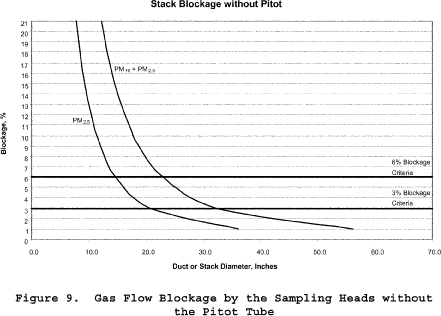
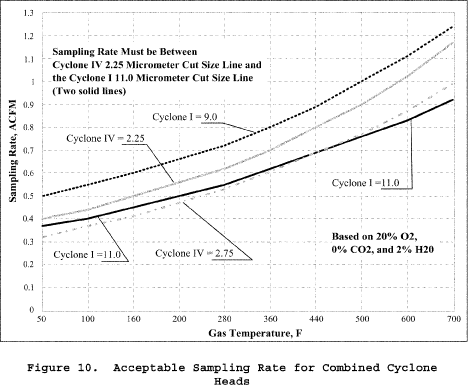
Method 202 - Dry Impinger Method for Determining Condensable Particulate Emissions From Stationary Sources
1.0 Scope and Applicability
1.1 Scope. The U.S. Environmental Protection Agency (U.S. EPA or "we") developed this method to describe the procedures that the stack tester ("you") must follow to measure condensable particulate matter (CPM) emissions from stationary sources. This method includes procedures for measuring both organic and inorganic CPM.
1.2 Applicability. This method addresses the equipment, preparation, and analysis necessary to measure only CPM. You can use this method only for stationary source emission measurements. You can use this method to measure CPM from stationary source emissions after filterable particulate matter (PM) has been removed. CPM is measured in the emissions after removal from the stack and after passing through a filter.
(a) If the gas filtration temperature exceeds 30°C (85°F) and you must measure both the filterable and condensable (material that condenses after passing through a filter) components of total primary (direct) PM emissions to the atmosphere, then you must combine the procedures in this method with the procedures in Method 201A of appendix M to this part for measuring filterable PM. However, if the gas filtration temperature never exceeds 30°C (85°F), then use of this method is not required to measure total primary PM.
(b) If Method 17 of appendix A-6 to part 60 is used in conjunction with this method and constant weight requirements for the in-stack filter cannot be met, the Method 17 filter and sampling nozzle rinse must be treated as described in Sections 8.5.4.4 and 11.2.1 of this method. (See Section 3.0 for a definition of constant weight.) Extracts resulting from the use of this procedure must be filtered to remove filter fragments before the filter is processed and weighed.
1.3 Responsibility. You are responsible for obtaining the equipment and supplies you will need to use this method. You should also develop your own procedures for following this method and any additional procedures to ensure accurate sampling and analytical measurements.
1.4 Additional Methods. To obtain reliable results, you should have a thorough knowledge of the following test methods that are found in appendices A-1 through A-3 and A-6 to part 60, and in appendix M to this part:
(a) Method 1 - Sample and velocity traverses for stationary sources.
(b) Method 2 - Determination of stack gas velocity and volumetric flow rate (Type S pitot tube).
(c) Method 3 - Gas analysis for the determination of dry molecular weight.
(d) Method 4 - Determination of moisture content in stack gases.
(e) Method 5 - Determination of particulate matter emissions from stationary sources.
(f) Method 17 - Determination of particulate matter emissions from stationary sources (in-stack filtration method).
(g) Method 201A - Determination of PM10 and PM2.5 emissions from stationary sources (Constant sampling rate procedure).
(h) You will need additional test methods to measure filterable PM. You may use Method 5 (including Method 5A, 5D and 5I but not 5B, 5E, 5F, 5G, or 5H) of appendix A-3 to part 60, or Method 17 of appendix A-6 to part 60, or Method 201A of appendix M to this part to collect filterable PM from stationary sources with temperatures above 30°C (85°F) in conjunction with this method. However, if the gas filtration temperature never exceeds 30°C (85°F), then use of this method is not required to measure total primary PM.
1.5 Limitations. You can use this method to measure emissions in stacks that have entrained droplets only when this method is combined with a filterable PM test method that operates at high enough temperatures to cause water droplets sampled through the probe to become vaporous.
1.6 Conditions. You must maintain isokinetic sampling conditions to meet the requirements of the filterable PM test method used in conjunction with this method. You must sample at the required number of sampling points specified in Method 5 of appendix A-3 to part 60, Method 17 of appendix A-6 to part 60, or Method 201A of appendix M to this part. Also, if you are using this method as an alternative to a required performance test method, you must receive approval from the regulatory authority that established the requirement to use this test method prior to conducting the test.
2.0 Summary of Method
2.1 Summary. The CPM is collected in dry impingers after filterable PM has been collected on a filter maintained as specified in either Method 5 of appendix A-3 to part 60, Method 17 of appendix A-6 to part 60, or Method 201A of appendix M to this part. The organic and aqueous fractions of the impingers and an out-of-stack CPM filter are then taken to dryness and weighed. The total of the impinger fractions and the CPM filter represents the CPM. Compared to the version of Method 202 that was promulgated on December 17, 1991, this method eliminates the use of water as the collection media in impingers and includes the addition of a condenser followed by a water dropout impinger immediately after the final in-stack or heated filter. This method also includes the addition of one modified Greenburg Smith impinger (backup impinger) and a CPM filter following the water dropout impinger. Figure 1 of Section 18 presents the schematic of the sampling train configured with these changes.
2.1.1 Condensable PM. CPM is collected in the water dropout impinger, the modified Greenburg Smith impinger, and the CPM filter of the sampling train as described in this method. The impinger contents are purged with nitrogen immediately after sample collection to remove dissolved sulfur dioxide (SO2) gases from the impinger. The CPM filter is extracted with water and hexane. The impinger solution is then extracted with hexane. The organic and aqueous fractions are dried and the residues are weighed. The total of the aqueous and organic fractions represents the CPM.
2.1.2 Dry Impinger and Additional Filter. The potential artifacts from SO2 are reduced using a condenser and water dropout impinger to separate CPM from reactive gases. No water is added to the impingers prior to the start of sampling. To improve the collection efficiency of CPM, an additional filter (the "CPM filter") is placed between the second and third impingers.
3.0 Definitions
3.1 Condensable PM (CPM) means material that is vapor phase at stack conditions, but condenses and/or reacts upon cooling and dilution in the ambient air to form solid or liquid PM immediately after discharge from the stack. Note that all condensable PM is assumed to be in the PM2.5 size fraction.
3.2 Constant weight means a difference of no more than 0.5 mg or one percent of total weight less tare weight, whichever is greater, between two consecutive weighings, with no less than six hours of desiccation time between weighings.
3.3 Field Train Proof Blank. A field train proof blank is recovered on site from a clean, fully-assembled sampling train prior to conducting the first emissions test.
3.4 Filterable PM means particles that are emitted directly by a source as a solid or liquid at stack or release conditions and captured on the filter of a stack test train.
3.5 Primary PM (also known as direct PM) means particles that enter the atmosphere as a direct emission from a stack or an open source. Primary PM comprises two components: filterable PM and condensable PM. These two PM components have no upper particle size limit.
3.6 Primary PM2.5 (also known as direct PM2.5, total PM2.5, PM2.5, or combined filterable PM2.5 and condensable PM) means PM with an aerodynamic diameter less than or equal to 2.5 micrometers. These solid particles are emitted directly from an air emissions source or activity, or are the gaseous emissions or liquid droplets from an air emissions source or activity that condense to form PM at ambient temperatures. Direct PM2.5 emissions include elemental carbon, directly emitted organic carbon, directly emitted sulfate, directly emitted nitrate, and other inorganic particles (including but not limited to crustal material, metals, and sea salt).
3.7 Primary PM10 (also known as direct PM10, total PM10, PM10, or the combination of filterable PM10 and condensable PM) means PM with an aerodynamic diameter equal to or less than 10 micrometers.
3.8 ASTM E617-13. ASTM E617-13 "Standard Specification for Laboratory Weights and Precisions Mass Standards," approved May 1, 2013, was developed and adopted by the American Society for Testing and Materials (ASTM). The standards cover weights and mass standards used in laboratories for specific classes. The ASTM E617-13 standard has been approved for incorporation by reference by the Director of the Office of the Federal Register in accordance with 5 U.S.C. 552(a) and 1 CFR part 51. The standard may be obtained from http://www.astm.org or from the ASTM at 100 Barr Harbor Drive, P.O. Box C700, West Conshohocken, PA 19428-2959. All approved material is available for inspection at EPA WJC West Building, Room 3334, 1301 Constitution Ave. NW., Washington, DC 20460, telephone number 202-566-1744. It is also available for inspection at the National Archives and Records Administration (NARA). For information on the availability of this material at NARA, call 202-741-6030 or go to http://www.archives.gov/federal_register/code_of_federal_regulattions/ibr_locations.html.
4.0 Interferences
[Reserved]
5.0 Safety
Disclaimer. Because the performance of this method may require the use of hazardous materials, operations, and equipment, you should develop a health and safety plan to ensure the safety of your employees who are on site conducting the particulate emission test. Your plan should conform with all applicable Occupational Safety and Health Administration, Mine Safety and Health Administration, and Department of Transportation regulatory requirements. Because of the unique situations at some facilities and because some facilities may have more stringent requirements than is required by State or federal laws, you may have to develop procedures to conform to the plant health and safety requirements.
6.0 Equipment and Supplies
The equipment used in the filterable particulate portion of the sampling train is described in Methods 5 and 17 of appendix A-1 through A-3 and A-6 to part 60 and Method 201A of appendix M to this part. The equipment used in the CPM portion of the train is described in this section.
6.1 Condensable Particulate Sampling Train Components. The sampling train for this method is used in addition to filterable particulate collection using Method 5 of appendix A-3 to part 60, Method 17 of appendix A-6 to part 60, or Method 201A of appendix M to this part. This method includes the following exceptions or additions:
6.1.1 Probe Extension and Liner. The probe extension between the filterable particulate filter and the condenser must be glass- or fluoropolymer-lined. Follow the specifications for the probe liner specified in Section 6.1.1.2 of Method 5 of appendix A-3 to part 60.
6.1.2 Condenser and Impingers. You must add the following components to the filterable particulate sampling train: A Method 23 type condenser as described in Section 2.1.2 of Method 23 of appendix A-8 to part 60, followed by a water dropout impinger or flask, followed by a modified Greenburg-Smith impinger (backup impinger) with an open tube tip as described in Section 6.1.1.8 of Method 5 of appendix A-3 to part 60.
6.1.3 CPM Filter Holder. The modified Greenburg-Smith impinger is followed by a filter holder that is either glass, stainless steel (316 or equivalent), or fluoropolymer-coated stainless steel. Commercial size filter holders are available depending on project requirements. Use a commercial filter holder capable of supporting 47 mm or greater diameter filters. Commercial size filter holders contain a fluoropolymer O-ring, stainless steel, ceramic or fluoropolymer filter support and a final fluoropolymer O-ring. A filter that meets the requirements specified in Section 7.1.1 may be placed behind the CPM filter to reduce the pressure drop across the CPM filter. This support filter is not part of the PM sample and is not recovered with the CPM filter. At the exit of the CPM filter, install a fluoropolymer-coated or stainless steel encased thermocouple that is in contact with the gas stream.
6.1.4 Long Stem Impinger Insert. You will need a long stem modified Greenburg Smith impinger insert for the water dropout impinger to perform the nitrogen purge of the sampling train.
6.2 Sample Recovery Equipment.
6.2.1 Condensable PM Recovery. Use the following equipment to quantitatively determine the amount of CPM recovered from the sampling train.
(a) Nitrogen purge line. You must use inert tubing and fittings capable of delivering at least 14 liters/min of nitrogen gas to the impinger train from a standard gas cylinder (see Figures 2 and 3 of Section 18). You may use standard 0.6 centimeters ( 1/4 inch) tubing and compression fittings in conjunction with an adjustable pressure regulator and needle valve.
(b) Rotameter. You must use a rotameter capable of measuring gas flow up to 20 L/min. The rotameter must be accurate to five percent of full scale.
(c) Nitrogen gas purging system. Compressed ultra-pure nitrogen, regulator, and filter must be capable of providing at least 14 L/min purge gas for one hour through the sampling train.
(d) Amber glass bottles (500 ml).
6.2.2 Analysis Equipment. The following equipment is necessary for CPM sample analysis:
(a) Separatory Funnel. Glass, 1 liter.
(b) Weighing Tins. 50 ml. Glass evaporation vials, fluoropolymer beaker liners, or aluminum weighing tins can be used.
(c) Glass Beakers. 300 to 500 ml.
(d) Drying Equipment. A desiccator containing anhydrous calcium sulfate that is maintained below 10 percent relative humidity, and a hot plate or oven equipped with temperature control.
(e) Glass Pipets. 5 ml.
(f) Burette. Glass, 0 to 100 ml in 0.1 ml graduations.
(g) Analytical Balance. Analytical balance capable of weighing at least 0.0001 g (0.1 mg).
(h) pH Meter or Colormetric pH Indicator. The pH meter or colormetric pH indicator (e.g., phenolphthalein) must be capable of determining the acidity of liquid within 0.1 pH units.
(i) Sonication Device. The device must have a minimum sonication frequency of 20 kHz and be approximately four to six inches deep to accommodate the sample extractor tube.
(j) Leak-Proof Sample Containers. Containers used for sample and blank recovery must not contribute more than 0.05 mg of residual mass to the CPM measurements.
(k) Wash bottles. Any container material is acceptable, but wash bottles used for sample and blank recovery must not contribute more than 0.1 mg of residual mass to the CPM measurements.
7.0 Reagents and Standards
7.1 Sample Collection. To collect a sample, you will need a CPM filter, crushed ice, and silica gel. You must also have water and nitrogen gas to purge the sampling train. You will find additional information on each of these items in the following summaries.
7.1.1 CPM Filter. You must use a nonreactive, nondisintegrating polymer filter that does not have an organic binder and does not contribute more than 0.5 mg of residual mass to the CPM measurements. The CPM filter must also have an efficiency of at least 99.95 percent (less than 0.05 percent penetration) on 0.3 micrometer dioctyl phthalate particles. You may use test data from the supplier's quality control program to document the CPM filter efficiency.
7.1.2 Silica Gel. Use an indicating-type silica gel of six to 16 mesh. You must obtain approval of the Administrator for other types of desiccants (equivalent or better) before you use them. Allow the silica gel to dry for two hours at 175°C (350°F) if it is being reused. You do not have to dry new silica gel if the indicator shows the silica gel is active for moisture collection.
7.1.3 Water. Use deionized, ultra-filtered water that contains 1.0 parts per million by weight (ppmw) (1 mg/L) residual mass or less to recover and extract samples.
7.1.4 Crushed Ice. Obtain from the best readily available source.
7.1.5 Nitrogen Gas. Use Ultra-High Purity compressed nitrogen or equivalent to purge the sampling train. The compressed nitrogen you use to purge the sampling train must contain no more than 1 parts per million by volume (ppmv) oxygen, 1 ppmv total hydrocarbons as carbon, and 2 ppmv moisture. The compressed nitrogen must not contribute more than 0.1 mg of residual mass per purge.
7.2 Sample Recovery and Analytical Reagents. You will need acetone, hexane, anhydrous calcium sulfate, ammonia hydroxide, and deionized water for the sample recovery and analysis. Unless otherwise indicated, all reagents must conform to the specifications established by the Committee on Analytical Reagents of the American Chemical Society. If such specifications are not available, then use the best available grade. Additional information on each of these items is in the following paragraphs:
7.2.1 Acetone. Use acetone that is stored in a glass bottle. Do not use acetone from a metal container because it normally produces a high residual mass in the laboratory and field reagent blanks. You must use acetone that has a blank value less than 1.0 ppmw (0.1 mg/100 g) residue.
7.2.2 Hexane, American Chemical Society grade. You must use hexane that has a blank residual mass value less than 1.0 ppmw (0.1 mg/100 g) residue.
7.2.3 Water. Use deionized, ultra-filtered water that contains 1 ppmw (1 mg/L) residual mass or less to recover material caught in the impinger.
7.2.4 Condensable Particulate Sample Desiccant. Use indicating-type anhydrous calcium sulfate to desiccate water and organic extract residue samples prior to weighing.
7.2.5 Ammonium Hydroxide. Use National Institute of Standards and Technology-traceable or equivalent (0.1 N) NH4OH.
7.2.6 Standard Buffer Solutions. Use one buffer solution with a neutral pH and a second buffer solution with an acid pH of no less than 4.
8.0 Sample Collection, Preservation, Storage, and Transport
8.1 Qualifications. This is a complex test method. To obtain reliable results, you should be trained and experienced with in-stack filtration systems (such as, cyclones, impactors, and thimbles) and impinger and moisture train systems.
8.2 Preparations. You must clean all glassware used to collect and analyze samples prior to field tests as described in Section 8.4 prior to use. Cleaned glassware must be used at the start of each new source category tested at a single facility. Analyze laboratory reagent blanks (water, acetone, and hexane) before field tests to verify low blank concentrations. Follow the pretest preparation instructions in Section 8.1 of Method 5.
8.3 Site Setup. You must follow the procedures required in Methods 5, 17, or 201A, whichever is applicable to your test requirements including:
(a) Determining the sampling site location and traverse points.
(b) Calculating probe/cyclone blockage (as appropriate).
(c) Verifying the absence of cyclonic flow.
(d) Completing a preliminary velocity profile, and selecting a nozzle(s) and sampling rate.
8.3.1 Sampling Site Location. Follow the standard procedures in Method 1 of appendix A-1 to part 60 to select the appropriate sampling site. Choose a location that maximizes the distance from upstream and downstream flow disturbances.
8.3.2 Traverse points. Use the required number of traverse points at any location, as found in Methods 5, 17, or 201A, whichever is applicable to your test requirements. You must prevent the disturbance and capture of any solids accumulated on the inner wall surfaces by maintaining a 1-inch distance from the stack wall (0.5 inch for sampling locations less than 24 inches in diameter).
8.4 Sampling Train Preparation. A schematic of the sampling train used in this method is shown in Figure 1 of Section 18. All glassware that is used to collect and analyze samples must be cleaned prior to the test with soap and water, and rinsed using tap water, deionized water, acetone, and finally, hexane. It is important to completely remove all silicone grease from areas that will be exposed to the hexane rinse during sample recovery. After cleaning, you must bake glassware at 300°C for six hours prior to beginning tests at each source category sampled at a facility. As an alternative to baking glassware, a field train proof blank, as specified in Section 8.5.4.10, can be performed on the sampling train glassware that is used to collect CPM samples. Prior to each sampling run, the train glassware used to collect condensable PM must be rinsed thoroughly with deionized, ultra-filtered water that that contains 1 ppmw (1 mg/L) residual mass or less.
8.4.1 Condenser and Water Dropout Impinger. Add a Method 23 type condenser and a condensate dropout impinger without bubbler tube after the final probe extension that connects the in-stack or out-of-stack hot filter assembly with the CPM sampling train. The Method 23 type stack gas condenser is described in Section 2.1.2 of Method 23. The condenser must be capable of cooling the stack gas to less than or equal to 30°C (85°F).
8.4.2 Backup Impinger. The water dropout impinger is followed by a modified Greenburg Smith impinger (backup impinger) with no taper (see Figure 1 of Section 18). Place the water dropout and backup impingers in an insulated box with water at less than or equal to 30°C (less than or equal to 85°F). At the start of the tests, the water dropout and backup impingers must be clean, without any water or reagent added.
8.4.3 CPM Filter. Place a filter holder with a filter meeting the requirements in Section 7.1.1 after the backup impinger. The connection between the CPM filter and the moisture trap impinger must include a thermocouple fitting that provides a leak-free seal between the thermocouple and the stack gas. (Note: A thermocouple well is not sufficient for this purpose because the fluoropolymer- or steel-encased thermocouple must be in contact with the sample gas.)
8.4.4 Moisture Traps. You must use a modified Greenburg-Smith impinger containing 100 ml of water, or the alternative described in Method 5 of appendix A-3 to part 60, followed by an impinger containing silica gel to collect moisture that passes through the CPM filter. You must maintain the gas temperature below 20°C (68°F) at the exit of the moisture traps.
8.4.5 Silica Gel Trap. Place 200 to 300 g of silica gel in each of several air-tight containers. Weigh each container, including silica gel, to the nearest 0.5 g, and record this weight on the filterable particulate data sheet. As an alternative, the silica gel need not be preweighed, but may be weighed directly in its impinger or sampling holder just prior to train assembly.
8.4.6 Leak-Check (Pretest). Use the procedures outlined in Method 5 of appendix A-3 to part 60, Method 17 of appendix A-6 to part 60, or Method 201A of appendix M to this part as appropriate to leak check the entire sampling system. Specifically, perform the following procedures:
8.4.6.1 Sampling train. You must pretest the entire sampling train for leaks. The pretest leak-check must have a leak rate of not more than 0.02 actual cubic feet per minute or 4 percent of the average sample flow during the test run, whichever is less. Additionally, you must conduct the leak-check at a vacuum equal to or greater than the vacuum anticipated during the test run. Enter the leak-check results on the field test data sheet for the filterable particulate method. (Note: Conduct leak-checks during port changes only as allowed by the filterable particulate method used with this method.)
8.4.6.2 Pitot tube assembly. After you leak-check the sample train, perform a leak-check of the pitot tube assembly. Follow the procedures outlined in Section 8.4.1 of Method 5.
8.5 Sampling Train Operation. Operate the sampling train as described in the filterable particulate sampling method (i.e., Method 5 of appendix A-3 to part 60, Method 17 of appendix A-6 to part 60, or Method 201A of appendix M to this part) with the following additions or exceptions:
8.5.1 Impinger and CPM Filter Assembly.
8.5.1.1 Monitor the moisture condensation in the knockout and backup impingers. If the accumulated water from moisture condensation overwhelms the knockout impinger, i.e., the water level is more than approximately one-half the capacity of the knockout impinger, or if water accumulates in the backup impinger sufficient to cover the impinger insert tip, then you may interrupt the sampling run, recover and weigh the moisture accumulated in the knockout and backup impinger, reassemble and leak check the sampling train, and resume the sampling run. You must purge the water collected during the test interruption as soon as practical following the procedures in Section 8.5.3.
8.5.1.2 You must include the weight or volume of the moisture in your moisture calculation and you must combine the recovered water with the appropriate sample fraction for subsequent CPM analysis.
8.5.1.3 Use the field data sheet for the filterable particulate method to record the CPM filter temperature readings at the beginning of each sample time increment and when sampling is halted. Maintain the CPM filter greater than 20°C (greater than 65°F) but less than or equal to 30°C (less than or equal to 85°F) during sample collection. (Note: Maintain the temperature of the CPM filter assembly as close to 30°C (85°F) as feasible.)
8.5.2 Leak-Check Probe/Sample Train Assembly (Post-Test). Conduct the leak rate check according to the filterable particulate sampling method used during sampling. If required, conduct the leak-check at a vacuum equal to or greater than the maximum vacuum achieved during the test run. If the leak rate of the sampling train exceeds 0.02 actual cubic feet per minute or four percent of the average sampling rate during the test run (whichever is less), then the run is invalid and you must repeat it.
8.5.3 Post-Test Nitrogen Purge. As soon as possible after the post-test leak-check, detach the probe, any cyclones, and in-stack or hot filters from the condenser and impinger train. If no water was collected before the CPM filter, then you may skip the remaining purge steps and proceed with sample recovery (see Section 8.5.4). You may purge the CPM sampling train using the sampling system meter box and vacuum pump or by passing nitrogen through the train under pressure. For either type of purge, you must first attach the nitrogen supply line to a purged inline filter.
8.5.3.1 If you choose to conduct a pressurized nitrogen purge at the completion of CPM sample collection, you may purge the entire CPM sample collection train from the condenser inlet to the CPM filter holder outlet or you may quantitatively transfer the water collected in the condenser and the water dropout impinger to the backup impinger and purge only the backup impinger and the CPM filter. You must measure the water in the knockout and backup impingers and record the volume or weight as part of the moisture collected during sampling as specified in Section 8.5.3.4.
8.5.3.1.1 If you choose to conduct a purge of the entire CPM sampling train, you must replace the short stem impinger insert in the knock out impinger with a standard modified Greenburg Smith impinger insert.
8.5.3.1.2 If you choose to combine the knockout and backup impinger catch prior to purge, you must purge the backup impinger and CPM filter holder.
8.5.3.1.3 If the tip of the impinger insert does not extend below the water level (including the water transferred from the first impinger if this option was chosen), you must add a measured amount of degassed, deionized ultra-filtered water that contains 1 ppmw (1 mg/L) residual mass or less until the impinger tip is at least 1 centimeter below the surface of the water. You must record the amount of water added to the water dropout impinger (Vp)(see Figure 4 of Section 18) to correct the moisture content of the effluent gas. (Note: Prior to use, water must be degassed using a nitrogen purge bubbled through the water for at least 15 minutes to remove dissolved oxygen).
8.5.3.1.4 To perform the nitrogen purge using positive pressure nitrogen flow, you must start with no flow of gas through the clean purge line and fittings. Connect the filter outlet to the input of the impinger train and disconnect the vacuum line from the exit of the silica moisture collection impinger (see Figure 3 of Section 18). You may purge only the CPM train by disconnecting the moisture train components if you measure moisture in the field prior to the nitrogen purge. You must increase the nitrogen flow gradually to avoid over-pressurizing the impinger array. You must purge the CPM train at a minimum of 14 liters per minute for at least one hour. At the conclusion of the purge, turn off the nitrogen delivery system.
8.5.3.2 If you choose to conduct a nitrogen purge on the complete CPM sampling train using the sampling system meter box and vacuum pump, replace the short stem impinger insert with a modified Greenberg Smith impinger insert. The impinger tip length must extend below the water level in the impinger catch.
(a) You must conduct the purge on the complete CPM sampling train starting at the inlet of the condenser. If insufficient water was collected, you must add a measured amount of degassed, deionized ultra-filtered water that contains 1 ppmw (1 mg/L) residual mass or less until the impinger tip is at least 1 centimeter below the surface of the water. You must record the amount of water added to the water dropout impinger (Vp) (see Figure 4 of Section 18) to correct the moisture content of the effluent gas. (Note: Prior to use, water must be degassed using a nitrogen purge bubbled through the water for at least 15 minutes to remove dissolved oxygen.)
(b) You must start the purge using the sampling train vacuum pump with no flow of gas through the clean purge line and fittings. Connect the filter outlet to the input of the impinger train (see Figure 2 of Section 18). To avoid over- or under-pressurizing the impinger array, slowly commence the nitrogen gas flow through the line while simultaneously opening the meter box pump valve(s). Adjust the pump bypass and/or nitrogen delivery rates to obtain the following conditions: 14 liters/min or ΔH@ and a positive overflow rate through the rotameter of less than 2 liters/min. The presence of a positive overflow rate guarantees that the nitrogen delivery system is operating at greater than ambient pressure and prevents the possibility of passing ambient air (rather than nitrogen) through the impingers. Continue the purge under these conditions for at least one hour, checking the rotameter and ΔH@ value(s) at least every 15 minutes. At the conclusion of the purge, simultaneously turn off the delivery and pumping systems.
8.5.3.3 During either purge procedure, continue operation of the condenser recirculation pump, and heat or cool the water surrounding the first two impingers to maintain the gas temperature measured at the exit of the CPM filter greater than 20°C (greater than 65°F), but less than or equal to 30°C (less than or equal to 85°F). If the volume of liquid collected in the moisture traps has not been determined prior to conducting the nitrogen purge, maintain the temperature of the moisture traps following the CPM filter to prevent removal of moisture during the purge. If necessary, add more ice during the purge to maintain the gas temperature measured at the exit of the silica gel impinger below 20°C (68°F). Continue the purge under these conditions for at least one hour, checking the rotameter and ΔH@ value(s) periodically. At the conclusion of the purge, simultaneously turn off the delivery and pumping systems.
8.5.3.4 Weigh the liquid, or measure the volume of the liquid collected in the dropout, impingers, and silica trap if this has not been done prior to purging the sampling train. Measure the liquid in the water dropout impinger to within 1 ml using a clean graduated cylinder or by weighing it to within 0.5 g using a balance. Record the volume or weight of liquid present to be used to calculate the moisture content of the effluent gas in the field log notebook.
8.5.3.5 If a balance is available in the field, weigh the silica impinger to within 0.5 g. Note the color of the indicating silica gel in the last impinger to determine whether it has been completely spent, and make a notation of its condition in the field log notebook.
8.5.4 Sample Recovery.
8.5.4.1 Recovery of filterable PM. Recovery of filterable PM involves the quantitative transfer of particles according to the filterable particulate sampling method (i.e., Method 5 of appendix A-3 to part 60, Method 17 of appendix A-6 to part 60, or Method 201A of appendix M to this part).
8.5.4.2 CPM Container #1, Aqueous liquid impinger contents. Quantitatively transfer liquid from the dropout and the backup impingers prior to the CPM filter into a clean, leak-proof container labeled with test identification and "CPM Container #1, Aqueous Liquid Impinger Contents." Rinse all sampling train components including the back half of the filterable PM filter holder, the probe extension, condenser, each impinger and the connecting glassware, and the front half of the CPM filter housing twice with water. Recover the rinse water, and add it to CPM Container #1. Mark the liquid level on the container.
8.5.4.3 CPM Container #2, Organic rinses. Follow the water rinses of the back half of the filterable PM filter holder, probe extension, condenser, each impinger, and all of the connecting glassware and front half of the CPM filter with an acetone rinse. Recover the acetone rinse into a clean, leak-proof container labeled with test identification and "CPM Container #2, Organic Rinses." Then repeat the entire rinse procedure with two rinses of hexane, and save the hexane rinses in the same container as the acetone rinse (CPM Container #2). Mark the liquid level on the jar.
8.5.4.4 CPM Container #3, CPM filter sample. Use tweezers and/or clean disposable surgical gloves to remove the filter from the CPM filter holder. Place the filter in the Petri dish labeled with test identification and "CPM Container #3, Filter Sample."
8.5.4.5 CPM Container #4, Cold impinger water. You must weigh or measure the volume of the contents of CPM Container #4 either in the field or during sample analysis (see Section 11.2.4). If the water from the cold impinger has been weighed in the field, it can be discarded. Otherwise, quantitatively transfer liquid from the cold impinger that follows the CPM filter into a clean, leak-proof container labeled with test identification and "CPM Container #4, Cold Water Impinger." Mark the liquid level on the container. CPM Container #4 holds the remainder of the liquid water from the emission gases.
8.5.4.6 CPM Container #5, Silica gel absorbent. You must weigh the contents of CPM Container #5 in the field or during sample analysis (see Section 11.2.5). If the silica gel has been weighed in the field to measure water content, then it can be discarded or recovered for reuse. Otherwise, transfer the silica gel to its original container labeled with test identification and "CPM Container #5, Silica Gel Absorbent" and seal. You may use a funnel to make it easier to pour the silica gel without spilling. You may also use a rubber policeman as an aid in removing the silica gel from the impinger. It is not necessary to remove the small amount of silica gel dust particles that may adhere to the impinger wall and are difficult to remove. Since the gain in weight is to be used for moisture calculations, do not use any water or other liquids to transfer the silica gel.
8.5.4.7 CPM Container #6, Acetone field reagent blank. Take approximately 200 ml of the acetone directly from the wash bottle you used for sample recovery and place it in a clean, leak-proof container labeled with test identification and "CPM Container #6, Acetone Field Reagent Blank" (see Section 11.2.6 for analysis). Mark the liquid level on the container. Collect one acetone field reagent blank from the lot(s) of solvent used for the test.
8.5.4.8 CPM Container #7, Water field reagent blank. Take approximately 200 ml of the water directly from the wash bottle you used for sample recovery and place it in a clean, leak-proof container labeled with test identification and "CPM Container #7, Water Field Reagent Blank" (see Section 11.2.7 for analysis). Mark the liquid level on the container. Collect one water field reagent blank from the lot(s) of water used for the test.
8.5.4.9 CPM Container #8, Hexane field reagent blank. Take approximately 200 ml of the hexane directly from the wash bottle you used for sample recovery and place it in a clean, leak-proof container labeled with test identification and "CPM Container #8, Hexane Field Reagent Blank" (see Section 11.2.8 for analysis). Mark the liquid level on the container. Collect one hexane field reagent blank from the lot(s) of solvent used for the test.
8.5.4.10 Field train proof blank. If you did not bake the sampling train glassware as specified in Section 8.4, you must conduct a field train proof blank as specified in Sections 8.5.4.11 and 8.5.4.12 to demonstrate the cleanliness of sampling train glassware.
8.5.4.11 CPM Container #9, Field train proof blank, inorganic rinses. Prior to conducting the emission test, rinse the probe extension, condenser, each impinger and the connecting glassware, and the front half of the CPM filter housing twice with water. Recover the rinse water and place it in a clean, leak-proof container labeled with test identification and "CPM Container #9, Field Train Proof Blank, Inorganic Rinses." Mark the liquid level on the container.
8.5.4.12 CPM Container #10, Field train proof blank, organic rinses. Follow the water rinse of the probe extension, condenser, each impinger and the connecting glassware, and the front half of the CPM filter housing with an acetone rinse. Recover the acetone rinse into a clean, leak-proof container labeled with test identification and "CPM Container #10, Field Train Proof Blank, Organic Rinses." Then repeat the entire rinse procedure with two rinses of hexane and save the hexane rinses in the same container as the acetone rinse (CPM Container #10). Mark the liquid level on the container.
8.5.5 Transport procedures. Containers must remain in an upright position at all times during shipping. You do not have to ship the containers under dry or blue ice. However, samples must be maintained at or below 30°C (85°F) during shipping.
9.0 Quality Control
9.1 Daily Quality Checks. You must perform daily quality checks of field log notebooks and data entries and calculations using data quality indicators from this method and your site-specific test plan. You must review and evaluate recorded and transferred raw data, calculations, and documentation of testing procedures. You must initial or sign log notebook pages and data entry forms that were reviewed.
9.2 Calculation Verification. Verify the calculations by independent, manual checks. You must flag any suspect data and identify the nature of the problem and potential effect on data quality. After you complete the test, prepare a data summary and compile all the calculations and raw data sheets.
9.3 Conditions. You must document data and information on the process unit tested, the particulate control system used to control emissions, any non-particulate control system that may affect particulate emissions, the sampling train conditions, and weather conditions. Discontinue the test if the operating conditions may cause non-representative particulate emissions.
9.4 Field Analytical Balance Calibration Check. Perform calibration check procedures on field analytical balances each day that they are used. You must use National Institute of Standards and Technology (NIST)-traceable weights at a mass approximately equal to the weight of the sample plus container you will weigh.
9.5 Glassware. Use class A volumetric glassware for titrations, or calibrate your equipment against NIST-traceable glassware.
9.6 Laboratory Analytical Balance Calibration Check. Check the calibration of your laboratory analytical balance each day that you weigh CPM samples. You must use NIST Class S weights at a mass approximately equal to the weight of the sample plus container you will weigh.
9.7 Laboratory Reagent Blanks. You should run blanks of water, acetone, and hexane used for field recovery and sample analysis. Analyze at least one sample (150 ml minimum) of each lot of reagents that you plan to use for sample recovery and analysis before you begin testing. These blanks are not required by the test method, but running blanks before field use is advisable to verify low blank concentrations, thereby reducing the potential for a high field blank on test samples.
9.8 Field Reagent Blanks. You should run at least one field reagent blank of water, acetone, and hexane you use for field recovery. These blanks are not required by the test method, but running independent field reagent blanks is advisable to verify that low blank concentrations were maintained during field solvent use and demonstrate that reagents have not been contaminated during field tests.
9.9 Field Train Proof Blank. If you are not baking glassware as specified in Section 8.4, you must recover a minimum of one field train proof blank for the sampling train used for testing each new source category at a single facility. You must assemble the sampling train as it will be used for testing. You must recover the field train proof blank samples as described in Section 8.5.4.11 and 8.5.4.12.
9.10 Field Train Recovery Blank. You must recover a minimum of one field train blank for each source category tested at the facility. You must recover the field train blank after the first or second run of the test. You must assemble the sampling train as it will be used for testing. Prior to the purge, you must add 100 ml of water to the first impinger and record this data on Figure 4. You must purge the assembled train as described in section 8.5.3. You must recover field train blank samples as described in section 8.5.4. From the field sample weight, you will subtract the condensable particulate mass you determine with this blank train or 0.002 g (2.0 mg), whichever is less.
10.0 Calibration and Standardization
Maintain a field log notebook of all condensable particulate sampling and analysis calibrations. Include copies of the relevant portions of the calibration and field logs in the final test report.
10.1 Thermocouple Calibration. You must calibrate the thermocouples using the procedures described in Section 10.3.1 of Method 2 of appendix A-1 to part 60 or Alternative Method 2, Thermocouple Calibration (ALT-011) (http://www.epa.gov/ttn/emc). Calibrate each temperature sensor at a minimum of three points over the anticipated range of use against a NIST-traceable thermometer. Alternatively, a reference thermocouple and potentiometer calibrated against NIST standards can be used.
10.2 Ammonium Hydroxide. The 0.1 N NH4OH used for titrations in this method is made as follows: Add 7 ml of concentrated (14.8 M) NH4OH to l liter of water. Standardize against standardized 0.1 N H2SO4, and calculate the exact normality using a procedure parallel to that described in Section 10.5 of Method 6 of appendix A-4 to 40 CFR part 60. Alternatively, purchase 0.1 N NH4OH that has been standardized against a NIST reference material. Record the normality on the CPM Work Table (see Figure 6 of Section 18).
10.3 Field Balance Calibration Check. Check the calibration of the balance used to weigh impingers with a weight that is at least 500g or within 50g of a loaded impinger. The weight must be ASTM E617-13 "Standard Specification for Laboratory Weights and Precision Mass Standards" Class 6 (or better). Daily before use, the field balance must measure the weight within ± 0.5g of the certified mass. If the daily balance calibration check fails, perform corrective measures and repeat the check before using balance.
10.4 Analytical Balance Calibration. Perform a multipoint calibration (at least five points spanning the operational range) of the analytical balance before the first use, and semiannually thereafter. The calibration of the analytical balance must be conducted using ASTM E617-13 "Standard Specification for Laboratory Weights and Precision Mass Standards" Class 2 (or better) tolerance weights. Audit the balance each day it is used for gravimetric measurements by weighing at least one ASTM E617-13 Class 2 tolerance (or better) calibration weight that corresponds to 50 to 150 percent of the weight of one filter or between 1g and 5g. If the scale cannot reproduce the value of the calibration weight to within 0.5mg of the certified mass, perform corrective measures, and conduct the multipoint calibration before use.
11.0 Analytical Procedures
11.1 Analytical Data Sheets. (a) Record the filterable particulate field data on the appropriate (i.e., Method 5, 17, or 201A) analytical data sheets. Alternatively, data may be recorded electronically using software applications such as the Electronic Reporting Tool available at http://www.epa.gov/ttn/chief/ert/ert_tool.html. Record the condensable particulate data on the CPM Work Table (see Figure 6 of Section 18).
(b) Measure the liquid in all containers either volumetrically to ±1 ml or gravimetrically to ±0.5 g. Confirm on the filterable particulate analytical data sheet whether leakage occurred during transport. If a noticeable amount of leakage has occurred, either void the sample or use methods (subject to the approval of the Administrator) to correct the final results.
11.2 Condensable PM Analysis. See the flow chart in Figure 7 of Section 18 for the steps to process and combine fractions from the CPM train.
11.2.1 Container #3, CPM Filter Sample. If the sample was collected by Method 17 or Method 201A with a stack temperature below 30°C (85°F), transfer the filter and any loose PM from the sample container to a tared glass weighing dish. (See Section 3.0 for a definition of constant weight.) Desiccate the sample for 24 hours in a desiccator containing anhydrous calcium sulfate. Weigh to a constant weight and report the results to the nearest 0.1 mg. [Note: In-stack filter samples collected at 30°C (85°F) may include both filterable insoluble particulate and condensable particulate. The nozzle and front half wash and filter collected at or below 30°C (85°F) may not be heated and must be maintained at or below 30°C (85°F).] If the sample was collected by Method 202, extract the CPM filter as follows:
11.2.1.1 Extract the water soluble (aqueous or inorganic) CPM from the CPM filter by folding the filter in quarters and placing it into a 50-ml extraction tube. Add sufficient deionized, ultra-filtered water to cover the filter (e.g., 10 ml of water). Place the extractor tube into a sonication bath and extract the water-soluble material for a minimum of two minutes. Combine the aqueous extract with the contents of Container #1. Repeat this extraction step twice for a total of three extractions.
11.2.1.2 Extract the organic soluble CPM from the CPM filter by adding sufficient hexane to cover the filter (e.g., 10 ml of hexane). Place the extractor tube into a sonication bath and extract the organic soluble material for a minimum of two minutes. Combine the organic extract with the contents of Container #2. Repeat this extraction step twice for a total of three extractions.
11.2.2 CPM Container #1, Aqueous Liquid Impinger Contents. Analyze the water soluble CPM in Container #1 as described in this section. Place the contents of Container #1 into a separatory funnel. Add approximately 30 ml of hexane to the funnel, mix well, and pour off the upper organic phase. Repeat this procedure twice with 30 ml of hexane each time combining the organic phase from each extraction. Each time, leave a small amount of the organic/hexane phase in the separatory funnel, ensuring that no water is collected in the organic phase. This extraction should yield about 90 ml of organic extract. Combine the organic extract from Container #1 with the organic train rinse in Container #2.
11.2.2.1 Determine the inorganic fraction weight. Transfer the aqueous fraction from the extraction to a clean 500-ml or smaller beaker. Evaporate to no less than 10 ml liquid on a hot plate or in the oven at 105°C and allow to dry at room temperature (not to exceed 30°C (85°F)). You must ensure that water and volatile acids have completely evaporated before neutralizing nonvolatile acids in the sample. Following evaporation, desiccate the residue for 24 hours in a desiccator containing anhydrous calcium sulfate. Weigh at intervals of at least 6 hours to a constant weight. (See section 3.0 for a definition of constant weight.) Report results to the nearest 0.1 mg on the CPM Work Table (see Figure 6 of section 18) and proceed directly to section 11.2.3. If the residue cannot be weighed to constant weight, re-dissolve the residue in 100 ml of deionized distilled ultra-filtered water that contains 1 ppmw (1 mg/L) residual mass or less and continue to section 11.2.2.2.
11.2.2.2 Use titration to neutralize acid in the sample and remove water of hydration. If used, calibrate the pH meter with the neutral and acid buffer solutions. Then titrate the sample with 0.1N NH4OH to a pH of 7.0, as indicated by the pH meter or colorimetric indicator. Record the volume of titrant used on the CPM Work Table (see Figure 6 of section 18).
11.2.2.3 Using a hot plate or an oven at 105°C, evaporate the aqueous phase to approximately 10 ml. Quantitatively transfer the beaker contents to a clean, 50-ml pre-tared weighing tin and evaporate to dryness at room temperature (not to exceed 30°C (85°F)) and pressure in a laboratory hood. Following evaporation, desiccate the residue for 24 hours in a desiccator containing anhydrous calcium sulfate. Weigh at intervals of at least 6 hours to a constant weight. (See section 3.0 for a definition of constant weight.) Report results to the nearest 0.1 mg on the CPM Work Table (see Figure 6 of section 18).
11.2.2.4 Calculate the correction factor to subtract the NH4+ retained in the sample using Equation 1 in section 12.
11.2.3 CPM Container #2, Organic Fraction Weight Determination. Analyze the organic soluble CPM in Container #2 as described in this section. Place the organic phase in a clean glass beaker. Evaporate the organic extract at room temperature (not to exceed 30°C (85°F)) and pressure in a laboratory hood to not less than 10 ml. Quantitatively transfer the beaker contents to a clean 50-ml pre-tared weighing tin and evaporate to dryness at room temperature (not to exceed 30°C (85°F)) and pressure in a laboratory hood. Following evaporation, desiccate the organic fraction for 24 hours in a desiccator containing anhydrous calcium sulfate. Weigh at intervals of at least six hours to a constant weight (i.e., less than or equal to 0.5 mg change from previous weighing), and report results to the nearest 0.1 mg on the CPM Work Table (see Figure 6 of Section 18).
11.2.4 CPM Container #4, Cold Impinger Water. If the amount of water has not been determined in the field, note the level of liquid in the container, and confirm on the filterable particulate analytical data sheet whether leakage occurred during transport. If a noticeable amount of leakage has occurred, either void the sample or use methods (subject to the approval of the Administrator) to correct the final results. Measure the liquid in Container #4 either volumetrically to ±1 ml or gravimetrically to ±0.5 g, and record the volume or weight on the filterable particulate analytical data sheet of the filterable PM test method.
11.2.5 CPM Container #5, Silica Gel Absorbent. Weigh the spent silica gel (or silica gel plus impinger) to the nearest 0.5 g using a balance. This step may be conducted in the field. Record the weight on the filterable particulate analytical data sheet of the filterable PM test method.
11.2.6 Container #6, Acetone Field Reagent Blank. Use 150 ml of acetone from the blank container used for this analysis. Transfer 150 ml of the acetone to a clean 250-ml beaker. Evaporate the acetone at room temperature (not to exceed 30°C (85°F)) and pressure in a laboratory hood to approximately 10 ml. Quantitatively transfer the beaker contents to a clean 50-ml pre-tared weighing tin, and evaporate to dryness at room temperature (not to exceed 30°C (85°F)) and pressure in a laboratory hood. Following evaporation, desiccate the residue for 24 hours in a desiccator containing anhydrous calcium sulfate. Weigh at intervals of at least six hours to a constant weight (i.e., less than or equal to 0.5 mg change from previous weighing), and report results to the nearest 0.1 mg on Figure 4 of Section 19.
11.2.7 Water Field Reagent Blank, Container #7. Use 150 ml of the water from the blank container for this analysis. Transfer the water to a clean 250-ml beaker, and evaporate to approximately 10 ml liquid in the oven at 105°C. Quantitatively transfer the beaker contents to a clean 50 ml pre-tared weighing tin and evaporate to dryness at room temperature (not to exceed 30°C (85°F)) and pressure in a laboratory hood. Following evaporation, desiccate the residue for 24 hours in a desiccator containing anhydrous calcium sulfate. Weigh at intervals of at least six hours to a constant weight (i.e., less than or equal to 0.5 mg change from previous weighing) and report results to the nearest 0.1 mg on Figure 4 of Section 18.
11.2.8 Hexane Field Reagent Blank, Container #8. Use 150 ml of hexane from the blank container for this analysis. Transfer 150 ml of the hexane to a clean 250-ml beaker. Evaporate the hexane at room temperature (not to exceed 30°C (85°F)) and pressure in a laboratory hood to approximately 10 ml. Quantitatively transfer the beaker contents to a clean 50-ml pre-tared weighing tin and evaporate to dryness at room temperature (not to exceed 30°C (85°F)) and pressure in a laboratory hood. Following evaporation, desiccate the residue for 24 hours in a desiccator containing anhydrous calcium sulfate. Weigh at intervals of at least six hours to a constant weight (i.e., less than or equal to 0.5 mg change from previous weighing), and report results to the nearest 0.1 mg on Figure 4 of Section 18.
12.0 Calculations and Data Analysis
12.1 Nomenclature. Report results in International System of Units (SI units) unless the regulatory authority for testing specifies English units. The following nomenclature is used.
ΔH@ = Pressure drop across orifice at flow rate of 0.75 SCFM at standard conditions, inches of water column (Note: Specific to each orifice and meter box).
17.03 = mg/milliequivalents for ammonium ion.
ACFM = Actual cubic feet per minute.
Ccpm = Concentration of the condensable PM in the stack gas, dry basis, corrected to standard conditions, milligrams/dry standard cubic foot.
mc = Mass of the NH4+ added to sample to form ammonium sulfate, mg.
mcpm = Mass of the total condensable PM, mg.
mfb = Mass of total CPM in field train recovery blank, mg.
mg = Milligrams.
mg/L = Milligrams per liter.
mi = Mass of inorganic CPM, mg.
mib = Mass of inorganic CPM in field train recovery blank, mg.
mo = Mass of organic CPM, mg.
mob = Mass of organic CPM in field train blank, mg.
mr = Mass of dried sample from inorganic fraction, mg.
N = Normality of ammonium hydroxide titrant.
ppmv = Parts per million by volume.
ppmw = Parts per million by weight.
Vm(std) = Volume of gas sample measured by the dry gas meter, corrected to standard conditions, dry standard cubic meter (dscm) or dry standard cubic foot (dscf) as defined in Equation 5-1 of Method 5.
Vt = Volume of NH4OH titrant, ml.
Vp = Volume of water added during train purge.
12.2 Calculations. Use the following equations to complete the calculations required in this test method. Enter the appropriate results from these calculations on the CPM Work Table (see Figure 6 of Section 18).
12.2.1 Mass of ammonia correction. Correction for ammonia added during titration of 100 ml aqueous CPM sample. This calculation assumes no waters of hydration.
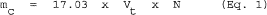
12.2.2 Mass of the Field Train Recovery Blank (mg). Per Section 9.10, the mass of the field train recovery blank, mfb, shall not exceed 2.0 mg.
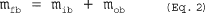
12.2.3 Mass of Inorganic CPM (mg).
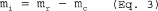
12.2.4 Total Mass of CPM (mg).
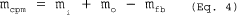
12.2.5 Concentration of CPM (mg/dscf).
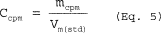
12.3 Emissions Test Report. You must prepare a test report following the guidance in EPA Guidance Document 043 (Preparation and Review of Test Reports. December 1998).
13.0 Method Performance
An EPA field evaluation of the revised Method 202 showed the following precision in the results: approximately 4 mg for total CPM, approximately 0.5 mg for organic CPM, and approximately 3.5 mg for inorganic CPM.
14.0 Pollution Prevention
[Reserved]
15.0 Waste Management
Solvent and water are evaporated in a laboratory hood during analysis. No liquid waste is generated in the performance of this method. Organic solvents used to clean sampling equipment should be managed as RCRA organic waste.
16.0 Alternative Procedures
Alternative Method 2, Thermocouple Calibration (ALT-011) for the thermocouple calibration can be found at http://www.epa.gov/ttn/emc/approalt.html.
17.0 References
(1) Commonwealth of Pennsylvania, Department of Environmental Resources. 1960. Chapter 139, Sampling and Testing (Title 25, Rules and Regulations, part I, Department of Environmental Resources, Subpart C, Protection of Natural Resources, Article III, Air Resources). January 8, 1960.
(2) DeWees, W.D. and K.C. Steinsberger. 1989. "Method Development and Evaluation of Draft Protocol for Measurement of Condensable Particulate Emissions." Draft Report. November 17, 1989.
(3) DeWees, W.D., K.C. Steinsberger, G.M. Plummer, L.T. Lay, G.D. McAlister, and R.T. Shigehara. 1989. "Laboratory and Field Evaluation of EPA Method 5 Impinger Catch for Measuring Condensable Matter from Stationary Sources." Paper presented at the 1989 EPA/AWMA International Symposium on Measurement of Toxic and Related Air Pollutants. Raleigh, North Carolina. May 1-5, 1989.
(4) Electric Power Research Institute (EPRI). 2008. "Laboratory Comparison of Methods to Sample and Analyze Condensable PM." EPRI Agreement EP-P24373/C11811 Condensable Particulate Methods: EPRI Collaboration with EPA, October 2008.
(5) Nothstein, Greg. Masters Thesis. University of Washington. Department of Environmental Health. Seattle, Washington.
(6) Richards, J., T. Holder, and D. Goshaw. 2005. "Optimized Method 202 Sampling Train to Minimize the Biases Associated with Method 202 Measurement of Condensable PM Emissions." Paper presented at Air & Waste Management Association Hazardous Waste Combustion Specialty Conference. St. Louis, Missouri. November 2-3, 2005.
(7) Texas Air Control Board, Laboratory Division. 1976. "Determination of Particulate in Stack Gases Containing Sulfuric Acid and/or Sulfur Dioxide." Laboratory Methods for Determination of Air Pollutants. Modified December 3, 1976.
(8) Puget Sound Air Pollution Control Agency, Engineering Division. 1983. "Particulate Source Test Procedures Adopted by Puget Sound Air Pollution Control Agency Board of Directors." Seattle, Washington. August 11, 1983.
(9) U.S. Environmental Protection Agency, Federal Reference Methods 1 through 5 and Method 17, 40 CFR 60, appendix A-1 through A-3 and A-6.
(10) U.S. Environmental Protection Agency. 2008. "Evaluation and Improvement of Condensable PM Measurement," EPA Contract No. EP-D-07-097, Work Assignment 2-03, October 2008.
(11) U.S. Environmental Protection Agency. 2005. "Laboratory Evaluation of Method 202 to Determine Fate of SO2 in Impinger Water," EPA Contract No. 68-D-02-061, Work Assignment 3-14, September 30, 2005.
(12) U.S. Environmental Protection Agency. 2010. Field valuation of an Improved Method for Sampling and Analysis of Filterable and Condensable Particulate Matter. Office of Air Quality Planning and Standards, Sector Policy and Program Division Monitoring Policy Group. Research Triangle Park, NC 27711.
(13) Wisconsin Department of Natural Resources. 1988. Air Management Operations Handbook, Revision 3. January 11, 1988.
18.0 Tables, Diagrams, Flowcharts, and Validation Data
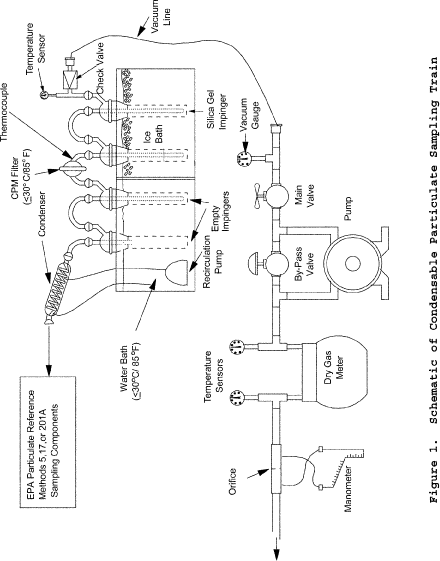
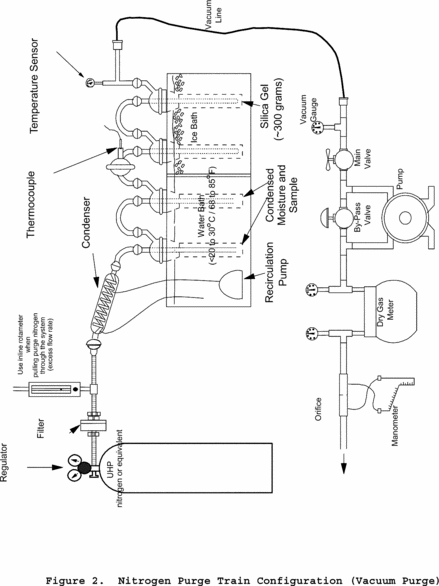
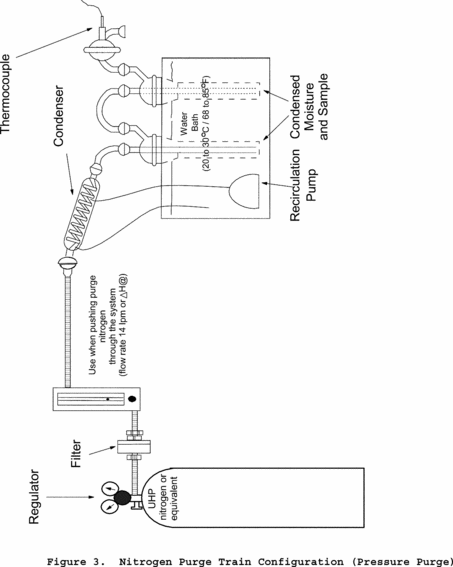


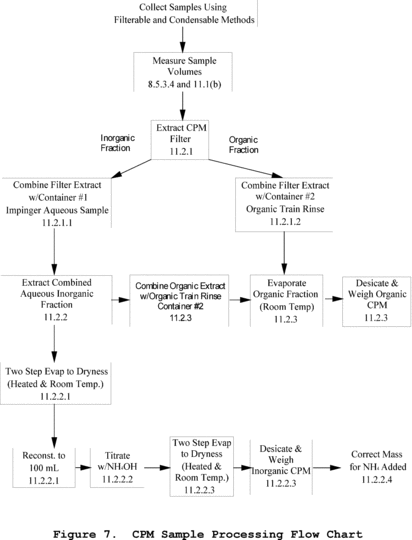
Method 203A - Visual Determination of Opacity of Emissions from Stationary Sources for Time-Averaged Regulations
1.0 Scope and Application
What is Method 203A?
Method 203A is an example test method suitable for State Implementation Plans (SIP) and is applicable to the determination of the opacity of emissions from sources of visible emissions for time-averaged regulations. A time-averaged regulation is any regulation that requires averaging visible emission data to determine the opacity of visible emissions over a specific time period.
Method 203A is virtually identical to EPA's Method 9 of 40 CFR Part 60, Appendix A, except for the data-reduction procedures, which provide for averaging times other than 6 minutes. Therefore, using Method 203A with a 6-minute averaging time would be the same as following EPA Method 9. The certification procedures for this method are identical to those provided in Method 9 and are provided here, in full, for clarity and convenience. An example visible emission observation form and instructions for its use can be found in reference 7 of Section 17 of Method 9.
2.0 Summary of Method
The opacity of emissions from sources of visible emissions is determined visually by an observer certified according to the procedures in Section 10 of this method. Readings taken every 15 seconds are averaged over a time period specified in the applicable regulation ranging from 2 minutes to 6 minutes.
3.0 Definitions [Reserved]
4.0 Interferences [Reserved]
5.0 Safety [Reserved]
6.0 Equipment and Supplies
What equipment and supplies are needed?
6.1 Stop Watch. Two watches are required that provide a continuous display of time to the nearest second.
6.2 Compass (optional). A compass is useful for determining the direction of the emission point from the spot where the visible emissions (VE) observer stands and for determining the wind direction at the source. For accurate readings, the compass should be magnetic with resolution better than 10 degrees. It is suggested that the compass be jewel-mounted and liquid-filled to dampen the needle swing; map reading compasses are excellent.
6.3 Range Finder (optional). Range finders determine distances from the observer to the emission point. The instrument should measure a distance of 1000 meters with a minimum accuracy of ±10 percent.
6.4 Abney Level (optional). This device for determining the vertical viewing angle should measure within 5 degrees.
6.5 Sling Psychrometer (optional). In case of the formation of a steam plume, a wet- and dry-bulb thermometer, accurate to 0.5°C, are mounted on a sturdy assembly and swung rapidly in the air in order to determine the relative humidity.
6.6 Binoculars (optional). Binoculars are recommended to help identify stacks and to characterize the plume. An 8 × 50 or 10 × 50 magnification, color-corrected coated lenses and rectilinear field of view is recommended.
6.7 Camera (optional). A camera is often used to document the emissions before and after the actual opacity determination.
6.8 Safety Equipment. The following safety equipment, which should be approved by the Occupational Safety and Health Association (OSHA), is recommended: orange or yellow hard hat, eye and ear protection, and steel-toed safety boots.
6.9 Clipboard and Accessories (optional). A clipboard, several ball-point pens (black ink recommended), a rubber band, and several visible emission observation forms facilitate documentation.
7.0 Reagents and Standards (Reserved]
8.0 Sample Collection, Preservation, Storage, and Transport
What is the Test Procedure?
An observer qualified in accordance with Section 10 of this method must use the following procedures to visually determine the opacity of emissions from stationary sources.
8.1 Procedure for Emissions from Stacks. These procedures are applicable for visually determining the opacity of stack emissions by a qualified observer.
8.1.1 Position. You must stand at a distance sufficient to provide a clear view of the emissions with the sun oriented in the 140-degree sector to your back. Consistent with maintaining the above requirement as much as possible, you must make opacity observations from a position such that the line of vision is approximately perpendicular to the plume direction, and when observing opacity of emissions from rectangular outlets (e.g., roof monitors, open baghouses, non-circular stacks), approximately perpendicular to the longer axis of the outlet. You should not include more than one plume in the line of sight at a time when multiple plumes are involved and, in any case, make opacity observations with the line of sight perpendicular to the longer axis of such a set of multiple stacks (e.g., stub stacks on baghouses).
8.1.2 Field Records. You must record the name of the plant, emission location, type of facility, observer's name and affiliation, a sketch of the observer's position relative to the source, and the date on a field data sheet. An example visible emission observation form can be found in reference 7 of Section 17 of this method. You must record the time, estimated distance to the emission location, approximate wind direction, estimated wind speed, description of the sky condition (presence and color of clouds), and plume background on the field data sheet at the time opacity readings are initiated and completed.
8.1.3 Observations. You must make opacity observations at the point of greatest opacity in that portion of the plume where condensed water vapor is not present. Do not look continuously at the plume but, instead, observe the plume momentarily at 15-second intervals.
8.1.3.1 Attached Steam Plumes. When condensed water vapor is present within the plume as it emerges from the emission outlet, you must make opacity observations beyond the point in the plume at which condensed water vapor is no longer visible. You must record the approximate distance from the emission outlet to the point in the plume at which the observations are made.
8.1.3.2 Detached Steam Plumes. When water vapor in the plume condenses and becomes visible at a distinct distance from the emission outlet, you must make the opacity observation at the emission outlet prior to the condensation of water vapor and the formation of the steam plume.
8.2 Recording Observations. You must record the opacity observations to the nearest 5 percent every 15 seconds on an observational record sheet such as the example visible emission observation form in reference 7 of Section 17 of this method. Each observation recorded represents the average opacity of emissions for a 15-second period. The overall length of time for which observations are recorded must be appropriate to the averaging time specified in the applicable regulation.
9.0 Quality Control [Reserved]
10.0 Calibration and Standardization
10.1 What are the Certification Requirements? To receive certification as a qualified observer, you must be trained and knowledgeable on the procedures in Section 8.0 of this method, be tested and demonstrate the ability to assign opacity readings in 5 percent increments to 25 different black plumes and 25 different white plumes, with an error not to exceed 15 percent opacity on any one reading and an average error not to exceed 7.5 percent opacity in each category. You must be tested according to the procedures described in Section 10.2 of this method. Any smoke generator used pursuant to Section 10.2 of this method must be equipped with a smoke meter which meets the requirements of Section 10.3 of this method. Certification tests that do not meet the requirements of Sections 10.2 and 10.3 of this method are not valid.
The certification must be valid for a period of 6 months, and after each 6-month period, the qualification procedures must be repeated by an observer in order to retain certification.
10.2 What is the Certification Procedure? The certification test consists of showing the candidate a complete run of 50 plumes, 25 black plumes and 25 white plumes, generated by a smoke generator. Plumes must be presented in random order within each set of 25 black and 25 white plumes. The candidate assigns an opacity value to each plume and records the observation on a suitable form. At the completion of each run of 50 readings, the score of the candidate is determined. If a candidate fails to qualify, the complete run of 50 readings must be repeated in any retest. The smoke test may be administered as part of a smoke school or training program, and may be preceded by training or familiarization runs of the smoke generator during which candidates are shown black and white plumes of known opacity.
10.3 Smoke Generator.
10.3.1 What are the Smoke Generator Specifications? Any smoke generator used for the purpose of Section 10.2 of this method must be equipped with a smoke meter installed to measure opacity across the diameter of the smoke generator stack. The smoke meter output must display in-stack opacity, based upon a path length equal to the stack exit diameter on a full 0 to 100 percent chart recorder scale. The smoke meter optical design and performance must meet the specifications shown in Table 203A-1 of this method. The smoke meter must be calibrated as prescribed in Section 10.3.2 of this method prior to conducting each smoke reading test. At the completion of each test, the zero and span drift must be checked and, if the drift exceeds ±1 percent opacity, the condition must be corrected prior to conducting any subsequent test runs. The smoke meter must be demonstrated at the time of installation to meet the specifications listed in Table 203A-1 of this method. This demonstration must be repeated following any subsequent repair or replacement of the photocell or associated electronic circuitry including the chart recorder or output meter, or every 6 months, whichever occurs first.
10.3.2 How is the Smoke Meter Calibrated? The smoke meter is calibrated after allowing a minimum of 30 minutes warm-up by alternately producing simulated opacity of 0 percent and 100 percent. When a stable response at 0 percent or 100 percent is noted, the smoke meter is adjusted to produce an output of 0 percent or 100 percent, as appropriate. This calibration must be repeated until stable 0 percent and 100 percent readings are produced without adjustment. Simulated 0 percent and 100 percent opacity values may be produced by alternately switching the power to the light source on and off while the smoke generator is not producing smoke.
10.3.3 How is the Smoke Meter Evaluated? The smoke meter design and performance are to be evaluated as follows:
10.3.3.1 Light Source. You must verify from manufacturer's data and from voltage measurements made at the lamp, as installed, that the lamp is operated within 5 percent of the nominal rated voltage.
10.3.3.2 Spectral Response of the Photocell. You must verify from manufacturer's data that the photocell has a photopic response; i.e., the spectral sensitivity of the cell must closely approximate the standard spectral-luminosity curve for photopic vision which is referenced in (b) of Table 203A-1 of this method.
10.3.3.3 Angle of View. You must check construction geometry to ensure that the total angle of view of the smoke plume, as seen by the photocell, does not exceed 15 degrees. Calculate the total angle of view as follows:
φv = 2 tanˆ’1 (d/2L)
Where:
φv = Total angle of view
d = The photocell diameter + the diameter of the limiting aperture
L = Distance from the photocell to the limiting aperture.
The limiting aperture is the point in the path between the photocell and the smoke plume where the angle of view is most restricted. In smoke generator smoke meters, this is normally an orifice plate.
10.3.3.4 Angle of Projection. You must check construction geometry to ensure that the total angle of projection of the lamp on the smoke plume does not exceed 15 degrees. Calculate the total angle of projection as follows:
φp = 2 tanˆ’1 (d/2L)
Where:
φp = Total angle of projection
d = The sum of the length of the lamp filament + the diameter of the limiting aperture
L = The distance from the lamp to the limiting aperture.
10.3.3.5 Calibration Error. Using neutral-density filters of known opacity, you must check the error between the actual response and the theoretical linear response of the smoke meter. This check is accomplished by first calibrating the smoke meter according to Section 10.3.2 of this method and then inserting a series of three neutral-density filters of nominal opacity of 20, 50, and 75 percent in the smoke meter path length. Use filters calibrated within 2 percent. Care should be taken when inserting the filters to prevent stray light from affecting the meter. Make a total of five non-consecutive readings for each filter. The maximum opacity error on any one reading shall be ±3 percent.
10.3.3.6 Zero and Span Drift. Determine the zero and span drift by calibrating and operating the smoke generator in a normal manner over a 1-hour period. The drift is measured by checking the zero and span at the end of this period.
10.3.3.7 Response Time. Determine the response time by producing the series of five simulated 0 percent and 100 percent opacity values and observing the time required to reach stable response. Opacity values of 0 percent and 100 percent may be simulated by alternately switching the power to the light source off and on while the smoke generator is not operating.
11.0 Analytical Procedures [Reserved]
12.0 Data Analysis and Calculations
12.1 Time-Averaged Regulations. A set of observations is composed of an appropriate number of consecutive observations determined by the averaging time specified (i.e., 8 observations for a two minute average). Divide the recorded observations into sets of appropriate time lengths for the specified averaging time. Sets must consist of consecutive observations; however, observations immediately preceding and following interrupted observations shall be deemed consecutive. Sets need not be consecutive in time and in no case shall two sets overlap. For each set of observations, calculate the average opacity by summing the opacity readings taken over the appropriate time period and dividing by the number of readings. For example, for a 2-minute average, eight consecutive readings would be averaged by adding the eight readings and dividing by eight.
13.0 Method Performance
13.1 Time-averaging Performances. The accuracy of test procedures for time-averaged regulations was evaluated through field studies that compare the opacity readings to a transmissometer. Analysis of these data shows that, as the time interval for averaging increases, the positive error decreases. For example, over a 2-minute time period, 90 percent of the results underestimated opacity or overestimated opacity by less than 9.5 percent opacity, while over a 6-minute time period, 90 percent of the data have less than a 7.5 percent positive error. Overall, the field studies demonstrated a negative bias. Over a 2-minute time period, 57 percent of the data have zero or negative error, and over a 6-minute time period, 58 percent of the data have zero or negative error. This means that observers are more likely to assign opacity values that are below, rather than above, the actual opacity value. Consequently, a larger percentage of noncompliance periods will be reported as compliant periods rather than compliant periods reported as violations. Table 203A-2 highlights the precision data results from the June 1985 report: "Opacity Errors for Averaging and Non Averaging Data Reduction and Reporting Techniques."
14.0 Pollution Prevention [Reserved]
15.0 Waste Management [Reserved]
16.0 Alternative Procedures [Reserved]
17.0 References
1. U.S. Environmental Protection Agency. Standards of Performance for New Stationary Sources; Appendix A; Method 9 for Visual Determination of the Opacity of Emissions from Stationary Sources. Final Rule. 39 FR 219. Washington, DC. U.S. Government Printing Office. November 12, 1974.
2. Office of Air and Radiation. "Quality Assurance Guideline for Visible Emission Training Programs." EPA-600/S4-83-011. Quality Assurance Division. Research Triangle Park, NC. May 1982.
3. Office of Research and Development. "Method 9 - Visible Determination of the Opacity of Emissions from Stationary Sources." February 1984. Quality Assurance Handbook for Air Pollution Measurement Systems. Volume III, Section 3.1.2. Stationary Source Specific Methods. EPA-600-4-77-027b. August 1977. Office of Research and Development Publications, 26 West Clair Street, Cincinnati, OH.
4. Office of Air Quality Planning and Standards. "Opacity Error for Averaging and Non-averaging Data Reduction and Reporting Techniques." Final Report-SR-1-6-85. Emission Measurement Branch, Research Triangle Park, NC. June 1985.
5. U.S. Environmental Protection Agency. Preparation, Adoption, and Submittal of State Implementation Plans. Methods for Measurement of PM10 Emissions from Stationary Sources. Final Rule. Federal Register. Washington, DC. U.S. Government Printing Office. Volume 55, No. 74. Pages 14246-14279. April 17, 1990.
6. Office of Air Quality Planning and Standards. "Collaborative Study of Opacity Observations of Fugitive Emissions from Unpaved Roads by Certified Observers." Emission Measurement Branch, Research Triangle Park, NC. October 1986.
7. Office of Air Quality Planning and Standards. "Field Data Forms and Instructions for EPA Methods 203A, 203B, and 203C." EPA 455/R-93-005. Stationary Source Compliance Division, Washington, DC, June 1993.
18.0 Tables, Diagrams, Flowcharts, and Validation Data
| Parameter | Specification |
|---|---|
| a. Light Source | Incandescent lamp operated at nominal rated voltage. |
| b. Spectral response of photocell | Photopic (daylight spectral response of the human eye - Citation 3). |
| c. Angle of view | 15° maximum total angle. |
| d. Angle of projection | 15° maximum total angle. |
| e. Calibration error | ±3% opacity, maximum. |
| f. Zero and span drift | ±1% opacity, 30 minutes |
| g. Response time | 5 seconds. |
| Averaging period |
Number of
observations |
Standard
deviation (% opacity) |
Amount with
<7.5% opacity difference |
|---|---|---|---|
| 15-second | 140,250 | 3.4 | 87 |
| 2 minutes | 17,694 | 2.6 | 92 |
| 3 minutes | 11,836 | 2.4 | 92 |
| 6 minutes | 5,954 | 2.1 | 93 |
Method 203B - Visual Determination of Opacity of Emissions From Stationary Sources for Time-Exception Regulations
1.0 Scope and Application
What is Method 203B?
Method 203B is an example test method suitable for State Implementation Plans (SIPs) and is applicable to the determination of the opacity of emissions from sources of visible emissions for time-exception regulations. A time-exception regulation means any regulation that allows predefined periods of opacity above the otherwise applicable opacity limit (e.g., allowing exceedances of 20 percent opacity for 3 minutes in 1 hour.)
Method 203B is virtually identical to EPA's Method 9 of 40 CFR part 60, Appendix A, except for the data-reduction procedures, which have been modified to apply to time-exception regulations. The certification procedures for this method are identical to those provided in Method 9. An example of a visible emission observation form and instructions for its use can be found in reference 7 of Section 17 of Method 203A.
2.0 Summary of Method
The opacity of emissions from sources of visible emissions is determined visually by a qualified observer.
3.0 Definitions [Reserved]
4.0 Interferences [Reserved]
5.0 Safety [Reserved]
6.0 Equipment and Supplies
What equipment and supplies are needed?
The same as specified in Section 6.0 of Method 203A.
7.0 Reagents and Standards [Reserved]
8.0 Sample Collection, Preservation, Storage, and Transport
What is the Test Procedure?
The observer qualified in accordance with Section 10 of Method 203A must use the following procedures for visually determining the opacity of emissions.
8.1 Procedures for Emissions From Stationary Sources. The procedures for emissions from stationary sources are the same as specified in 8.1 of Method 203A.
8.2 Recording Observations. You must record opacity observations to the nearest 5 percent at 15-second intervals on an observational record sheet. Each observation recorded represents the average opacity of emissions for a 15-second period. The overall length of time for which observations are recorded must be appropriate to the applicable regulation.
9.0 Quality Control [Reserved]
10.0 Calibration and Standardization
The Calibration and Standardization requirements are the same as specified in Section 10 of Method 203A.
11.0 Analytical Procedures [Reserved]
12.0 Data Analysis and Calculations
Data Reduction for Time-Exception Regulations. For a time-exception regulation, reduce opacity observations as follows: Count the number of observations above the applicable standard and multiply that number by 0.25 to determine the minutes of emissions above the target opacity.
13.0 Method Performance
13.1 Time-Exception Regulations. "Opacity Errors for Averaging and Non-Averaging Data Reduction and Reporting Techniques" analyzed the time errors associated with false compliance or false non-compliance determinations resulting from a sample of 1110 opacity readings with 6-minute observation periods. The study applied a 20 percent opacity standard. Fifty-one percent of the data showed zero error in time determinations. The standard deviation was 97.5 seconds for the 6-minute time period.
13.1.1 Overall, the study showed a negative bias. Each reading is associated with a 15-second block of time. The readings were multiplied by 15 seconds and the resulting time spent above the standard was compared to the transmissometer results. The average amount of time that observations deviated from the transmissometer's determinations was ˆ’8.3 seconds. Seventy percent of the time determinations were either correct or underestimated the time of excess emissions. Consequently, a larger percentage of noncompliance periods would be reported as compliant periods rather than compliant periods reported as violations.
13.1.2 Some time-exception regulations reduce the data by averaging over 1-minute periods and then counting those minutes above the standard. This data reduction procedure results in a less stringent standard than determinations resulting from data reduction procedures of Method 203B.
14.0 Pollution Prevention [Reserved]
15.0 Waste Management [Reserved]
16.0 Alternative Procedures [Reserved]
17.0 References
The references are the same as specified in Section 17 of Method 203A.
18.0 Tables, Diagrams, Flowcharts, and Validation Data [Reserved]
Method 203C - Visual Determination of Opacity of Emissions From Stationary Sources for Instantaneous Limitation Regulations
1.0 Scope and Application
What is Method 203C?
Method 203C is an example test method suitable for State Implementation Plans (SIPs) and is applicable to the determination of the opacity of emissions from sources of visible emissions for regulations with an instantaneous opacity limitation. An instantaneous opacity limitation is an opacity limit which is never to be exceeded.
Method 203C is virtually identical to EPA's Method 9 of 40 CFR Part 60, Appendix A, except for 5-second reading intervals and the data-reduction procedures, which have been modified for instantaneous limitation regulations. The certification procedures for this method are virtually identical to Method 9. An example visible emission observation form and instructions for its use can be found in reference 7 of Section 17 of Method 203A.
2.0 Summary of Method
The opacity of emissions from sources of visible emissions is determined visually by an observer certified according to the procedures in Section 10 of Method 203A.
3.0 Definitions [Reserved]
4.0 Interferences [Reserved]
5.0 Safety [Reserved]
6.0 Equipment and Supplies
The equipment and supplies used are the same as Section 6.0 of Method 203A.
7.0 Reagents and Standards [Reserved]
8.0 Sample Collection, Preservation, Storage, and Transport
What is the Test Procedure?
The qualified observer must use the following procedures for visually determining the opacity of emissions.
8.1 Procedures for Emissions From Stationary Sources. These are the same as Section 8.1 of Method 203A.
8.1.1 Position. Same as Section 8.1.1 of Method 203A.
8.1.2 Field Records. Same as Section 8.1.2 of Method 203A.
8.1.3 Observations. Make opacity observations at the point of greatest opacity in that portion of the plume where condensed water vapor is not present. Do not look continuously at the plume, instead, observe the plume momentarily at 5-second intervals.
8.1.3.1 Attached Steam Plumes. Same as Section 8.1.3.1 of Method 203A.
8.1.3.2 Detached Steam Plumes. Same as Section 8.1.3.2 of Method 203A.
8.2 Recording Observations. You must record opacity observations to the nearest 5 percent at 5-second intervals on an observational record sheet. Each observation recorded represents the average of emissions for the 5-second period. The overall time for which recordings are made must be of a length appropriate to the applicable regulation for which opacity is being measured.
9.0 Quality Control [Reserved]
10.0 Calibration and Standardization
The calibration and standardization procedures are the same as Section 10 of Method 203A.
11.0 Analytical Procedures [Reserved]
12.0 Data Analysis and Calculations
12.1 Data Reduction for Instantaneous Limitation Regulations. For an instantaneous limitation regulation, a 1-minute averaging time will be used. You must divide the observations recorded on the record sheet into sets of consecutive observations. A set is composed of the consecutive observations made in 1 minute. Sets need not be consecutive in time, and in no case must two sets overlap. You must reduce opacity observations by dividing the sum of all observations recorded in a set by the number of observations recorded in each set.
12.2 Reduce opacity observations by averaging 12 consecutive observations recorded at 5-second intervals. Divide the observations recorded on the record sheet into sets of 12 consecutive observations. For each set of 12 observations, calculate the average by summing the opacity of the 12 observations and dividing this sum by 12.
13.0 Method Performance
The results of the "Collaborative Study of Opacity Observations at Five-second Intervals by Certified Observers" are almost identical to those of previous studies of Method 9 observations taken at 15-second intervals and indicate that observers can make valid observations at 5-second intervals. The average difference of all observations from the transmissometer values was 8.8 percent opacity, which shows a fairly high negative bias. Underestimating the opacity of the visible emissions is more likely than overestimating the opacity of the emissions.
14.0 Pollution Prevention [Reserved]
15.0 Waste Management [Reserved]
16.0 Alternative Procedures [Reserved]
17.0 References
The references are the same as references 1-7 in Method 203A in addition to the following:
1. Office of Air Quality Planning and Standards. "Collaborative Study of Opacity Observations at Five-second Intervals by Certified Observers." Docket A-84-22, IV-A-2. Emission Measurement Branch, Research Triangle Park, N.C. September 1990.
18.0 Tables, Diagrams, Flowcharts, and Validation Data
Method 204 - Criteria for and Verification of a Permanent or Temporary Total Enclosure
1. Scope and Application
This procedure is used to determine whether a permanent or temporary enclosure meets the criteria for a total enclosure. An existing building may be used as a temporary or permanent enclosure as long as it meets the appropriate criteria described in this method.
2. Summary of Method
An enclosure is evaluated against a set of criteria. If the criteria are met and if all the exhaust gases from the enclosure are ducted to a control device, then the volatile organic compounds (VOC) capture efficiency (CE) is assumed to be 100 percent, and CE need not be measured. However, if part of the exhaust gas stream is not ducted to a control device, CE must be determined.
3. Definitions
3.1 Natural Draft Opening (NDO). Any permanent opening in the enclosure that remains open during operation of the facility and is not connected to a duct in which a fan is installed.
3.2 Permanent Total Enclosure (PE). A permanently installed enclosure that completely surrounds a source of emissions such that all VOC emissions are captured and contained for discharge to a control device.
3.3 Temporary Total Enclosure (TTE). A temporarily installed enclosure that completely surrounds a source of emissions such that all VOC emissions that are not directed through the control device (i.e., uncaptured) are captured by the enclosure and contained for discharge through ducts that allow for the accurate measurement of the uncaptured VOC emissions.
3.4 Building Enclosure (BE). An existing building that is used as a TTE.
4. Safety
An evaluation of the proposed building materials and the design for the enclosure is recommended to minimize any potential hazards.
5. Criteria for Temporary Total Enclosure
5.1 Any NDO shall be at least four equivalent opening diameters from each VOC emitting point unless otherwise specified by the Administrator.
5.2 Any exhaust point from the enclosure shall be at least four equivalent duct or hood diameters from each NDO.
5.3 The total area of all NDO's shall not exceed 5 percent of the surface area of the enclosure's four walls, floor, and ceiling.
5.4 The average facial velocity (FV) of air through all NDO's shall be at least 3,600 m/hr (200 fpm). The direction of air flow through all NDO's shall be into the enclosure.
5.5 All access doors and windows whose areas are not included in section 5.3 and are not included in the calculation in section 5.4 shall be closed during routine operation of the process.
6. Criteria for a Permanent Total Enclosure
6.1 Same as sections 5.1 and 5.3 through 5.5.
6.2 All VOC emissions must be captured and contained for discharge through a control device.
7. Quality Control
7.1 The success of this method lies in designing the TTE to simulate the conditions that exist without the TTE (i.e., the effect of the TTE on the normal flow patterns around the affected facility or the amount of uncaptured VOC emissions should be minimal). The TTE must enclose the application stations, coating reservoirs, and all areas from the application station to the oven. The oven does not have to be enclosed if it is under negative pressure. The NDO's of the temporary enclosure and an exhaust fan must be properly sized and placed.
7.2 Estimate the ventilation rate of the TTE that best simulates the conditions that exist without the TTE (i.e., the effect of the TTE on the normal flow patterns around the affected facility or the amount of uncaptured VOC emissions should be minimal). Figure 204-1 or the following equation may be used as an aid.
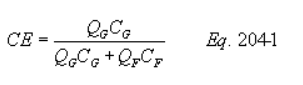
Measure the concentration (CG) and flow rate (QG) of the captured gas stream, specify a safe concentration (CF) for the uncaptured gas stream, estimate the CE, and then use the plot in Figure 204-1 or Equation 204-1 to determine the volumetric flow rate of the uncaptured gas stream (QF). An exhaust fan that has a variable flow control is desirable.
7.3 Monitor the VOC concentration of the captured gas steam in the duct before the capture device without the TTE. To minimize the effect of temporal variation on the captured emissions, the baseline measurement should be made over as long a time period as practical. However, the process conditions must be the same for the measurement in section 7.5 as they are for this baseline measurement. This may require short measuring times for this quality control check before and after the construction of the TTE.
7.4 After the TTE is constructed, monitor the VOC concentration inside the TTE. This concentration should not continue to increase, and must not exceed the safe level according to Occupational Safety and Health Administration requirements for permissible exposure limits. An increase in VOC concentration indicates poor TTE design.
7.5 Monitor the VOC concentration of the captured gas stream in the duct before the capture device with the TTE. To limit the effect of the TTE on the process, the VOC concentration with and without the TTE must be within 10 percent. If the measurements do not agree, adjust the ventilation rate from the TTE until they agree within 10 percent.
8. Procedure
8.1 Determine the equivalent diameters of the NDO's and determine the distances from each VOC emitting point to all NDO's. Determine the equivalent diameter of each exhaust duct or hood and its distance to all NDO's. Calculate the distances in terms of equivalent diameters. The number of equivalent diameters shall be at least four.
8.2 Measure the total surface area (AT) of the enclosure and the total area (AN) of all NDO's in the enclosure. Calculate the NDO to enclosure area ratio (NEAR) as follows:
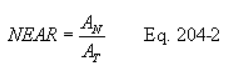
The NEAR must be ‰¤0.05.
8.3 Measure the volumetric flow rate, corrected to standard conditions, of each gas stream exiting the enclosure through an exhaust duct or hood using EPA Method 2. In some cases (e.g., when the building is the enclosure), it may be necessary to measure the volumetric flow rate, corrected to standard conditions, of each gas stream entering the enclosure through a forced makeup air duct using Method 2. Calculate FV using the following equation:
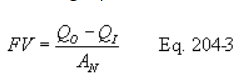
where:
QO = the sum of the volumetric flow from all gas streams exiting the enclosure through an exhaust duct or hood.
QI = the sum of the volumetric flow from all gas streams into the enclosure through a forced makeup air duct; zero, if there is no forced makeup air into the enclosure.
AN = total area of all NDO's in enclosure.
The FV shall be at least 3,600 m/hr (200 fpm). Alternatively, measure the pressure differential across the enclosure. A pressure drop of 0.013 mm Hg (0.007 in. H2O) corresponds to an FV of 3,600 m/hr (200 fpm).
8.4 Verify that the direction of air flow through all NDO's is inward. If FV is less than 9,000 m/hr (500 fpm), the continuous inward flow of air shall be verified using streamers, smoke tubes, or tracer gases. Monitor the direction of air flow for at least 1 hour, with checks made no more than 10 minutes apart. If FV is greater than 9,000 m/hr (500 fpm), the direction of air flow through the NDOs shall be presumed to be inward at all times without verification.
9. Diagrams
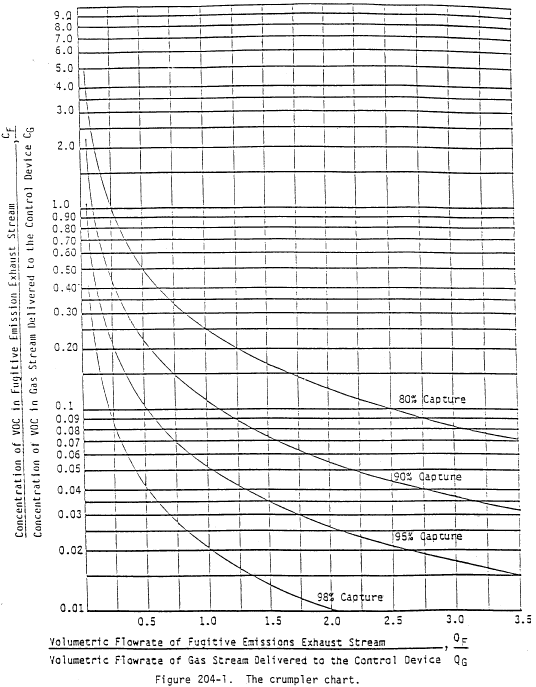
Method 204A - Volatile Organic Compounds Content in Liquid Input Stream
1. Scope and Application
1.1 Applicability. This procedure is applicable for determining the input of volatile organic compounds (VOC). It is intended to be used in the development of liquid/gas protocols for determining VOC capture efficiency (CE) for surface coating and printing operations.
1.2 Principle. The amount of VOC introduced to the process (L) is the sum of the products of the weight (W) of each VOC containing liquid (ink, paint, solvent, etc.) used and its VOC content (V).
1.3 Sampling Requirements. A CE test shall consist of at least three sampling runs. Each run shall cover at least one complete production cycle, but shall be at least 3 hours long. The sampling time for each run need not exceed 8 hours, even if the production cycle has not been completed. Alternative sampling times may be used with the approval of the Administrator.
2. Summary of Method
The amount of VOC containing liquid introduced to the process is determined as the weight difference of the feed material before and after each sampling run. The VOC content of the liquid input material is determined by volatilizing a small aliquot of the material and analyzing the volatile material using a flame ionization analyzer (FIA). A sample of each VOC containing liquid is analyzed with an FIA to determine V.
3. Safety
Because this procedure is often applied in highly explosive areas, caution and care should be exercised in choosing, installing, and using the appropriate equipment.
4. Equipment and Supplies
Mention of trade names or company products does not constitute endorsement. All gas concentrations (percent, ppm) are by volume, unless otherwise noted.
4.1 Liquid Weight.
4.1.1 Balances/Digital Scales. To weigh drums of VOC containing liquids to within 0.2 lb or 1.0 percent of the total weight of VOC liquid used.
4.1.2 Volume Measurement Apparatus (Alternative). Volume meters, flow meters, density measurement equipment, etc., as needed to achieve the same accuracy as direct weight measurements.
4.2 VOC Content (FIA Technique). The liquid sample analysis system is shown in Figures 204A-1 and 204A-2. The following equipment is required:
4.2.1 Sample Collection Can. An appropriately-sized metal can to be used to collect VOC containing materials. The can must be constructed in such a way that it can be grounded to the coating container.
4.2.2 Needle Valves. To control gas flow.
4.2.3 Regulators. For carrier gas and calibration gas cylinders.
4.2.4 Tubing. Teflon or stainless steel tubing with diameters and lengths determined by connection requirements of equipment. The tubing between the sample oven outlet and the FIA shall be heated to maintain a temperature of 120 ±5°C.
4.2.5 Atmospheric Vent. A tee and 0- to 0.5-liter/min rotameter placed in the sampling line between the carrier gas cylinder and the VOC sample vessel to release the excess carrier gas. A toggle valve placed between the tee and the rotameter facilitates leak tests of the analysis system.
4.2.6 Thermometer. Capable of measuring the temperature of the hot water bath to within 1°C.
4.2.7 Sample Oven. Heated enclosure, containing calibration gas coil heaters, critical orifice, aspirator, and other liquid sample analysis components, capable of maintaining a temperature of 120 ±5°C.
4.2.8 Gas Coil Heaters. Sufficient lengths of stainless steel or Teflon tubing to allow zero and calibration gases to be heated to the sample oven temperature before entering the critical orifice or aspirator.
4.2.9 Water Bath. Capable of heating and maintaining a sample vessel temperature of 100 ±5°C.
4.2.10 Analytical Balance. To measure ±0.001 g.
4.2.11 Disposable Syringes. 2-cc or 5-cc.
4.2.12 Sample Vessel. Glass, 40-ml septum vial. A separate vessel is needed for each sample.
4.2.13 Rubber Stopper. Two-hole stopper to accommodate 3.2-mm ( 1/8-in.) Teflon tubing, appropriately sized to fit the opening of the sample vessel. The rubber stopper should be wrapped in Teflon tape to provide a tighter seal and to prevent any reaction of the sample with the rubber stopper. Alternatively, any leak-free closure fabricated of nonreactive materials and accommodating the necessary tubing fittings may be used.
4.2.14 Critical Orifices. Calibrated critical orifices capable of providing constant flow rates from 50 to 250 ml/min at known pressure drops. Sapphire orifice assemblies (available from O'Keefe Controls Company) and glass capillary tubing have been found to be adequate for this application.
4.2.15 Vacuum Gauge. Zero to 760-mm (0- to 30-in.) Hg U-Tube manometer or vacuum gauge.
4.2.16 Pressure Gauge. Bourdon gauge capable of measuring the maximum air pressure at the aspirator inlet (e.g., 100 psig).
4.2.17 Aspirator. A device capable of generating sufficient vacuum at the sample vessel to create critical flow through the calibrated orifice when sufficient air pressure is present at the aspirator inlet. The aspirator must also provide sufficient sample pressure to operate the FIA. The sample is also mixed with the dilution gas within the aspirator.
4.2.18 Soap Bubble Meter. Of an appropriate size to calibrate the critical orifices in the system.
4.2.19 Organic Concentration Analyzer. An FIA with a span value of 1.5 times the expected concentration as propane; however, other span values may be used if it can be demonstrated that they would provide more accurate measurements. The FIA instrument should be the same instrument used in the gaseous analyses adjusted with the same fuel, combustion air, and sample back-pressure (flow rate) settings. The system shall be capable of meeting or exceeding the following specifications:
4.2.19.1 Zero Drift. Less than ±3.0 percent of the span value.
4.2.19.2 Calibration Drift. Less than ±3.0 percent of the span value.
4.2.19.3 Calibration Error. Less than ±5.0 percent of the calibration gas value.
4.2.20 Integrator/Data Acquisition System. An analog or digital device or computerized data acquisition system used to integrate the FIA response or compute the average response and record measurement data. The minimum data sampling frequency for computing average or integrated values is one measurement value every 5 seconds. The device shall be capable of recording average values at least once per minute.
4.2.21 Chart Recorder (Optional). A chart recorder or similar device is recommended to provide a continuous analog display of the measurement results during the liquid sample analysis.
5. Reagents and Standards
5.1 Calibration and Other Gases. Gases used for calibration, fuel, and combustion air (if required) are contained in compressed gas cylinders. All calibration gases shall be traceable to National Institute of Standards and Technology standards and shall be certified by the manufacturer to ±1 percent of the tag value. Additionally, the manufacturer of the cylinder should provide a recommended shelf life for each calibration gas cylinder over which the concentration does not change more than ±2 percent from the certified value. For calibration gas values not generally available, dilution systems calibrated using Method 205 may be used. Alternative methods for preparing calibration gas mixtures may be used with the approval of the Administrator.
5.1.1 Fuel. The FIA manufacturer's recommended fuel should be used. A 40 percent H2/60 percent He or 40 percent H2/60 percent N2 gas mixture is recommended to avoid an oxygen synergism effect that reportedly occurs when oxygen concentration varies significantly from a mean value. Other mixtures may be used provided the tester can demonstrate to the Administrator that there is no oxygen synergism effect.
5.1.2 Carrier Gas. High purity air with less than 1 ppm of organic material (as propane) or less than 0.1 percent of the span value, whichever is greater.
5.1.3 FIA Linearity Calibration Gases. Low-, mid-, and high-range gas mixture standards with nominal propane concentrations of 20-30, 45-55, and 70-80 percent of the span value in air, respectively. Other calibration values and other span values may be used if it can be shown to the Administrator's satisfaction that equally accurate measurements would be achieved.
5.1.4 System Calibration Gas. Gas mixture standard containing propane in air, approximating the undiluted VOC concentration expected for the liquid samples.
6. Sample Collection, Preservation and Storage
6.1 Samples must be collected in a manner that prevents or minimizes loss of volatile components and that does not contaminate the coating reservoir.
6.2 Collect a 100-ml or larger sample of the VOC containing liquid mixture at each application location at the beginning and end of each test run. A separate sample should be taken of each VOC containing liquid added to the application mixture during the test run. If a fresh drum is needed during the sampling run, then obtain a sample from the fresh drum.
6.3 When collecting the sample, ground the sample container to the coating drum. Fill the sample container as close to the rim as possible to minimize the amount of headspace.
6.4 After the sample is collected, seal the container so the sample cannot leak out or evaporate.
6.5 Label the container to clearly identify the contents.
7. Quality Control
7.1 Required instrument quality control parameters are found in the following sections:
7.1.1 The FIA system must be calibrated as specified in section 8.1.
7.1.2 The system drift check must be performed as specified in section 8.2.
8. Calibration and Standardization
8.1 FIA Calibration and Linearity Check. Make necessary adjustments to the air and fuel supplies for the FIA and ignite the burner. Allow the FIA to warm up for the period recommended by the manufacturer. Inject a calibration gas into the measurement system and adjust the back-pressure regulator to the value required to achieve the flow rates specified by the manufacturer. Inject the zero- and the high-range calibration gases and adjust the analyzer calibration to provide the proper responses. Inject the low- and mid-range gases and record the responses of the measurement system. The calibration and linearity of the system are acceptable if the responses for all four gases are within 5 percent of the respective gas values. If the performance of the system is not acceptable, repair or adjust the system and repeat the linearity check. Conduct a calibration and linearity check after assembling the analysis system and after a major change is made to the system.
8.2 Systems Drift Checks. After each sample, repeat the system calibration checks in section 9.2.7 before any adjustments to the FIA or measurement system are made. If the zero or calibration drift exceeds ±3 percent of the span value, discard the result and repeat the analysis.
Alternatively, recalibrate the FIA as in section 8.1 and report the results using both sets of calibration data (i.e., data determined prior to the test period and data determined following the test period). The data that results in the lowest CE value shall be reported as the results for the test run.
8.3 Critical Orifice Calibration.
8.3.1 Each critical orifice must be calibrated at the specific operating conditions under which it will be used. Therefore, assemble all components of the liquid sample analysis system as shown in Figure 204A-3. A stopwatch is also required.
8.3.2 Turn on the sample oven, sample line, and water bath heaters, and allow the system to reach the proper operating temperature. Adjust the aspirator to a vacuum of 380 mm (15 in.) Hg vacuum. Measure the time required for one soap bubble to move a known distance and record barometric pressure.
8.3.3 Repeat the calibration procedure at a vacuum of 406 mm (16 in.) Hg and at 25-mm (1-in.) Hg intervals until three consecutive determinations provide the same flow rate. Calculate the critical flow rate for the orifice in ml/min at standard conditions. Record the vacuum necessary to achieve critical flow.
9. Procedure
9.1 Determination of Liquid Input Weight.
9.1.1 Weight Difference. Determine the amount of material introduced to the process as the weight difference of the feed material before and after each sampling run. In determining the total VOC containing liquid usage, account for:
(a) The initial (beginning) VOC containing liquid mixture.
(b) Any solvent added during the test run.
(c) Any coating added during the test run.
(d) Any residual VOC containing liquid mixture remaining at the end of the sample run.
9.1.1.1 Identify all points where VOC containing liquids are introduced to the process. To obtain an accurate measurement of VOC containing liquids, start with an empty fountain (if applicable). After completing the run, drain the liquid in the fountain back into the liquid drum (if possible) and weigh the drum again. Weigh the VOC containing liquids to ±0.5 percent of the total weight (full) or ±1.0 percent of the total weight of VOC containing liquid used during the sample run, whichever is less. If the residual liquid cannot be returned to the drum, drain the fountain into a preweighed empty drum to determine the final weight of the liquid.
9.1.1.2 If it is not possible to measure a single representative mixture, then weigh the various components separately (e.g., if solvent is added during the sampling run, weigh the solvent before it is added to the mixture). If a fresh drum of VOC containing liquid is needed during the run, then weigh both the empty drum and fresh drum.
9.1.2 Volume Measurement (Alternative). If direct weight measurements are not feasible, the tester may use volume meters or flow rate meters and density measurements to determine the weight of liquids used if it can be demonstrated that the technique produces results equivalent to the direct weight measurements. If a single representative mixture cannot be measured, measure the components separately.
9.2 Determination of VOC Content in Input Liquids
9.2.1 Assemble the liquid VOC content analysis system as shown in Figure 204A-1.
9.2.2 Permanently identify all of the critical orifices that may be used. Calibrate each critical orifice under the expected operating conditions (i.e., sample vacuum and temperature) against a volume meter as described in section 8.3.
9.2.3 Label and tare the sample vessels (including the stoppers and caps) and the syringes.
9.2.4 Install an empty sample vessel and perform a leak test of the system. Close the carrier gas valve and atmospheric vent and evacuate the sample vessel to 250 mm (10 in.) Hg absolute or less using the aspirator. Close the toggle valve at the inlet to the aspirator and observe the vacuum for at least 1 minute. If there is any change in the sample pressure, release the vacuum, adjust or repair the apparatus as necessary, and repeat the leak test.
9.2.5 Perform the analyzer calibration and linearity checks according to the procedure in section 5.1. Record the responses to each of the calibration gases and the back-pressure setting of the FIA.
9.2.6 Establish the appropriate dilution ratio by adjusting the aspirator air supply or substituting critical orifices. Operate the aspirator at a vacuum of at least 25 mm (1 in.) Hg greater than the vacuum necessary to achieve critical flow. Select the dilution ratio so that the maximum response of the FIA to the sample does not exceed the high-range calibration gas.
9.2.7 Perform system calibration checks at two levels by introducing compressed gases at the inlet to the sample vessel while the aspirator and dilution devices are operating. Perform these checks using the carrier gas (zero concentration) and the system calibration gas. If the response to the carrier gas exceeds ±0.5 percent of span, clean or repair the apparatus and repeat the check. Adjust the dilution ratio as necessary to achieve the correct response to the upscale check, but do not adjust the analyzer calibration. Record the identification of the orifice, aspirator air supply pressure, FIA back-pressure, and the responses of the FIA to the carrier and system calibration gases.
9.2.8 After completing the above checks, inject the system calibration gas for approximately 10 minutes. Time the exact duration of the gas injection using a stopwatch. Determine the area under the FIA response curve and calculate the system response factor based on the sample gas flow rate, gas concentration, and the duration of the injection as compared to the integrated response using Equations 204A-2 and 204A-3.
9.2.9 Verify that the sample oven and sample line temperatures are 120 ±5°C and that the water bath temperature is 100 ±5°C.
9.2.10 Fill a tared syringe with approximately 1 g of the VOC containing liquid and weigh it. Transfer the liquid to a tared sample vessel. Plug the sample vessel to minimize sample loss. Weigh the sample vessel containing the liquid to determine the amount of sample actually received. Also, as a quality control check, weigh the empty syringe to determine the amount of material delivered. The two coating sample weights should agree within 0.02 g. If not, repeat the procedure until an acceptable sample is obtained.
9.2.11 Connect the vessel to the analysis system. Adjust the aspirator supply pressure to the correct value. Open the valve on the carrier gas supply to the sample vessel and adjust it to provide a slight excess flow to the atmospheric vent. As soon as the initial response of the FIA begins to decrease, immerse the sample vessel in the water bath. (Applying heat to the sample vessel too soon may cause the FIA response to exceed the calibrated range of the instrument and, thus, invalidate the analysis.)
9.2.12 Continuously measure and record the response of the FIA until all of the volatile material has been evaporated from the sample and the instrument response has returned to the baseline (i.e., response less than 0.5 percent of the span value). Observe the aspirator supply pressure, FIA back-pressure, atmospheric vent, and other system operating parameters during the run; repeat the analysis procedure if any of these parameters deviate from the values established during the system calibration checks in section 9.2.7. After each sample, perform the drift check described in section 8.2. If the drift check results are acceptable, calculate the VOC content of the sample using the equations in section 11.2. Alternatively, recalibrate the FIA as in section 8.1 and report the results using both sets of calibration data (i.e., data determined prior to the test period and data determined following the test period). The data that results in the lowest CE value shall be reported as the results for the test run. Integrate the area under the FIA response curve, or determine the average concentration response and the duration of sample analysis.
10. Data Analysis and Calculations
10.1 Nomenclature.
AL = area under the response curve of the liquid sample, area count.
AS = area under the response curve of the calibration gas, area count.
CS = actual concentration of system calibration gas, ppm propane.
K = 1.830 × 10ˆ’9 g/(ml-ppm).
L = total VOC content of liquid input, kg.
ML = mass of liquid sample delivered to the sample vessel, g.
q = flow rate through critical orifice, ml/min.
RF = liquid analysis system response factor, g/area count.
θS = total gas injection time for system calibration gas during integrator calibration, min.
VFj = final VOC fraction of VOC containing liquid j.
VIj = initial VOC fraction of VOC containing liquid j.
VAj = VOC fraction of VOC containing liquid j added during the run.
V = VOC fraction of liquid sample.
WFj = weight of VOC containing liquid j remaining at end of the run, kg.
WIj = weight of VOC containing liquid j at beginning of the run, kg.
WAj = weight of VOC containing liquid j added during the run, kg.
10.2 Calculations
10.2.1 Total VOC Content of the Input VOC Containing Liquid.
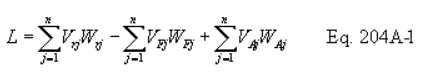
10.2.2 Liquid Sample Analysis System Response Factor for Systems Using Integrators, Grams/Area Count.
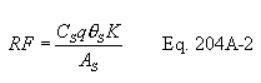
10.2.3 VOC Content of the Liquid Sample.
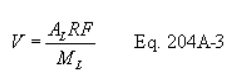
11. Method Performance
The measurement uncertainties are estimated for each VOC containing liquid as follows: W = ±2.0 percent and V = ±4.0 percent. Based on these numbers, the probable uncertainty for L is estimated at about ±4.5 percent for each VOC containing liquid.
12. Diagrams
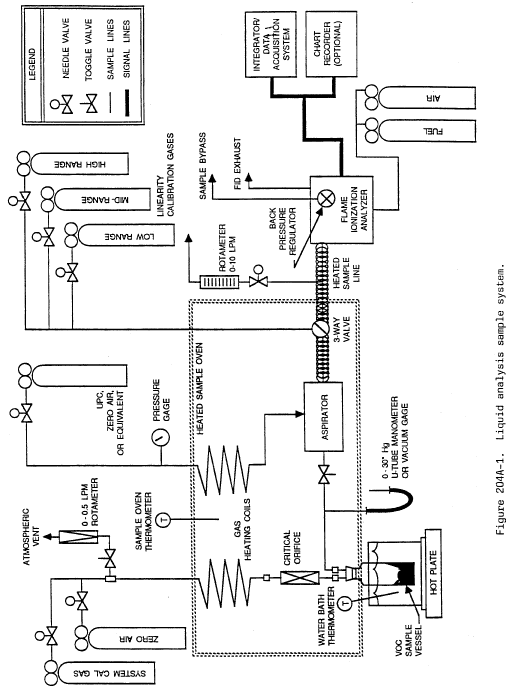
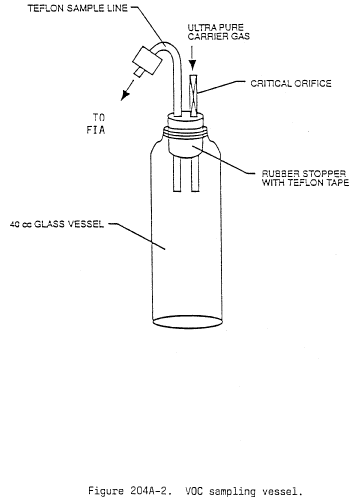
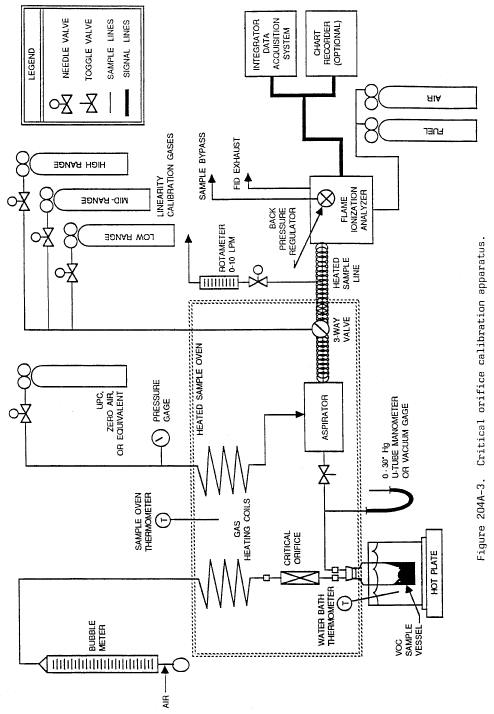
Method 204B - Volatile Organic Compounds Emissions in Captured Stream
1. Scope and Application
1.1 Applicability. This procedure is applicable for determining the volatile organic compounds (VOC) content of captured gas streams. It is intended to be used in the development of a gas/gas protocol for determining VOC capture efficiency (CE) for surface coating and printing operations. The procedure may not be acceptable in certain site-specific situations [e.g., when: (1) direct-fired heaters or other circumstances affect the quantity of VOC at the control device inlet; and (2) particulate organic aerosols are formed in the process and are present in the captured emissions].
1.2 Principle. The amount of VOC captured (G) is calculated as the sum of the products of the VOC content (CGj), the flow rate (QGj), and the sample time (ΘC) from each captured emissions point.
1.3 Sampling Requirements. A CE test shall consist of at least three sampling runs. Each run shall cover at least one complete production cycle, but shall be at least 3 hours long. The sampling time for each run need not exceed 8 hours, even if the production cycle has not been completed. Alternative sampling times may be used with the approval of the Administrator.
2. Summary of Method
A gas sample is extracted from the source though a heated sample line and, if necessary, a glass fiber filter to a flame ionization analyzer (FIA).
3. Safety
Because this procedure is often applied in highly explosive areas, caution and care should be exercised in choosing, installing, and using the appropriate equipment.
4. Equipment and Supplies
Mention of trade names or company products does not constitute endorsement. All gas concentrations (percent, ppm) are by volume, unless otherwise noted.
4.1 Gas VOC Concentration. A schematic of the measurement system is shown in Figure 204B-1. The main components are as follows:
4.1.1 Sample Probe. Stainless steel or equivalent. The probe shall be heated to prevent VOC condensation.
4.1.2 Calibration Valve Assembly. Three-way valve assembly at the outlet of the sample probe to direct the zero and calibration gases to the analyzer. Other methods, such as quick-connect lines, to route calibration gases to the outlet of the sample probe are acceptable.
4.1.3 Sample Line. Stainless steel or Teflon tubing to transport the sample gas to the analyzer. The sample line must be heated to prevent condensation.
4.1.4 Sample Pump. A leak-free pump, to pull the sample gas through the system at a flow rate sufficient to minimize the response time of the measurement system. The components of the pump that contact the gas stream shall be constructed of stainless steel or Teflon. The sample pump must be heated to prevent condensation.
4.1.5 Sample Flow Rate Control. A sample flow rate control valve and rotameter, or equivalent, to maintain a constant sampling rate within 10 percent. The flow rate control valve and rotameter must be heated to prevent condensation. A control valve may also be located on the sample pump bypass loop to assist in controlling the sample pressure and flow rate.
4.1.6 Organic Concentration Analyzer. An FIA with a span value of 1.5 times the expected concentration as propane; however, other span values may be used if it can be demonstrated to the Administrator's satisfaction that they would provide equally accurate measurements. The system shall be capable of meeting or exceeding the following specifications:
4.1.6.1 Zero Drift. Less than ±3.0 percent of the span value.
4.1.6.2 Calibration Drift. Less than ±3.0 percent of the span value.
4.1.6.3 Calibration Error. Less than ±5.0 percent of the calibration gas value.
4.1.6.4 Response Time. Less than 30 seconds.
4.1.7 Integrator/Data Acquisition System. An analog or digital device, or computerized data acquisition system used to integrate the FIA response or compute the average response and record measurement data. The minimum data sampling frequency for computing average or integrated values is one measurement value every 5 seconds. The device shall be capable of recording average values at least once per minute.
4.2 Captured Emissions Volumetric Flow Rate.
4.2.1 Method 2 or 2A Apparatus. For determining volumetric flow rate.
4.2.2 Method 3 Apparatus and Reagents. For determining molecular weight of the gas stream. An estimate of the molecular weight of the gas stream may be used if approved by the Administrator.
4.2.3 Method 4 Apparatus and Reagents. For determining moisture content, if necessary.
5. Reagents and Standards
5.1 Calibration and Other Gases. Gases used for calibration, fuel, and combustion air (if required) are contained in compressed gas cylinders. All calibration gases shall be traceable to National Institute of Standards and Technology standards and shall be certified by the manufacturer to ±1 percent of the tag value. Additionally, the manufacturer of the cylinder should provide a recommended shelf life for each calibration gas cylinder over which the concentration does not change more than ±2 percent from the certified value. For calibration gas values not generally available, dilution systems calibrated using Method 205 may be used. Alternative methods for preparing calibration gas mixtures may be used with the approval of the Administrator.
5.1.1 Fuel. The FIA manufacturer's recommended fuel should be used. A 40 percent H2/60 percent He or 40 percent H2/60 percent N2 gas mixture is recommended to avoid an oxygen synergism effect that reportedly occurs when oxygen concentration varies significantly from a mean value. Other mixtures may be used provided the tester can demonstrate to the Administrator that there is no oxygen synergism effect.
5.1.2 Carrier Gas. High purity air with less than 1 ppm of organic material (as propane or carbon equivalent) or less than 0.1 percent of the span value, whichever is greater.
5.1.3 FIA Linearity Calibration Gases. Low-, mid-, and high-range gas mixture standards with nominal propane concentrations of 20-30, 45-55, and 70-80 percent of the span value in air, respectively. Other calibration values and other span values may be used if it can be shown to the Administrator's satisfaction that equally accurate measurements would be achieved.
5.2 Particulate Filter. An in-stack or an out-of-stack glass fiber filter is recommended if exhaust gas particulate loading is significant. An out-of-stack filter must be heated to prevent any condensation unless it can be demonstrated that no condensation occurs.
6. Quality Control
6.1 Required instrument quality control parameters are found in the following sections:
6.1.1 The FIA system must be calibrated as specified in section 7.1.
6.1.2 The system drift check must be performed as specified in section 7.2.
6.1.3 The system check must be conducted as specified in section 7.3.
7. Calibration and Standardization
7.1 FIA Calibration and Linearity Check. Make necessary adjustments to the air and fuel supplies for the FIA and ignite the burner. Allow the FIA to warm up for the period recommended by the manufacturer. Inject a calibration gas into the measurement system and adjust the back-pressure regulator to the value required to achieve the flow rates specified by the manufacturer. Inject the zero-and the high-range calibration gases and adjust the analyzer calibration to provide the proper responses. Inject the low- and mid-range gases and record the responses of the measurement system. The calibration and linearity of the system are acceptable if the responses for all four gases are within 5 percent of the respective gas values. If the performance of the system is not acceptable, repair or adjust the system and repeat the linearity check. Conduct a calibration and linearity check after assembling the analysis system and after a major change is made to the system.
7.2 Systems Drift Checks. Select the calibration gas that most closely approximates the concentration of the captured emissions for conducting the drift checks. Introduce the zero and calibration gases at the calibration valve assembly and verify that the appropriate gas flow rate and pressure are present at the FIA. Record the measurement system responses to the zero and calibration gases. The performance of the system is acceptable if the difference between the drift check measurement and the value obtained in section 7.1 is less than 3 percent of the span value. Alternatively, recalibrate the FIA as in section 7.1 and report the results using both sets of calibration data (i.e., data determined prior to the test period and data determined following the test period). The data that results in the lowest CE value shall be reported as the results for the test run. Conduct the system drift checks at the end of each run.
7.3 System Check. Inject the high-range calibration gas at the inlet of the sampling probe and record the response. The performance of the system is acceptable if the measurement system response is within 5 percent of the value obtained in section 7.1 for the high-range calibration gas. Conduct a system check before and after each test run.
8. Procedure
8.1. Determination of Volumetric Flow Rate of Captured Emissions.
8.1.1 Locate all points where emissions are captured from the affected facility. Using Method 1, determine the sampling points. Be sure to check each site for cyclonic or swirling flow.
8.1.2 Measure the velocity at each sampling site at least once every hour during each sampling run using Method 2 or 2A.
8.2 Determination of VOC Content of Captured Emissions.
8.2.1 Analysis Duration. Measure the VOC responses at each captured emissions point during the entire test run or, if applicable, while the process is operating. If there are multiple captured emission locations, design a sampling system to allow a single FIA to be used to determine the VOC responses at all sampling locations.
8.2.2 Gas VOC Concentration.
8.2.2.1 Assemble the sample train as shown in Figure 204B-1. Calibrate the FIA according to the procedure in section 7.1.
8.2.2.2 Conduct a system check according to the procedure in section 7.3.
8.2.2.3 Install the sample probe so that the probe is centrally located in the stack, pipe, or duct, and is sealed tightly at the stack port connection.
8.2.2.4 Inject zero gas at the calibration valve assembly. Allow the measurement system response to reach zero. Measure the system response time as the time required for the system to reach the effluent concentration after the calibration valve has been returned to the effluent sampling position.
8.2.2.5 Conduct a system check before, and a system drift check after, each sampling run according to the procedures in sections 7.2 and 7.3. If the drift check following a run indicates unacceptable performance (see section 7.3), the run is not valid. Alternatively, recalibrate the FIA as in section 7.1 and report the results using both sets of calibration data (i.e., data determined prior to the test period and data determined following the test period). The data that results in the lowest CE value shall be reported as the results for the test run. The tester may elect to perform system drift checks during the run not to exceed one drift check per hour.
8.2.2.6 Verify that the sample lines, filter, and pump temperatures are 120 ±5°C.
8.2.2.7 Begin sampling at the start of the test period and continue to sample during the entire run. Record the starting and ending times and any required process information as appropriate. If multiple captured emission locations are sampled using a single FIA, sample at each location for the same amount of time (e.g., 2 minutes) and continue to switch from one location to another for the entire test run. Be sure that total sampling time at each location is the same at the end of the test run. Collect at least four separate measurements from each sample point during each hour of testing. Disregard the measurements at each sampling location until two times the response time of the measurement system has elapsed. Continue sampling for at least 1 minute and record the concentration measurements.
8.2.3 Background Concentration.
Note:
Not applicable when the building is used as the temporary total enclosure (TTE).
8.2.3.1 Locate all natural draft openings (NDO's) of the TTE. A sampling point shall be at the center of each NDO, unless otherwise specified by the Administrator. If there are more than six NDO's, choose six sampling points evenly spaced among the NDO's.
8.2.3.2 Assemble the sample train as shown in Figure 204B-2. Calibrate the FIA and conduct a system check according to the procedures in sections 7.1 and 7.3.
Note:
This sample train shall be separate from the sample train used to measure the captured emissions.
8.2.3.3 Position the probe at the sampling location.
8.2.3.4 Determine the response time, conduct the system check, and sample according to the procedures described in sections 8.2.2.4 through 8.2.2.7.
8.2.4 Alternative Procedure. The direct interface sampling and analysis procedure described in section 7.2 of Method 18 may be used to determine the gas VOC concentration. The system must be designed to collect and analyze at least one sample every 10 minutes. If the alternative procedure is used to determine the VOC concentration of the captured emissions, it must also be used to determine the VOC concentration of the uncaptured emissions.
9. Data Analysis and Calculations
9.1 Nomenclature.
Ai = area of NDO i, ft 2.
AN = total area of all NDO's in the enclosure, ft 2.
CBi = corrected average VOC concentration of background emissions at point i, ppm propane.
CB = average background concentration, ppm propane.
CGj = corrected average VOC concentration of captured emissions at point j, ppm propane.
CDH = average measured concentration for the drift check calibration gas, ppm propane.
CDO = average system drift check concentration for zero concentration gas, ppm propane.
CH = actual concentration of the drift check calibration gas, ppm propane.
Ci = uncorrected average background VOC concentration measured at point i, ppm propane.
Cj = uncorrected average VOC concentration measured at point j, ppm propane.
G = total VOC content of captured emissions, kg.
K1 = 1.830 × 10ˆ’6 kg/(m 3-ppm).
n = number of measurement points.
QGj = average effluent volumetric flow rate corrected to standard conditions at captured emissions point j, m 3/min.
ΘC = total duration of captured emissions.
9.2 Calculations.
9.2.1 Total VOC Captured Emissions.
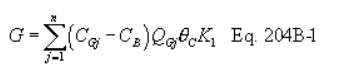
9.2.2 VOC Concentration of the Captured Emissions at Point j.
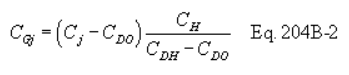
9.2.3 Background VOC Concentration at Point i.
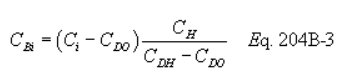
9.2.4 Average Background Concentration.
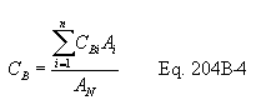
Note:
If the concentration at each point is within 20 percent of the average concentration of all points, then use the arithmetic average.
10. Method Performance
The measurement uncertainties are estimated for each captured or uncaptured emissions point as follows: QGj=±5.5 percent and CGj=±5.0 percent. Based on these numbers, the probable uncertainty for G is estimated at about ±7.4 percent.
11. Diagrams
Method 204C - Volatile Organic Compounds Emissions in Captured Stream (Dilution Technique)
1. Scope and Application
1.1 Applicability. This procedure is applicable for determining the volatile organic compounds (VOC) content of captured gas streams. It is intended to be used in the development of a gas/gas protocol in which uncaptured emissions are also measured for determining VOC capture efficiency (CE) for surface coating and printing operations. A dilution system is used to reduce the VOC concentration of the captured emissions to about the same concentration as the uncaptured emissions. The procedure may not be acceptable in certain site-specific situations [e.g., when: (1) direct-fired heaters or other circumstances affect the quantity of VOC at the control device inlet; and (2) particulate organic aerosols are formed in the process and are present in the captured emissions].
1.2 Principle. The amount of VOC captured (G) is calculated as the sum of the products of the VOC content (CGj), the flow rate (QGj), and the sampling time (ΘC) from each captured emissions point.
1.3 Sampling Requirements. A CE test shall consist of at least three sampling runs. Each run shall cover at least one complete production cycle, but shall be at least 3 hours long. The sampling time for each run need not exceed 8 hours, even if the production cycle has not been completed. Alternative sampling times may be used with the approval of the Administrator.
2. Summary of Method
A gas sample is extracted from the source using an in-stack dilution probe through a heated sample line and, if necessary, a glass fiber filter to a flame ionization analyzer (FIA). The sample train contains a sample gas manifold which allows multiple points to be sampled using a single FIA.
3. Safety
Because this procedure is often applied in highly explosive areas, caution and care should be exercised in choosing, installing, and using the appropriate equipment.
4. Equipment and Supplies
Mention of trade names or company products does not constitute endorsement. All gas concentrations (percent, ppm) are by volume, unless otherwise noted.
4.1 Gas VOC Concentration. A schematic of the measurement system is shown in Figure 204C-1. The main components are as follows:
4.1.1 Dilution System. A Kipp in-stack dilution probe and controller or similar device may be used. The dilution rate may be changed by substituting different critical orifices or adjustments of the aspirator supply pressure. The dilution system shall be heated to prevent VOC condensation. Note: An out-of-stack dilution device may be used.
4.1.2 Calibration Valve Assembly. Three-way valve assembly at the outlet of the sample probe to direct the zero and calibration gases to the analyzer. Other methods, such as quick-connect lines, to route calibration gases to the outlet of the sample probe are acceptable.
4.1.3 Sample Line. Stainless steel or Teflon tubing to transport the sample gas to the analyzer. The sample line must be heated to prevent condensation.
4.1.4 Sample Pump. A leak-free pump, to pull the sample gas through the system at a flow rate sufficient to minimize the response time of the measurement system. The components of the pump that contact the gas stream shall be constructed of stainless steel or Teflon. The sample pump must be heated to prevent condensation.
4.1.5 Sample Flow Rate Control. A sample flow rate control valve and rotameter, or equivalent, to maintain a constant sampling rate within 10 percent. The flow control valve and rotameter must be heated to prevent condensation. A control valve may also be located on the sample pump bypass loop to assist in controlling the sample pressure and flow rate.
4.1.6 Sample Gas Manifold. Capable of diverting a portion of the sample gas stream to the FIA, and the remainder to the bypass discharge vent. The manifold components shall be constructed of stainless steel or Teflon. If captured or uncaptured emissions are to be measured at multiple locations, the measurement system shall be designed to use separate sampling probes, lines, and pumps for each measurement location and a common sample gas manifold and FIA. The sample gas manifold and connecting lines to the FIA must be heated to prevent condensation.
Note:
Depending on the number of sampling points and their location, it may not be possible to use only one FIA. However to reduce the effect of calibration error, the number of FIA's used during a test should be keep as small as possible.
4.1.7 Organic Concentration Analyzer. An FIA with a span value of 1.5 times the expected concentration as propane; however, other span values may be used if it can be demonstrated to the Administrator's satisfaction that they would provide equally accurate measurements. The system shall be capable of meeting or exceeding the following specifications:
4.1.7.1 Zero Drift. Less than ±3.0 percent of the span value.
4.1.7.2 Calibration Drift. Less than ±3.0 percent of the span value.
4.1.7.3 Calibration Error. Less than ±5.0 percent of the calibration gas value.
4.1.7.4 Response Time. Less than 30 seconds.
4.1.8 Integrator/Data Acquisition System. An analog or digital device or computerized data acquisition system used to integrate the FIA response or compute the average response and record measurement data. The minimum data sampling frequency for computing average or integrated values is one measurement value every 5 seconds. The device shall be capable of recording average values at least once per minute.
4.2 Captured Emissions Volumetric Flow Rate.
4.2.1 Method 2 or 2A Apparatus. For determining volumetric flow rate.
4.2.2 Method 3 Apparatus and Reagents. For determining molecular weight of the gas stream. An estimate of the molecular weight of the gas stream may be used if approved by the Administrator.
4.2.3 Method 4 Apparatus and Reagents. For determining moisture content, if necessary.
5. Reagents and Standards
5.1 Calibration and Other Gases. Gases used for calibration, fuel, and combustion air (if required) are contained in compressed gas cylinders. All calibration gases shall be traceable to National Institute of Standards and Technology standards and shall be certified by the manufacturer to ±1 percent of the tag value. Additionally, the manufacturer of the cylinder should provide a recommended shelf life for each calibration gas cylinder over which the concentration does not change more than ±2 percent from the certified value. For calibration gas values not generally available, dilution systems calibrated using Method 205 may be used. Alternative methods for preparing calibration gas mixtures may be used with the approval of the Administrator.
5.1.1 Fuel. The FIA manufacturer's recommended fuel should be used. A 40 percent H2/60 percent He or 40 percent H2/60 percent N2 gas mixture is recommended to avoid an oxygen synergism effect that reportedly occurs when oxygen concentration varies significantly from a mean value. Other mixtures may be used provided the tester can demonstrate to the Administrator that there is no oxygen synergism effect
5.1.2 Carrier Gas and Dilution Air Supply. High purity air with less than 1 ppm of organic material (as propane or carbon equivalent), or less than 0.1 percent of the span value, whichever is greater.
5.1.3 FIA Linearity Calibration Gases. Low-, mid-, and high-range gas mixture standards with nominal propane concentrations of 20-30, 45-55, and 70-80 percent of the span value in air, respectively. Other calibration values and other span values may be used if it can be shown to the Administrator's satisfaction that equally accurate measurements would be achieved.
5.1.4 Dilution Check Gas. Gas mixture standard containing propane in air, approximately half the span value after dilution.
5.2 Particulate Filter. An in-stack or an out-of-stack glass fiber filter is recommended if exhaust gas particulate loading is significant. An out-of-stack filter must be heated to prevent any condensation unless it can be demonstrated that no condensation occurs.
6. Quality Control
6.1 Required instrument quality control parameters are found in the following sections:
6.1.1 The FIA system must be calibrated as specified in section 7.1.
6.1.2 The system drift check must be performed as specified in section 7.2.
6.1.3 The dilution factor must be determined as specified in section 7.3.
6.1.4 The system check must be conducted as specified in section 7.4.
7. Calibration and Standardization
7.1 FIA Calibration and Linearity Check. Make necessary adjustments to the air and fuel supplies for the FIA and ignite the burner. Allow the FIA to warm up for the period recommended by the manufacturer. Inject a calibration gas into the measurement system after the dilution system and adjust the back-pressure regulator to the value required to achieve the flow rates specified by the manufacturer. Inject the zero-and the high-range calibration gases and adjust the analyzer calibration to provide the proper responses. Inject the low-and mid-range gases and record the responses of the measurement system. The calibration and linearity of the system are acceptable if the responses for all four gases are within 5 percent of the respective gas values. If the performance of the system is not acceptable, repair or adjust the system and repeat the linearity check. Conduct a calibration and linearity check after assembling the analysis system and after a major change is made to the system.
7.2 Systems Drift Checks. Select the calibration gas that most closely approximates the concentration of the diluted captured emissions for conducting the drift checks. Introduce the zero and calibration gases at the calibration valve assembly, and verify that the appropriate gas flow rate and pressure are present at the FIA. Record the measurement system responses to the zero and calibration gases. The performance of the system is acceptable if the difference between the drift check measurement and the value obtained in section 7.1 is less than 3 percent of the span value. Alternatively, recalibrate the FIA as in section 7.1 and report the results using both sets of calibration data (i.e., data determined prior to the test period and data determined following the test period). The data that results in the lowest CE value shall be reported as the results for the test run. Conduct the system drift check at the end of each run.
7.3 Determination of Dilution Factor. Inject the dilution check gas into the measurement system before the dilution system and record the response. Calculate the dilution factor using Equation 204C-3.
7.4 System Check. Inject the high-range calibration gas at the inlet to the sampling probe while the dilution air is turned off. Record the response. The performance of the system is acceptable if the measurement system response is within 5 percent of the value obtained in section 7.1 for the high-range calibration gas. Conduct a system check before and after each test run.
8. Procedure
8.1 Determination of Volumetric Flow Rate of Captured Emissions
8.1.1 Locate all points where emissions are captured from the affected facility. Using Method 1, determine the sampling points. Be sure to check each site for cyclonic or swirling flow.
8.2.2 Measure the velocity at each sampling site at least once every hour during each sampling run using Method 2 or 2A.
8.2 Determination of VOC Content of Captured Emissions
8.2.1 Analysis Duration. Measure the VOC responses at each captured emissions point during the entire test run or, if applicable, while the process is operating. If there are multiple captured emissions locations, design a sampling system to allow a single FIA to be used to determine the VOC responses at all sampling locations.
8.2.2 Gas VOC Concentration.
8.2.2.1 Assemble the sample train as shown in Figure 204C-1. Calibrate the FIA according to the procedure in section 7.1.
8.2.2.2 Set the dilution ratio and determine the dilution factor according to the procedure in section 7.3.
8.2.2.3 Conduct a system check according to the procedure in section 7.4.
8.2.2.4 Install the sample probe so that the probe is centrally located in the stack, pipe, or duct, and is sealed tightly at the stack port connection.
8.2.2.5 Inject zero gas at the calibration valve assembly. Measure the system response time as the time required for the system to reach the effluent concentration after the calibration valve has been returned to the effluent sampling position.
8.2.2.6 Conduct a system check before, and a system drift check after, each sampling run according to the procedures in sections 7.2 and 7.4. If the drift check following a run indicates unacceptable performance (see section 7.4), the run is not valid. Alternatively, recalibrate the FIA as in section 7.1 and report the results using both sets of calibration data (i.e., data determined prior to the test period and data determined following the test period). The data that results in the lowest CE value shall be reported as the results for the test run. The tester may elect to perform system drift checks during the run not to exceed one drift check per hour.
8.2.2.7 Verify that the sample lines, filter, and pump temperatures are 120 ±5°C.
8.2.2.8 Begin sampling at the start of the test period and continue to sample during the entire run. Record the starting and ending times and any required process information as appropriate. If multiple captured emission locations are sampled using a single FIA, sample at each location for the same amount of time (e.g., 2 min.) and continue to switch from one location to another for the entire test run. Be sure that total sampling time at each location is the same at the end of the test run. Collect at least four separate measurements from each sample point during each hour of testing. Disregard the measurements at each sampling location until two times the response time of the measurement system has elapsed. Continue sampling for at least 1 minute and record the concentration measurements.
8.2.3 Background Concentration.
Note:
Not applicable when the building is used as the temporary total enclosure (TTE).
8.2.3.1 Locate all natural draft openings (NDO's) of the TTE. A sampling point shall be at the center of each NDO, unless otherwise approved by the Administrator. If there are more than six NDO's, choose six sampling points evenly spaced among the NDO's.
8.2.3.2 Assemble the sample train as shown in Figure 204C-2. Calibrate the FIA and conduct a system check according to the procedures in sections 7.1 and 7.4.
8.2.3.3 Position the probe at the sampling location.
8.2.3.4 Determine the response time, conduct the system check, and sample according to the procedures described in sections 8.2.2.4 through 8.2.2.8.
8.2.4 Alternative Procedure. The direct interface sampling and analysis procedure described in section 7.2 of Method 18 may be used to determine the gas VOC concentration. The system must be designed to collect and analyze at least one sample every 10 minutes. If the alternative procedure is used to determine the VOC concentration of the captured emissions, it must also be used to determine the VOC concentration of the uncaptured emissions.
9. Data Analysis and Calculations
9.1 Nomenclature.
Ai = area of NDO i, ft 2.
AN = total area of all NDO's in the enclosure, ft 2.
CA = actual concentration of the dilution check gas, ppm propane.
CBi = corrected average VOC concentration of background emissions at point i, ppm propane.
CB = average background concentration, ppm propane.
CDH = average measured concentration for the drift check calibration gas, ppm propane.
CD0 = average system drift check concentration for zero concentration gas, ppm propane.
CH = actual concentration of the drift check calibration gas, ppm propane.
Ci = uncorrected average background VOC concentration measured at point i, ppm propane.
Cj = uncorrected average VOC concentration measured at point j, ppm propane.
CM = measured concentration of the dilution check gas, ppm propane.
DF = dilution factor.
G = total VOC content of captured emissions, kg.
K1 = 1.830 × 10ˆ’6 kg/(m 3ˆ’ppm).
n = number of measurement points.
QGj = average effluent volumetric flow rate corrected to standard conditions at captured emissions point j, m 3/min.
ΘC = total duration of CE sampling run, min.
9.2 Calculations.
9.2.1 Total VOC Captured Emissions.
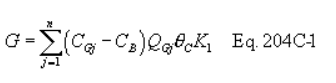
9.2.2 VOC Concentration of the Captured Emissions at Point j.
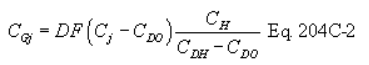
9.2.3 Dilution Factor.
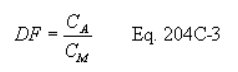
9.2.4 Background VOC Concentration at Point i.
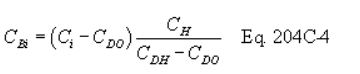
9.2.5 Average Background Concentration.
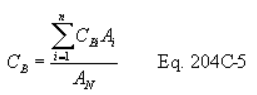
Note:
If the concentration at each point is within 20 percent of the average concentration of all points, then use the arithmetic average.
10. Method Performance
The measurement uncertainties are estimated for each captured or uncaptured emissions point as follows: QGj=±5.5 percent and CGj= ±5 percent. Based on these numbers, the probable uncertainty for G is estimated at about ±7.4 percent.
11. Diagrams
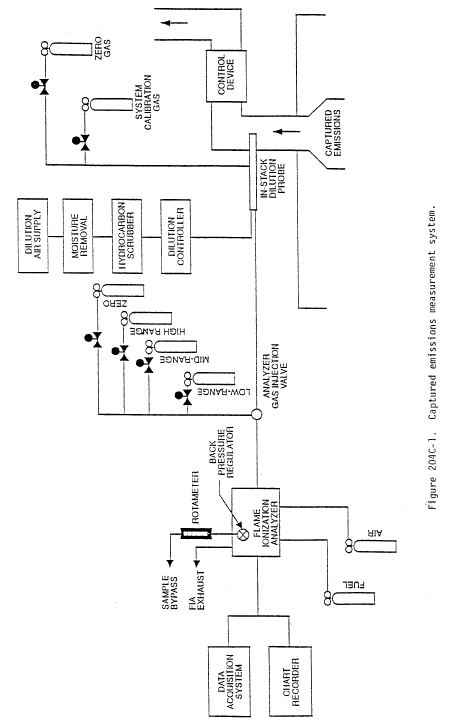
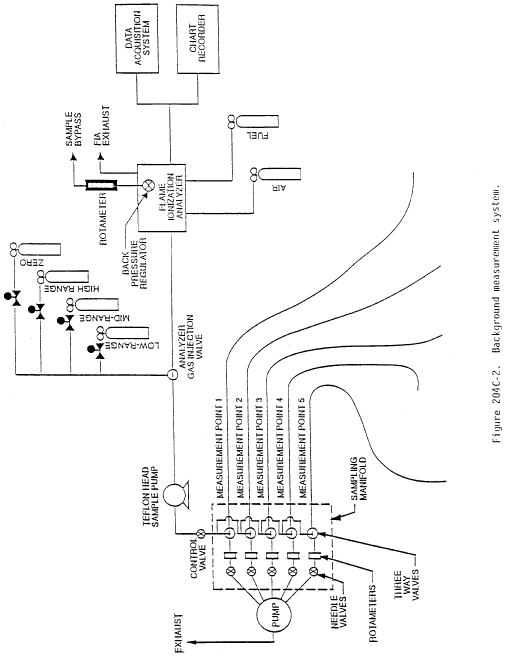
Method 204D - Volatile Organic Compounds Emissions in Uncaptured Stream From Temporary Total Enclosure
1. Scope and Application
1.1 Applicability. This procedure is applicable for determining the uncaptured volatile organic compounds (VOC) emissions from a temporary total enclosure (TTE). It is intended to be used as a segment in the development of liquid/gas or gas/gas protocols for determining VOC capture efficiency (CE) for surface coating and printing operations.
1.2 Principle. The amount of uncaptured VOC emissions (F) from the TTE is calculated as the sum of the products of the VOC content (CFj), the flow rate (QFj) from each uncaptured emissions point, and the sampling time (ΘF).
1.3 Sampling Requirements. A CE test shall consist of at least three sampling runs. Each run shall cover at least one complete production cycle, but shall be at least 3 hours long. The sampling time for each run need not exceed 8 hours, even if the production cycle has not been completed. Alternative sampling times may be used with the approval of the Administrator.
2. Summary of Method
A gas sample is extracted from the uncaptured exhaust duct of a TTE through a heated sample line and, if necessary, a glass fiber filter to a flame ionization analyzer (FIA).
3. Safety
Because this procedure is often applied in highly explosive areas, caution and care should be exercised in choosing, installing, and using the appropriate equipment.
4. Equipment and Supplies
Mention of trade names or company products does not constitute endorsement. All gas concentrations (percent, ppm) are by volume, unless otherwise noted.
4.1 Gas VOC Concentration. A schematic of the measurement system is shown in Figure 204D-1. The main components are as follows:
4.1.1 Sample Probe. Stainless steel or equivalent. The probe shall be heated to prevent VOC condensation.
4.1.2 Calibration Valve Assembly. Three-way valve assembly at the outlet of the sample probe to direct the zero and calibration gases to the analyzer. Other methods, such as quick-connect lines, to route calibration gases to the outlet of the sample probe are acceptable.
4.1.3 Sample Line. Stainless steel or Teflon tubing to transport the sample gas to the analyzer. The sample line must be heated to prevent condensation.
4.1.4 Sample Pump. A leak-free pump, to pull the sample gas through the system at a flow rate sufficient to minimize the response time of the measurement system. The components of the pump that contact the gas stream shall be constructed of stainless steel or Teflon. The sample pump must be heated to prevent condensation.
4.1.5 Sample Flow Rate Control. A sample flow rate control valve and rotameter, or equivalent, to maintain a constant sampling rate within 10 percent. The flow control valve and rotameter must be heated to prevent condensation. A control valve may also be located on the sample pump bypass loop to assist in controlling the sample pressure and flow rate.
4.1.6 Sample Gas Manifold. Capable of diverting a portion of the sample gas stream to the FIA, and the remainder to the bypass discharge vent. The manifold components shall be constructed of stainless steel or Teflon. If emissions are to be measured at multiple locations, the measurement system shall be designed to use separate sampling probes, lines, and pumps for each measurement location and a common sample gas manifold and FIA. The sample gas manifold and connecting lines to the FIA must be heated to prevent condensation.
4.1.7 Organic Concentration Analyzer. An FIA with a span value of 1.5 times the expected concentration as propane; however, other span values may be used if it can be demonstrated to the Administrator's satisfaction that they would provide more accurate measurements. The system shall be capable of meeting or exceeding the following specifications:
4.1.7.1 Zero Drift. Less than ±3.0 percent of the span value.
4.1.7.2 Calibration Drift. Less than ±3.0 percent of the span value.
4.1.7.3 Calibration Error. Less than ±5.0 percent of the calibration gas value.
4.1.7.4 Response Time. Less than 30 seconds.
4.1.8 Integrator/Data Acquisition System. An analog or digital device or computerized data acquisition system used to integrate the FIA response or compute the average response and record measurement data. The minimum data sampling frequency for computing average or integrated values is one measurement value every 5 seconds. The device shall be capable of recording average values at least once per minute.
4.2 Uncaptured Emissions Volumetric Flow Rate.
4.2.1 Method 2 or 2A Apparatus. For determining volumetric flow rate.
4.2.2 Method 3 Apparatus and Reagents. For determining molecular weight of the gas stream. An estimate of the molecular weight of the gas stream may be used if approved by the Administrator.
4.2.3 Method 4 Apparatus and Reagents. For determining moisture content, if necessary.
4.3 Temporary Total Enclosure. The criteria for designing an acceptable TTE are specified in Method 204.
5. Reagents and Standards
5.1 Calibration and Other Gases. Gases used for calibration, fuel, and combustion air (if required) are contained in compressed gas cylinders. All calibration gases shall be traceable to National Institute of Standards and Technology standards and shall be certified by the manufacturer to ±1 percent of the tag value. Additionally, the manufacturer of the cylinder should provide a recommended shelf life for each calibration gas cylinder over which the concentration does not change more than ±2 percent from the certified value. For calibration gas values not generally available, dilution systems calibrated using Method 205 may be used. Alternative methods for preparing calibration gas mixtures may be used with the approval of the Administrator.
5.1.1 Fuel. The FIA manufacturer's recommended fuel should be used. A 40 percent H2/60 percent He or 40 percent H2/60 percent N2 gas mixture is recommended to avoid an oxygen synergism effect that reportedly occurs when oxygen concentration varies significantly from a mean value. Other mixtures may be used provided the tester can demonstrate to the Administrator that there is no oxygen synergism effect.
5.1.2 Carrier Gas. High purity air with less than 1 ppm of organic material (as propane or carbon equivalent) or less than 0.1 percent of the span value, whichever is greater.
5.1.3 FIA Linearity Calibration Gases. Low-, mid-, and high-range gas mixture standards with nominal propane concentrations of 20-30, 45-55, and 70-80 percent of the span value in air, respectively. Other calibration values and other span values may be used if it can be shown to the Administrator's satisfaction that equally accurate measurements would be achieved.
5.2 Particulate Filter. An in-stack or an out-of-stack glass fiber filter is recommended if exhaust gas particulate loading is significant. An out-of-stack filter must be heated to prevent any condensation unless it can be demonstrated that no condensation occurs.
6. Quality Control
6.1 Required instrument quality control parameters are found in the following sections:
6.1.1 The FIA system must be calibrated as specified in section 7.1.
6.1.2 The system drift check must be performed as specified in section 7.2.
6.1.3 The system check must be conducted as specified in section 7.3.
7. Calibration and Standardization
7.1 FIA Calibration and Linearity Check. Make necessary adjustments to the air and fuel supplies for the FIA and ignite the burner. Allow the FIA to warm up for the period recommended by the manufacturer. Inject a calibration gas into the measurement system and adjust the back-pressure regulator to the value required to achieve the flow rates specified by the manufacturer. Inject the zero-and the high-range calibration gases and adjust the analyzer calibration to provide the proper responses. Inject the low-and mid-range gases and record the responses of the measurement system. The calibration and linearity of the system are acceptable if the responses for all four gases are within 5 percent of the respective gas values. If the performance of the system is not acceptable, repair or adjust the system and repeat the linearity check. Conduct a calibration and linearity check after assembling the analysis system and after a major change is made to the system.
7.2 Systems Drift Checks. Select the calibration gas concentration that most closely approximates that of the uncaptured gas emissions concentration to conduct the drift checks. Introduce the zero and calibration gases at the calibration valve assembly and verify that the appropriate gas flow rate and pressure are present at the FIA. Record the measurement system responses to the zero and calibration gases. The performance of the system is acceptable if the difference between the drift check measurement and the value obtained in section 7.1 is less than 3 percent of the span value. Alternatively, recalibrate the FIA as in section 7.1 and report the results using both sets of calibration data (i.e., data determined prior to the test period and data determined following the test period). The data that results in the lowest CE value shall be reported as the results for the test run. Conduct a system drift check at the end of each run.
7.3 System Check. Inject the high-range calibration gas at the inlet of the sampling probe and record the response. The performance of the system is acceptable if the measurement system response is within 5 percent of the value obtained in section 7.1 for the high-range calibration gas. Conduct a system check before each test run.
8. Procedure
8.1 Determination of Volumetric Flow Rate of Uncaptured Emissions
8.1.1 Locate all points where uncaptured emissions are exhausted from the TTE. Using Method 1, determine the sampling points. Be sure to check each site for cyclonic or swirling flow.
8.1.2 Measure the velocity at each sampling site at least once every hour during each sampling run using Method 2 or 2A.
8.2 Determination of VOC Content of Uncaptured Emissions.
8.2.1 Analysis Duration. Measure the VOC responses at each uncaptured emission point during the entire test run or, if applicable, while the process is operating. If there are multiple emission locations, design a sampling system to allow a single FIA to be used to determine the VOC responses at all sampling locations.
8.2.2 Gas VOC Concentration.
8.2.2.1 Assemble the sample train as shown in Figure 204D-1. Calibrate the FIA and conduct a system check according to the procedures in sections 7.1 and 7.3, respectively.
8.2.2.2 Install the sample probe so that the probe is centrally located in the stack, pipe, or duct, and is sealed tightly at the stack port connection.
8.2.2.3 Inject zero gas at the calibration valve assembly. Allow the measurement system response to reach zero. Measure the system response time as the time required for the system to reach the effluent concentration after the calibration valve has been returned to the effluent sampling position.
8.2.2.4 Conduct a system check before, and a system drift check after, each sampling run according to the procedures in sections 7.2 and 7.3. If the drift check following a run indicates unacceptable performance (see section 7.3), the run is not valid. Alternatively, recalibrate the FIA as in section 7.1 and report the results using both sets of calibration data (i.e., data determined prior to the test period and data determined following the test period). The data that results in the lowest CE value shall be reported as the results for the test run. The tester may elect to perform system drift checks during the run not to exceed one drift check per hour.
8.2.2.5 Verify that the sample lines, filter, and pump temperatures are 120 ±5°C.
8.2.2.6 Begin sampling at the start of the test period and continue to sample during the entire run. Record the starting and ending times and any required process information, as appropriate. If multiple emission locations are sampled using a single FIA, sample at each location for the same amount of time (e.g., 2 min.) and continue to switch from one location to another for the entire test run. Be sure that total sampling time at each location is the same at the end of the test run. Collect at least four separate measurements from each sample point during each hour of testing. Disregard the response measurements at each sampling location until 2 times the response time of the measurement system has elapsed. Continue sampling for at least 1 minute and record the concentration measurements.
8.2.3 Background Concentration.
8.2.3.1 Locate all natural draft openings (NDO's) of the TTE. A sampling point shall be at the center of each NDO, unless otherwise approved by the Administrator. If there are more than six NDO's, choose six sampling points evenly spaced among the NDO's.
8.2.3.2 Assemble the sample train as shown in Figure 204D-2. Calibrate the FIA and conduct a system check according to the procedures in sections 7.1 and 7.3.
8.2.3.3 Position the probe at the sampling location.
8.2.3.4 Determine the response time, conduct the system check, and sample according to the procedures described in sections 8.2.2.3 through 8.2.2.6.
8.2.4 Alternative Procedure. The direct interface sampling and analysis procedure described in section 7.2 of Method 18 may be used to determine the gas VOC concentration. The system must be designed to collect and analyze at least one sample every 10 minutes. If the alternative procedure is used to determine the VOC concentration of the uncaptured emissions in a gas/gas protocol, it must also be used to determine the VOC concentration of the captured emissions. If a tester wishes to conduct a liquid/gas protocol using a gas chromatograph, the tester must use Method 204F for the liquid steam. A gas chromatograph is not an acceptable alternative to the FIA in Method 204A.
9. Data Analysis and Calculations
9.1 Nomenclature.
Ai = area of NDO i, ft 2.
AN = total area of all NDO's in the enclosure, ft 2.
CBi = corrected average VOC concentration of background emissions at point i, ppm propane.
CB = average background concentration, ppm propane.
CDH = average measured concentration for the drift check calibration gas, ppm propane.
CD0 = average system drift check concentration for zero concentration gas, ppm propane.
CFj = corrected average VOC concentration of uncaptured emissions at point j, ppm propane.
CH = actual concentration of the drift check calibration gas, ppm propane.
Ci = uncorrected average background VOC concentration at point i, ppm propane.
Cj = uncorrected average VOC concentration measured at point j, ppm propane.
F = total VOC content of uncaptured emissions, kg.
K1 = 1.830 × 10ˆ’6 kg/(m 3-ppm).
n = number of measurement points.
QFj = average effluent volumetric flow rate corrected to standard conditions at uncaptured emissions point j, m 3/min.
ΘF = total duration of uncaptured emissions sampling run, min.
9.2 Calculations.
9.2.1 Total Uncaptured VOC Emissions.
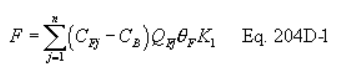
9.2.2 VOC Concentration of the Uncaptured Emissions at Point j.
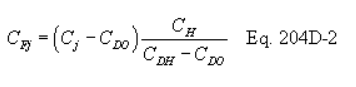
9.2.3 Background VOC Concentration at Point i.
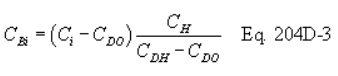
9.2.4 Average Background Concentration.
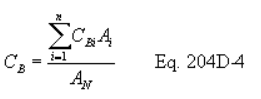
Note:
If the concentration at each point is within 20 percent of the average concentration of all points, use the arithmetic average.
10. Method Performance
The measurement uncertainties are estimated for each uncaptured emission point as follows: QFj=±5.5 percent and CFj=±5.0 percent. Based on these numbers, the probable uncertainty for F is estimated at about ±7.4 percent.
11. Diagrams
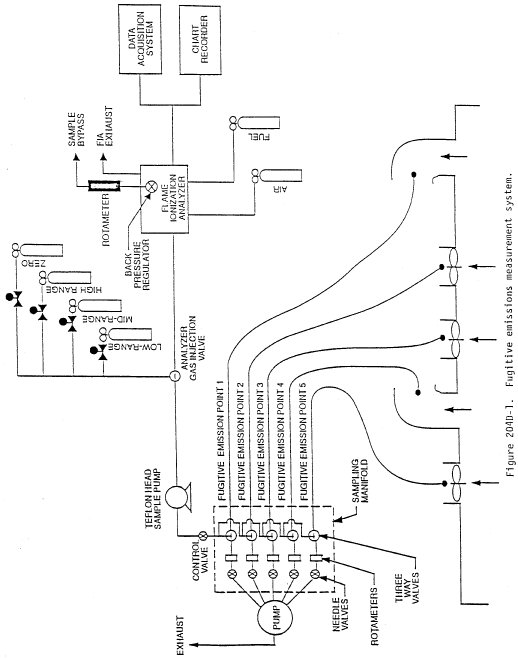
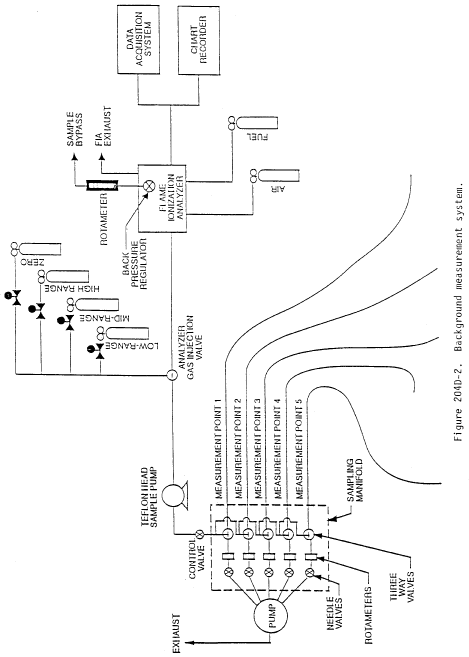
Method 204E - Volatile Organic Compounds Emissions in Uncaptured Stream From Building Enclosure
1. Scope and Application
1.1 Applicability. This procedure is applicable for determining the uncaptured volatile organic compounds (VOC) emissions from a building enclosure (BE). It is intended to be used in the development of liquid/gas or gas/gas protocols for determining VOC capture efficiency (CE) for surface coating and printing operations.
1.2 Principle. The total amount of uncaptured VOC emissions (FB) from the BE is calculated as the sum of the products of the VOC content (CFj) of each uncaptured emissions point, the flow rate (QFj) at each uncaptured emissions point, and time (ΘF).
1.3 Sampling Requirements. A CE test shall consist of at least three sampling runs. Each run shall cover at least one complete production cycle, but shall be at least 3 hours long. The sampling time for each run need not exceed 8 hours, even if the production cycle has not been completed. Alternative sampling times may be used with the approval of the Administrator.
2. Summary of Method
A gas sample is extracted from the uncaptured exhaust duct of a BE through a heated sample line and, if necessary, a glass fiber filter to a flame ionization analyzer (FIA).
3. Safety
Because this procedure is often applied in highly explosive areas, caution and care should be exercised in choosing, installing, and using the appropriate equipment.
4. Equipment and Supplies
Mention of trade names or company products does not constitute endorsement. All gas concentrations (percent, ppm) are by volume, unless otherwise noted.
4.1 Gas VOC Concentration. A schematic of the measurement system is shown in Figure 204E-1. The main components are as follows:
4.1.1 Sample Probe. Stainless steel or equivalent. The probe shall be heated to prevent VOC condensation.
4.1.2 Calibration Valve Assembly. Three-way valve assembly at the outlet of the sample probe to direct the zero and calibration gases to the analyzer. Other methods, such as quick-connect lines, to route calibration gases to the outlet of the sample probe are acceptable.
4.1.3 Sample Line. Stainless steel or Teflon tubing to transport the sample gas to the analyzer. The sample line must be heated to prevent condensation.
4.1.4 Sample Pump. A leak-free pump, to pull the sample gas through the system at a flow rate sufficient to minimize the response time of the measurement system. The components of the pump that contact the gas stream shall be constructed of stainless steel or Teflon. The sample pump must be heated to prevent condensation.
4.1.5 Sample Flow Rate Control. A sample flow rate control valve and rotameter, or equivalent, to maintain a constant sampling rate within 10 percent. The flow rate control valve and rotameter must be heated to prevent condensation. A control valve may also be located on the sample pump bypass loop to assist in controlling the sample pressure and flow rate.
4.1.6 Sample Gas Manifold. Capable of diverting a portion of the sample gas stream to the FIA, and the remainder to the bypass discharge vent. The manifold components shall be constructed of stainless steel or Teflon. If emissions are to be measured at multiple locations, the measurement system shall be designed to use separate sampling probes, lines, and pumps for each measurement location, and a common sample gas manifold and FIA. The sample gas manifold must be heated to prevent condensation.
4.1.7 Organic Concentration Analyzer. An FIA with a span value of 1.5 times the expected concentration as propane; however, other span values may be used if it can be demonstrated to the Administrator's satisfaction that they would provide equally accurate measurements. The system shall be capable of meeting or exceeding the following specifications:
4.1.7.1 Zero Drift. Less than ±3.0 percent of the span value.
4.1.7.2 Calibration Drift. Less than ±3.0 percent of the span value.
4.1.7.3 Calibration Error. Less than ±5.0 percent of the calibration gas value.
4.1.7.4 Response Time. Less than 30 seconds.
4.1.8 Integrator/Data Acquisition System. An analog or digital device or computerized data acquisition system used to integrate the FIA response or compute the average response and record measurement data. The minimum data sampling frequency for computing average or integrated values is one measurement value every 5 seconds. The device shall be capable of recording average values at least once per minute.
4.2 Uncaptured Emissions Volumetric Flow Rate.
4.2.1 Flow Direction Indicators. Any means of indicating inward or outward flow, such as light plastic film or paper streamers, smoke tubes, filaments, and sensory perception.
4.2.2 Method 2 or 2A Apparatus. For determining volumetric flow rate. Anemometers or similar devices calibrated according to the manufacturer's instructions may be used when low velocities are present. Vane anemometers (Young-maximum response propeller), specialized pitots with electronic manometers (e.g., Shortridge Instruments Inc., Airdata Multimeter 860) are commercially available with measurement thresholds of 15 and 8 mpm (50 and 25 fpm), respectively.
4.2.3 Method 3 Apparatus and Reagents. For determining molecular weight of the gas stream. An estimate of the molecular weight of the gas stream may be used if approved by the Administrator.
4.2.4 Method 4 Apparatus and Reagents. For determining moisture content, if necessary.
4.3 Building Enclosure. The criteria for an acceptable BE are specified in Method 204.
5. Reagents and Standards
5.1 Calibration and Other Gases. Gases used for calibration, fuel, and combustion air (if required) are contained in compressed gas cylinders. All calibration gases shall be traceable to National Institute of Standards and Technology standards and shall be certified by the manufacturer to ±1 percent of the tag value. Additionally, the manufacturer of the cylinder should provide a recommended shelf life for each calibration gas cylinder over which the concentration does not change more than ±2 percent from the certified value. For calibration gas values not generally available, dilution systems calibrated using Method 205 may be used. Alternative methods for preparing calibration gas mixtures may be used with the approval of the Administrator.
5.1.1 Fuel. The FIA manufacturer's recommended fuel should be used. A 40 percent H2/60 percent He or 40 percent H2/60 percent N2 gas mixture is recommended to avoid an oxygen synergism effect that reportedly occurs when oxygen concentration varies significantly from a mean value. Other mixtures may be used provided the tester can demonstrate to the Administrator that there is no oxygen synergism effect.
5.1.2 Carrier Gas. High purity air with less than 1 ppm of organic material (propane or carbon equivalent) or less than 0.1 percent of the span value, whichever is greater.
5.1.3 FIA Linearity Calibration Gases. Low-, mid-, and high-range gas mixture standards with nominal propane concentrations of 20-30, 45-55, and 70-80 percent of the span value in air, respectively. Other calibration values and other span values may be used if it can be shown to the Administrator's satisfaction that equally accurate measurements would be achieved.
5.2 Particulate Filter. An in-stack or an out-of-stack glass fiber filter is recommended if exhaust gas particulate loading is significant. An out-of-stack filter must be heated to prevent any condensation unless it can be demonstrated that no condensation occurs.
6. Quality Control
6.1 Required instrument quality control parameters are found in the following sections:
6.1.1 The FIA system must be calibrated as specified in section 7.1.
6.1.2 The system drift check must be performed as specified in section 7.2.
6.1.3 The system check must be conducted as specified in section 7.3.
7. Calibration and Standardization
7.1 FIA Calibration and Linearity Check. Make necessary adjustments to the air and fuel supplies for the FIA and ignite the burner. Allow the FIA to warm up for the period recommended by the manufacturer. Inject a calibration gas into the measurement system and adjust the back-pressure regulator to the value required to achieve the flow rates specified by the manufacturer. Inject the zero-and the high-range calibration gases, and adjust the analyzer calibration to provide the proper responses. Inject the low-and mid-range gases and record the responses of the measurement system. The calibration and linearity of the system are acceptable if the responses for all four gases are within 5 percent of the respective gas values. If the performance of the system is not acceptable, repair or adjust the system and repeat the linearity check. Conduct a calibration and linearity check after assembling the analysis system and after a major change is made to the system.
7.2 Systems Drift Checks. Select the calibration gas that most closely approximates the concentration of the captured emissions for conducting the drift checks. Introduce the zero and calibration gases at the calibration valve assembly and verify that the appropriate gas flow rate and pressure are present at the FIA. Record the measurement system responses to the zero and calibration gases. The performance of the system is acceptable if the difference between the drift check measurement and the value obtained in section 7.1 is less than 3 percent of the span value. Alternatively, recalibrate the FIA as in section 7.1 and report the results using both sets of calibration data (i.e., data determined prior to the test period and data determined following the test period). The data that results in the lowest CE value shall be reported as the results for the test run. Conduct a system drift check at the end of each run.
7.3 System Check. Inject the high-range calibration gas at the inlet of the sampling probe and record the response. The performance of the system is acceptable if the measurement system response is within 5 percent of the value obtained in section 7.1 for the high-range calibration gas. Conduct a system check before each test run.
8. Procedure
8.1 Preliminary Determinations. The following points are considered exhaust points and should be measured for volumetric flow rates and VOC concentrations:
8.1.1 Forced Draft Openings. Any opening in the facility with an exhaust fan. Determine the volumetric flow rate according to Method 2.
8.1.2 Roof Openings. Any openings in the roof of a facility which does not contain fans are considered to be exhaust points. Determine volumetric flow rate from these openings. Use the appropriate velocity measurement devices (e.g., propeller anemometers).
8.2 Determination of Flow Rates.
8.2.1 Measure the volumetric flow rate at all locations identified as exhaust points in section 8.1. Divide each exhaust opening into nine equal areas for rectangular openings and into eight equal areas for circular openings.
8.2.2 Measure the velocity at each site at least once every hour during each sampling run using Method 2 or 2A, if applicable, or using the low velocity instruments in section 4.2.2.
8.3 Determination of VOC Content of Uncaptured Emissions.
8.3.1 Analysis Duration. Measure the VOC responses at each uncaptured emissions point during the entire test run or, if applicable, while the process is operating. If there are multiple emissions locations, design a sampling system to allow a single FIA to be used to determine the VOC responses at all sampling locations.
8.3.2 Gas VOC Concentration.
8.3.2.1 Assemble the sample train as shown in Figure 204E-1. Calibrate the FIA and conduct a system check according to the procedures in sections 7.1 and 7.3, respectively.
8.3.2.2 Install the sample probe so that the probe is centrally located in the stack, pipe, or duct, and is sealed tightly at the stack port connection.
8.3.2.3 Inject zero gas at the calibration valve assembly. Allow the measurement system response to reach zero. Measure the system response time as the time required for the system to reach the effluent concentration after the calibration valve has been returned to the effluent sampling position.
8.3.2.4 Conduct a system check before, and a system drift check after, each sampling run according to the procedures in sections 7.2 and 7.3. If the drift check following a run indicates unacceptable performance (see section 7.3), the run is not valid. Alternatively, recalibrate the FIA as in section 7.1 and report the results using both sets of calibration data (i.e., data determined prior to the test period and data determined following the test period). The data that results in the lowest CE value shall be reported as the results for the test run. The tester may elect to perform drift checks during the run, not to exceed one drift check per hour.
8.3.2.5 Verify that the sample lines, filter, and pump temperatures are 120 ±5°C.
8.3.2.6 Begin sampling at the start of the test period and continue to sample during the entire run. Record the starting and ending times, and any required process information, as appropriate. If multiple emission locations are sampled using a single FIA, sample at each location for the same amount of time (e.g., 2 minutes) and continue to switch from one location to another for the entire test run. Be sure that total sampling time at each location is the same at the end of the test run. Collect at least four separate measurements from each sample point during each hour of testing. Disregard the response measurements at each sampling location until 2 times the response time of the measurement system has elapsed. Continue sampling for at least 1 minute, and record the concentration measurements.
8.4 Alternative Procedure. The direct interface sampling and analysis procedure described in section 7.2 of Method 18 may be used to determine the gas VOC concentration. The system must be designed to collect and analyze at least one sample every 10 minutes. If the alternative procedure is used to determine the VOC concentration of the uncaptured emissions in a gas/gas protocol, it must also be used to determine the VOC concentration of the captured emissions. If a tester wishes to conduct a liquid/gas protocol using a gas chromatograph, the tester must use Method 204F for the liquid steam. A gas chromatograph is not an acceptable alternative to the FIA in Method 204A.
9. Data Analysis and Calculations
9.1 Nomenclature.
CDH = average measured concentration for the drift check calibration gas, ppm propane.
CD0 = average system drift check concentration for zero concentration gas, ppm propane.
CFj = corrected average VOC concentration of uncaptured emissions at point j, ppm propane.
CH = actual concentration of the drift check calibration gas, ppm propane.
Cj = uncorrected average VOC concentration measured at point j, ppm propane.
FB = total VOC content of uncaptured emissions from the building, kg.
K1 = 1.830 × 10ˆ’6 kg/(m 3-ppm).
n = number of measurement points.
QFj = average effluent volumetric flow rate corrected to standard conditions at uncaptured emissions point j, m 3/min.
ΘF = total duration of CE sampling run, min.
9.2 Calculations
9.2.1 Total VOC Uncaptured Emissions from the Building.
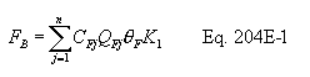
9.2.2 VOC Concentration of the Uncaptured Emissions at Point j.
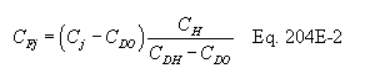
10. Method Performance
The measurement uncertainties are estimated for each uncaptured emissions point as follows: QFj=±10.0 percent and CFj=±5.0 percent. Based on these numbers, the probable uncertainty for FB is estimated at about ±11.2 percent.
11. Diagrams
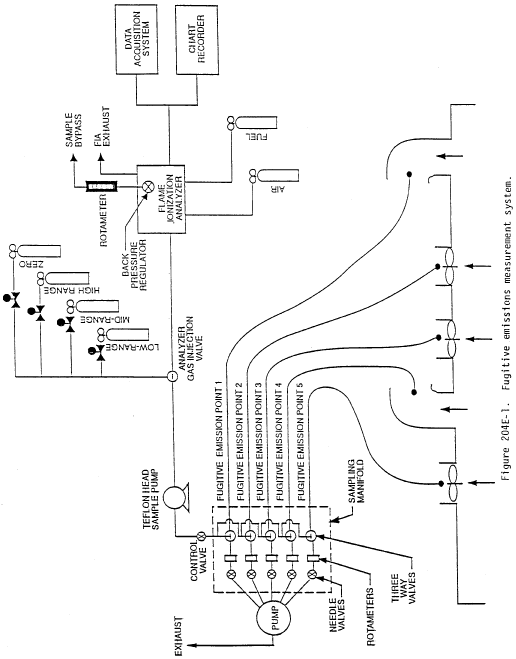
Method 204F - Volatile Organic Compounds Content in Liquid Input Stream (Distillation Approach)
1. Introduction
1.1 Applicability. This procedure is applicable for determining the input of volatile organic compounds (VOC). It is intended to be used as a segment in the development of liquid/gas protocols for determining VOC capture efficiency (CE) for surface coating and printing operations.
1.2 Principle. The amount of VOC introduced to the process (L) is the sum of the products of the weight (W) of each VOC containing liquid (ink, paint, solvent, etc.) used, and its VOC content (V), corrected for a response factor (RF).
1.3 Sampling Requirements. A CE test shall consist of at least three sampling runs. Each run shall cover at least one complete production cycle, but shall be at least 3 hours long. The sampling time for each run need not exceed 8 hours, even if the production cycle has not been completed. Alternative sampling times may be used with the approval of the Administrator.
2. Summary of Method
A sample of each coating used is distilled to separate the VOC fraction. The distillate is used to prepare a known standard for analysis by a flame ionization analyzer (FIA), calibrated against propane, to determine its RF.
3. Safety
Because this procedure is often applied in highly explosive areas, caution and care should be exercised in choosing, installing, and using the appropriate equipment.
4. Equipment and Supplies
Mention of trade names or company products does not constitute endorsement. All gas concentrations (percent, ppm) are by volume, unless otherwise noted.
4.1 Liquid Weight.
4.1.1 Balances/Digital Scales. To weigh drums of VOC containing liquids to within 0.2 lb or 1.0 percent of the total weight of VOC liquid used.
4.1.2 Volume Measurement Apparatus (Alternative). Volume meters, flow meters, density measurement equipment, etc., as needed to achieve the same accuracy as direct weight measurements.
4.2 Response Factor Determination (FIA Technique). The VOC distillation system and Tedlar gas bag generation system apparatuses are shown in Figures 204F-1 and 204F-2, respectively. The following equipment is required:
4.2.1 Sample Collection Can. An appropriately-sized metal can to be used to collect VOC containing materials. The can must be constructed in such a way that it can be grounded to the coating container.
4.2.2 Needle Valves. To control gas flow.
4.2.3 Regulators. For calibration, dilution, and sweep gas cylinders.
4.2.4 Tubing and Fittings. Teflon and stainless steel tubing and fittings with diameters, lengths, and sizes determined by the connection requirements of the equipment.
4.2.5 Thermometer. Capable of measuring the temperature of the hot water and oil baths to within 1°C.
4.2.6 Analytical Balance. To measure ±0.01 mg.
4.2.7 Microliter Syringe. 10-µl size.
4.2.8 Vacuum Gauge or Manometer. 0- to 760-mm (0- to 30-in.) Hg U-Tube manometer or vacuum gauge.
4.2.9 Hot Oil Bath, With Stirring Hot Plate. Capable of heating and maintaining a distillation vessel at 110 ±3°C.
4.2.10 Ice Water Bath. To cool the distillation flask.
4.2.11 Vacuum/Water Aspirator. A device capable of drawing a vacuum to within 20 mm Hg from absolute.
4.2.12 Rotary Evaporator System. Complete with folded inner coil, vertical style condenser, rotary speed control, and Teflon sweep gas delivery tube with valved inlet. Buchi Rotavapor or equivalent.
4.2.13 Ethylene Glycol Cooling/Circulating Bath. Capable of maintaining the condenser coil fluid at ˆ’10°C.
4.2.14 Dry Gas Meter (DGM). Capable of measuring the dilution gas volume within 2 percent, calibrated with a spirometer or bubble meter, and equipped with a temperature gauge capable of measuring temperature within 3°C.
4.2.15 Activated Charcoal/Mole Sieve Trap. To remove any trace level of organics picked up from the DGM.
4.2.16 Gas Coil Heater. Sufficient length of 0.125-inch stainless steel tubing to allow heating of the dilution gas to near the water bath temperature before entering the volatilization vessel.
4.2.17 Water Bath, With Stirring Hot Plate. Capable of heating and maintaining a volatilization vessel and coil heater at a temperature of 100 ±5°C.
4.2.18 Volatilization Vessel. 50-ml midget impinger fitted with a septum top and loosely filled with glass wool to increase the volatilization surface.
4.2.19 Tedlar Gas Bag. Capable of holding 30 liters of gas, flushed clean with zero air, leak tested, and evacuated.
4.2.20 Organic Concentration Analyzer. An FIA with a span value of 1.5 times the expected concentration as propane; however, other span values may be used if it can be demonstrated that they would provide equally accurate measurements. The FIA instrument should be the same instrument used in the gaseous analyses adjusted with the same fuel, combustion air, and sample back-pressure (flow rate) settings. The system shall be capable of meeting or exceeding the following specifications:
4.2.20.1 Zero Drift. Less than ±3.0 percent of the span value.
4.2.20.2 Calibration Drift. Less than ±3.0 percent of the span value.
4.2.20.3 Calibration Error. Less than ±3.0 percent of the calibration gas value.
4.2.21 Integrator/Data Acquisition System. An analog or digital device or computerized data acquisition system used to integrate the FIA response or compute the average response and record measurement data. The minimum data sampling frequency for computing average or integrated value is one measurement value every 5 seconds. The device shall be capable of recording average values at least once per minute.
4.2.22 Chart Recorder (Optional). A chart recorder or similar device is recommended to provide a continuous analog display of the measurement results during the liquid sample analysis.
5. Reagents and Standards
5.1 Zero Air. High purity air with less than 1 ppm of organic material (as propane) or less than 0.1 percent of the span value, whichever is greater. Used to supply dilution air for making the Tedlar bag gas samples.
5.2 THC Free N2. High purity N2 with less than 1 ppm THC. Used as sweep gas in the rotary evaporator system.
5.3 Calibration and Other Gases. Gases used for calibration, fuel, and combustion air (if required) are contained in compressed gas cylinders. All calibration gases shall be traceable to National Institute of Standards and Technology standards and shall be certified by the manufacturer to ±1 percent of the tag value. Additionally, the manufacturer of the cylinder should provide a recommended shelf life for each calibration gas cylinder over which the concentration does not change more than ±2 percent from the certified value. For calibration gas values not generally available, dilution systems calibrated using Method 205 may be used. Alternative methods for preparing calibration gas mixtures may be used with the approval of the Administrator.
5.3.1 Fuel. The FIA manufacturer's recommended fuel should be used. A 40 percent H2/60 percent He, or 40 percent H2/60 percent N2 mixture is recommended to avoid fuels with oxygen to avoid an oxygen synergism effect that reportedly occurs when oxygen concentration varies significantly from a mean value. Other mixtures may be used provided the tester can demonstrate to the Administrator that there is no oxygen synergism effect.
5.3.2 Combustion Air. High purity air with less than 1 ppm of organic material (as propane) or less than 0.1 percent of the span value, whichever is greater.
5.3.3 FIA Linearity Calibration Gases. Low-, mid-, and high-range gas mixture standards with nominal propane concentration of 20-30, 45-55, and 70-80 percent of the span value in air, respectively. Other calibration values and other span values may be used if it can be shown that equally accurate measurements would be achieved.
5.3.4 System Calibration Gas. Gas mixture standard containing propane in air, approximating the VOC concentration expected for the Tedlar gas bag samples.
6. Quality Control
6.1 Required instrument quality control parameters are found in the following sections:
6.1.1 The FIA system must be calibrated as specified in section 7.1.
6.1.2 The system drift check must be performed as specified in section 7.2.
6.2 Precision Control. A minimum of one sample in each batch must be distilled and analyzed in duplicate as a precision control. If the results of the two analyses differ by more than ±10 percent of the mean, then the system must be reevaluated and the entire batch must be redistilled and analyzed.
7. Calibration and Standardization
7.1 FIA Calibration and Linearity Check. Make necessary adjustments to the air and fuel supplies for the FIA and ignite the burner. Allow the FIA to warm up for the period recommended by the manufacturer. Inject a calibration gas into the measurement system and adjust the back-pressure regulator to the value required to achieve the flow rates specified by the manufacturer. Inject the zero-and the high-range calibration gases and adjust the analyzer calibration to provide the proper responses. Inject the low-and mid-range gases and record the responses of the measurement system. The calibration and linearity of the system are acceptable if the responses for all four gases are within 5 percent of the respective gas values. If the performance of the system is not acceptable, repair or adjust the system and repeat the linearity check. Conduct a calibration and linearity check after assembling the analysis system and after a major change is made to the system. A calibration curve consisting of zero gas and two calibration levels must be performed at the beginning and end of each batch of samples.
7.2 Systems Drift Checks. After each sample, repeat the system calibration checks in section 7.1 before any adjustments to the FIA or measurement system are made. If the zero or calibration drift exceeds ±3 percent of the span value, discard the result and repeat the analysis. Alternatively, recalibrate the FIA as in section 7.1 and report the results using both sets of calibration data (i.e., data determined prior to the test period and data determined following the test period). The data that results in the lowest CE value shall be reported as the results for the test run.
8. Procedures
8.1 Determination of Liquid Input Weight
8.1.1 Weight Difference. Determine the amount of material introduced to the process as the weight difference of the feed material before and after each sampling run. In determining the total VOC containing liquid usage, account for: (a) The initial (beginning) VOC containing liquid mixture; (b) any solvent added during the test run; (c) any coating added during the test run; and (d) any residual VOC containing liquid mixture remaining at the end of the sample run.
8.1.1.1 Identify all points where VOC containing liquids are introduced to the process. To obtain an accurate measurement of VOC containing liquids, start with an empty fountain (if applicable). After completing the run, drain the liquid in the fountain back into the liquid drum (if possible), and weigh the drum again. Weigh the VOC containing liquids to ±0.5 percent of the total weight (full) or ±1.0 percent of the total weight of VOC containing liquid used during the sample run, whichever is less. If the residual liquid cannot be returned to the drum, drain the fountain into a preweighed empty drum to determine the final weight of the liquid.
8.1.1.2 If it is not possible to measure a single representative mixture, then weigh the various components separately (e.g., if solvent is added during the sampling run, weigh the solvent before it is added to the mixture). If a fresh drum of VOC containing liquid is needed during the run, then weigh both the empty drum and fresh drum.
8.1.2 Volume Measurement (Alternative). If direct weight measurements are not feasible, the tester may use volume meters and flow rate meters (and density measurements) to determine the weight of liquids used if it can be demonstrated that the technique produces results equivalent to the direct weight measurements. If a single representative mixture cannot be measured, measure the components separately.
8.2 Determination of VOC Content in Input Liquids
8.2.1 Collection of Liquid Samples.
8.2.1.1 Collect a 1-pint or larger sample of the VOC containing liquid mixture at each application location at the beginning and end of each test run. A separate sample should be taken of each VOC containing liquid added to the application mixture during the test run. If a fresh drum is needed during the sampling run, then obtain a sample from the fresh drum.
8.2.1.2 When collecting the sample, ground the sample container to the coating drum. Fill the sample container as close to the rim as possible to minimize the amount of headspace.
8.2.1.3 After the sample is collected, seal the container so the sample cannot leak out or evaporate.
8.2.1.4 Label the container to identify clearly the contents.
8.2.2 Distillation of VOC.
8.2.2.1 Assemble the rotary evaporator as shown in Figure 204F-1.
8.2.2.2 Leak check the rotary evaporation system by aspirating a vacuum of approximately 20 mm Hg from absolute. Close up the system and monitor the vacuum for approximately 1 minute. If the vacuum falls more than 25 mm Hg in 1 minute, repair leaks and repeat. Turn off the aspirator and vent vacuum.
8.2.2.3 Deposit approximately 20 ml of sample (inks, paints, etc.) into the rotary evaporation distillation flask.
8.2.2.4 Install the distillation flask on the rotary evaporator.
8.2.2.5 Immerse the distillate collection flask into the ice water bath.
8.2.2.6 Start rotating the distillation flask at a speed of approximately 30 rpm.
8.2.2.7 Begin heating the vessel at a rate of 2 to 3°C per minute.
8.2.2.8 After the hot oil bath has reached a temperature of 50°C or pressure is evident on the mercury manometer, turn on the aspirator and gradually apply a vacuum to the evaporator to within 20 mm Hg of absolute. Care should be taken to prevent material burping from the distillation flask.
8.2.2.9 Continue heating until a temperature of 110°C is achieved and maintain this temperature for at least 2 minutes, or until the sample has dried in the distillation flask.
8.2.2.10 Slowly introduce the N2 sweep gas through the purge tube and into the distillation flask, taking care to maintain a vacuum of approximately 400-mm Hg from absolute.
8.2.2.11 Continue sweeping the remaining solvent VOC from the distillation flask and condenser assembly for 2 minutes, or until all traces of condensed solvent are gone from the vessel. Some distillate may remain in the still head. This will not affect solvent recovery ratios.
8.2.2.12 Release the vacuum, disassemble the apparatus and transfer the distillate to a labeled, sealed vial.
8.2.3 Preparation of VOC standard bag sample.
8.2.3.1 Assemble the bag sample generation system as shown in Figure 204F-2 and bring the water bath up to near boiling temperature.
8.2.3.2 Inflate the Tedlar bag and perform a leak check on the bag.
8.2.3.3 Evacuate the bag and close the bag inlet valve.
8.2.3.4 Record the current barometric pressure.
8.2.3.5 Record the starting reading on the dry gas meter, open the bag inlet valve, and start the dilution zero air flowing into the Tedlar bag at approximately 2 liters per minute.
8.2.3.6 The bag sample VOC concentration should be similar to the gaseous VOC concentration measured in the gas streams. The amount of liquid VOC required can be approximated using equations in section 9.2. Using Equation 204F-4, calculate CVOC by assuming RF is 1.0 and selecting the desired gas concentration in terms of propane, CC3. Assuming BV is 20 liters, ML, the approximate amount of liquid to be used to prepare the bag gas sample, can be calculated using Equation 204F-2.
8.2.3.7 Quickly withdraw an aliquot of the approximate amount calculated in section 8.2.3.6 from the distillate vial with the microliter syringe and record its weight from the analytical balance to the nearest 0.01 mg.
8.2.3.8 Inject the contents of the syringe through the septum of the volatilization vessel into the glass wool inside the vessel.
8.2.3.9 Reweigh and record the tare weight of the now empty syringe.
8.2.3.10 Record the pressure and temperature of the dilution gas as it is passed through the dry gas meter.
8.2.3.11 After approximately 20 liters of dilution gas have passed into the Tedlar bag, close the valve to the dilution air source and record the exact final reading on the dry gas meter.
8.2.3.12 The gas bag is then analyzed by FIA within 1 hour of bag preparation in accordance with the procedure in section 8.2.4.
8.2.4 Determination of VOC response factor.
8.2.4.1 Start up the FIA instrument using the same settings as used for the gaseous VOC measurements.
8.2.4.2 Perform the FIA analyzer calibration and linearity checks according to the procedure in section 7.1. Record the responses to each of the calibration gases and the back-pressure setting of the FIA.
8.2.4.3 Connect the Tedlar bag sample to the FIA sample inlet and record the bag concentration in terms of propane. Continue the analyses until a steady reading is obtained for at least 30 seconds. Record the final reading and calculate the RF.
8.2.5 Determination of coating VOC content as VOC (VIJ).
8.2.5.1 Determine the VOC content of the coatings used in the process using EPA Method 24 or 24A as applicable.
9. Data Analysis and Calculations
9.1. Nomenclature.
BV = Volume of bag sample volume, liters.
CC3 = Concentration of bag sample as propane, mg/liter.
CVOC = Concentration of bag sample as VOC, mg/liter.
K = 0.00183 mg propane/(liter-ppm propane)
L = Total VOC content of liquid input, kg propane.
ML = Mass of VOC liquid injected into the bag, mg.
MV = Volume of gas measured by DGM, liters.
PM = Absolute DGM gas pressure, mm Hg.
PSTD = Standard absolute pressure, 760 mm Hg.
RC3 = FIA reading for bag gas sample, ppm propane.
RF = Response factor for VOC in liquid, weight VOC/weight propane.
RFJ = Response factor for VOC in liquid J, weight VOC/weight propane.
TM = DGM temperature,°K.
TSTD = Standard absolute temperature, 293°K.
VIJ = Initial VOC weight fraction of VOC liquid J.
VFJ = Final VOC weight fraction of VOC liquid J.
VAJ = VOC weight fraction of VOC liquid J added during the run.
WIJ = Weight of VOC containing liquid J at beginning of run, kg.
WFJ = Weight of VOC containing liquid J at end of run, kg.
WAJ = Weight of VOC containing liquid J added during the run, kg.
9.2 Calculations.
9.2.1 Bag sample volume.
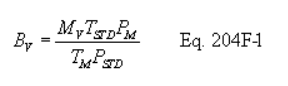
9.2.2 Bag sample VOC concentration.
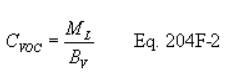
9.2.3 Bag sample VOC concentration as propane.
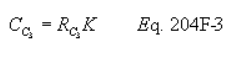
9.2.4 Response Factor.
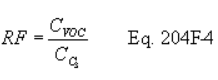
9.2.5 Total VOC Content of the Input VOC Containing Liquid.
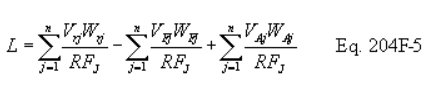
10. Diagrams
Method 205 - Verification of Gas Dilution Systems for Field Instrument Calibrations
1. Introduction
1.1 Applicability. A gas dilution system can provide known values of calibration gases through controlled dilution of high-level calibration gases with an appropriate dilution gas. The instrumental test methods in 40 CFR part 60 - e.g., Methods 3A, 6C, 7E, 10, 15, 16, 20, 25A and 25B - require on-site, multi-point calibration using gases of known concentrations. A gas dilution system that produces known low-level calibration gases from high-level calibration gases, with a degree of confidence similar to that for Protocol 1 gases, may be used for compliance tests in lieu of multiple calibration gases when the gas dilution system is demonstrated to meet the requirements of this method. The Administrator may also use a gas dilution system in order to produce a wide range of Cylinder Gas Audit concentrations when conducting performance specifications according to appendix F, 40 CFR part 60. As long as the acceptance criteria of this method are met, this method is applicable to gas dilution systems using any type of dilution technology, not solely the ones mentioned in this method.
1.2 Principle. The gas dilution system shall be evaluated on one analyzer once during each field test. A precalibrated analyzer is chosen, at the discretion of the source owner or operator, to demonstrate that the gas dilution system produces predictable gas concentrations spanning a range of concentrations. After meeting the requirements of this method, the remaining analyzers may be calibrated with the dilution system in accordance to the requirements of the applicable method for the duration of the field test. In Methods 15 and 16, 40 CFR part 60, appendix A, reactive compounds may be lost in the gas dilution system. Also, in Methods 25A and 25B, 40 CFR part 60, appendix A, calibration with target compounds other than propane is allowed. In these cases, a laboratory evaluation is required once per year in order to assure the Administrator that the system will dilute these reactive gases without significant loss.
Note:
The laboratory evaluation is required only if the source owner or operator plans to utilize the dilution system to prepare gases mentioned above as being reactive.
2. Specifications
2.1 Gas Dilution System. The gas dilution system shall produce calibration gases whose measured values are within ±2 percent of the predicted values. The predicted values are calculated based on the certified concentration of the supply gas (Protocol gases, when available, are recommended for their accuracy) and the gas flow rates (or dilution ratios) through the gas dilution system.
2.1.1 The gas dilution system shall be recalibrated once per calendar year using NIST-traceable flow standards with an uncertainty ‰¤0.25 percent. You shall report the results of the calibration by the person or manufacturer who carried out the calibration whenever the dilution system is used, listing the date of the most recent calibration, the due date for the next calibration, calibration point, reference flow device (ID, S/N), and acceptance criteria. Follow the manufacturer's instructions for the operation and use of the gas dilution system. A copy of the manufacturer's instructions for the operation of the instrument, as well as the most recent calibration documentation, shall be made available for inspection at the test site.
2.1.2 Some manufacturers of mass flow controllers recommend that flow rates below 10 percent of flow controller capacity be avoided; check for this recommendation and follow the manufacturer's instructions. One study has indicated that silicone oil from a positive displacement pump produces an interference in SO2 analyzers utilizing ultraviolet fluorescence; follow laboratory procedures similar to those outlined in Section 3.1 in order to demonstrate the significance of any resulting effect on instrument performance.
2.2 High-Level Supply Gas. An EPA Protocol calibration gas is recommended, due to its accuracy, as the high-level supply gas.
2.3 Mid-Level Supply Gas. An EPA Protocol gas shall be used as an independent check of the dilution system. The concentration of the mid-level supply gas shall be within 10 percent of one of the dilution levels tested in Section 3.2.
3. Performance Tests
3.1 Laboratory Evaluation (Optional). If the gas dilution system is to be used to formulate calibration gases with reactive compounds (Test Methods 15, 16, and 25A/25B (only if using a calibration gas other than propane during the field test) in 40 CFR part 60, appendix A), a laboratory certification must be conducted once per calendar year for each reactive compound to be diluted. In the laboratory, carry out the procedures in Section 3.2 on the analyzer required in each respective test method to be laboratory certified (15, 16, or 25A and 25B for compounds other than propane). For each compound in which the gas dilution system meets the requirements in Section 3.2, the source must provide the laboratory certification data for the field test and in the test report.
3.2 Field Evaluation (Required). The gas dilution system shall be evaluated at the test site with an analyzer or monitor chosen by the source owner or operator. It is recommended that the source owner or operator choose a precalibrated instrument with a high level of precision and accuracy for the purposes of this test. This method is not meant to replace the calibration requirements of test methods. In addition to the requirements in this method, all the calibration requirements of the applicable test method must also be met.
3.2.1 Prepare the gas dilution system according to the manufacturer's instructions. Using the high-level supply gas, prepare, at a minimum, two dilutions within the range of each dilution device utilized in the dilution system (unless, as in critical orifice systems, each dilution device is used to make only one dilution; in that case, prepare one dilution for each dilution device). Dilution device in this method refers to each mass flow controller, critical orifice, capillary tube, positive displacement pump, or any other device which is used to achieve gas dilution.
3.2.2 Calculate the predicted concentration for each of the dilutions based on the flow rates through the gas dilution system (or the dilution ratios) and the certified concentration of the high-level supply gas.
3.2.3 Introduce each of the dilutions from Section 3.2.1 into the analyzer or monitor one at a time and determine the instrument response for each of the dilutions.
3.2.4 Repeat the procedure in Section 3.2.3 two times, i.e., until three injections are made at each dilution level. Calculate the average instrument response for each triplicate injection at each dilution level. No single injection shall differ by more than ±2 percent from the average instrument response for that dilution.
3.2.5 For each level of dilution, calculate the difference between the average concentration output recorded by the analyzer and the predicted concentration calculated in Section 3.2.2. The average concentration output from the analyzer shall be within ±2 percent of the predicted value.
3.2.6 Introduce the mid-level supply gas directly into the analyzer, bypassing the gas dilution system. Repeat the procedure twice more, for a total of three mid-level supply gas injections. Calculate the average analyzer output concentration for the mid-level supply gas. The difference between the certified concentration of the mid-level supply gas and the average instrument response shall be within ±2 percent.
3.3 If the gas dilution system meets the criteria listed in Section 3.2, the gas dilution system may be used throughout that field test. If the gas dilution system fails any of the criteria listed in Section 3.2, and the tester corrects the problem with the gas dilution system, the procedure in Section 3.2 must be repeated in its entirety and all the criteria in Section 3.2 must be met in order for the gas dilution system to be utilized in the test.
4. References
1. "EPA Traceability Protocol for Assay and Certification of Gaseous Calibration Standards," EPA-600/R93/224, Revised September 1993.
Method 207 - Pre-survey Procedure for Corn Wet-milling Facility Emission Sources
1.0 Scope and Application
1.1 Analyte. Total gaseous organic compounds.
1.2 Applicability. This pre-survey method is intended for use at corn wet-milling (CWM) facilities to satisfy the requirements of Method 18, Section 16 (Pre-survey). This procedure establishes the analytes for subsequent Method 18 testing to determine the total mass emissions of VOCs from sources at CWM facilities. The specific objectives of the pre-survey procedure are:
1.2.1 Identify the physical characteristics of the VOC contained in the effluent.
1.2.2 Determine the appropriate Method 18 sampling approach to ensure efficient collection of all VOC present in the effluent.
1.2.3 Develop a specific list of target compounds to be quantified during the subsequent total VOC test program.
1.2.4 Qualify the list of target compounds as being a true representation of the total VOC.
1.3 Range. The lower and upper ranges of this procedure are determined by the sensitivity of the flame ionization detector (FID) instruments used. Typically, gas detection limits for the VOCs will be on the order of 1-5 ppmv, with the upper limit on the order of 100,000 ppmv.
2.0 Summary of Method
Note:
Method 6, Method 18, and Method 25A as cited in this method refer to the methods in 40 CFR Part 60, Appendix A.
This procedure calls for using an FIA in conjunction with various configurations of impingers, and other absorbents, or adsorbents to determine the best EPA Method 18 sampling train configuration for the assessment and capture of VOCs. VOC compounds present in the exhaust gas from processes located at CWM facilities fall into five general categories: Alcohols, aldehydes, acetate esters, ketones, and carboxylic acids, and typically contain fewer than six carbon atoms. This pre-survey protocol characterizes and identifies the VOC species present. Since it is qualitative in nature, quantitative performance criteria do not apply.
3.0 Definitions
3.1 Calibration drift means the difference in the measurement system response to a mid-level calibration gas before and after a stated period of operation during which no unscheduled maintenance, repair, or adjustment took place.
3.2 Calibration error means the difference between the gas concentration indicated by the measurement system and the known concentration of the calibration gas.
3.3 Calibration gas means a known concentration of a gas in an appropriate diluent gas.
3.4 Measurement system means the equipment required for the determination of the gas concentration. The system consists of the following major subsystems:
3.4.1 Sample interface means that portion of a system used for one or more of the following: Sample acquisition, sample transportation, sample conditioning, or protection of the analyzer(s) from the effects of the stack effluent.
3.4.2 Organic analyzer means that portion of the measurement system that senses the gas to be measured and generates an output proportional to its concentration.
3.5 Response time means the time interval from a step change in pollutant concentration at the inlet to the emission measurement system to the time at which 95 percent of the corresponding final value is reached as displayed on the recorder.
3.6 Span Value means the upper limit of a gas concentration measurement range that is specified for affected source categories in the applicable part of the regulations. The span value is established in the applicable regulation and is usually 1.5 to 2.5 times the applicable emission limit. If no span value is provided, use a span value equivalent to 1.5 to 2.5 times the expected concentration. For convenience, the span value should correspond to 100 percent of the recorder scale.
3.7 Zero drift means the difference in the measurement system response to a zero level calibration gas before or after a stated period of operation during which no unscheduled maintenance, repair, or adjustment took place.
4.0 Interferences [Reserved]
5.0 Safety [Reserved]
6.0 Equipment and Supplies
6.1 Organic Concentration Analyzer. A flame ionization analyzer (FIA) with heated detector block and sample handling system, meeting the requirements of USEPA Method 25A.
6.2 Heated Sampling System. A sampling system consisting of a stainless steel probe with particulate filter, Teflon ® sample line, and sampling pump capable of moving 1.0 l/min through the sample probe and line. The entire system from probe tip to FIA analyzer must have the capability to maintain all sample-wetted parts at a temperature >120°C. A schematic of the heated sampling system and impinger train is shown in Figure 1 of this method.
6.3 Impinger Train. EPA Method 6 type, comprised of three midget impingers with appropriate connections to the sampling system and FIA system. The impinger train may be chilled in an ice bath or maintained at a set temperature in a water bath as indicated by the operator's knowledge of the source and the compounds likely to be present. Additional impingers or larger impingers may be used for high moisture sources.
6.4 Adsorbent tubes.
6.4.1 Silica gel, SKC Type 226-22 or equivalent, with appropriate end connectors and holders.
6.4.2 Activated carbon, SKC Type 226-84 or equivalent, with appropriate end connectors and holders.
6.5 Tedlar bag. 24 liter, w/ Roberts valve, for GC/MS analysis of "breakthrough" VOC fraction as needed.
7.0 Reagents and Standards
7.1 Organic-free water, HPLC, or pharmaceutical grade.
7.2 Calibration Gases. The calibration gases for the gas analyzer shall be propane in air or propane in nitrogen. If organic compounds other than propane are used, the appropriate corrections for response factor must be available and applied to the results. Calibration gases shall be prepared in accordance with the procedure listed in Citation 2 of section 16. Additionally, the manufacturer of the cylinder must provide a recommended shelf life for each calibration gas cylinder over which the concentration does not change more than ±2 percent from the certified value. For calibration gas values not generally available (i.e., organics between 1 and 10 percent by volume), alternative methods for preparing calibration gas mixtures, such as dilution systems (Test Method 205, 40 CFR Part 51, Appendix M), may be used with prior approval of the Administrator.
7.3 Fuel. A 40 percent H2/60 percent N2 or He gas mixture is recommended to avoid an oxygen synergism effect that reportedly occurs when oxygen concentration varies significantly from a mean value.
7.4 Zero Gas. High purity air with less than 0.1 parts per million by volume (ppmv) of organic material (propane or carbon equivalent) or less than 0.1 percent of the span value, whichever is greater.
7.5 Low-level Calibration Gas. An organic calibration gas with a concentration equivalent to 25 to 35 percent of the applicable span value.
7.6 Mid-level Calibration Gas. An organic calibration gas with a concentration equivalent to 45 to 55 percent of the applicable span value.
7.7 High-level Calibration Gas. An organic calibration gas with a concentration equivalent to 80 to 90 percent of the applicable span value.
8.0 Sample Collection, Preservation and Storage
8.1 Configuration. The configuration of the pre-survey sampling system is provided in Figure 1. This figure shows the primary components of the sampling system needed to conduct a VOC survey. A dual-channel analyzer is beneficial, but not necessary. Only a single channel is indicated in the figure.
8.2 Sampling. The pre-survey system should be set up and calibrated with the targeted sampling flow rate that will be used during Method 18 VOC sampling. The targeted flow rate for capture of most expected VOC species is 400 cc/min. Since most FIA analyzers do not specifically allow for adjusting the total sample flow rate (only the back pressure), it may be necessary to insert a flow control valve at the sample inlet to the FIA. The total sample flow can be measured at the FIA bypass, since only a small fraction of the sample flow is diverted to analysis portion of the instrument.
The sampling system configuration shown in Figure 1 is operated using the process flow diagram provided in Figure 2. As noted in the process flowchart, the initial sampling media consists of the three midget impingers. The attenuation of the VOC sample stream is evaluated to determine if 95 percent or greater attenuation (capture) of the VOCs present has been achieved. The flow diagram specifies successive adjustments to the sampling media that are utilized to increase VOC capture.
A one-hour test of the final sampling configuration is performed using fresh media to ensure that significant breakthrough does not occur. Additional sampling media (more water, silica or carbon tubes) may be added to ensure that breakthrough is not occurring for the full duration of a test run.
If 95 percent or greater attenuation has not been achieved after inserting all indicated media, the most likely scenario is that methane is present. This is easily checked by collecting a sample of this final bypass sample stream and analyzing for methane. There are other VOC compounds which could also penetrate the media. Their identification by gas chromatography followed by mass spectrometry would be required if the breakthrough cannot be accounted for by the presence of methane.
9.0 Quality Control
9.1 Blanks. A minimum of one method blank shall be prepared and analyzed for each sample medium employed during a pre-survey testing field deployment to assess the effect of media contamination. Method blanks are prepared by assembling and charging the sample train with reagents, then recovering and preserving the blanks in the same manner as the test samples. Method blanks and test samples are stored, transported and analyzed in identical fashion as the test samples.
9.2 Synthetic Sample (optional). A synthetic sample may be used to assess the performance of the VOC characterization apparatus with respect to specific compounds. The synthetic sample is prepared by injecting appropriate volume(s) of the compounds of interest into a Tedlar bag containing a known volume of zero air or nitrogen. The contents of the bag are allowed to equilibrate, and the bag is connected to the sampling system. The sampling system, VOC characterization apparatus and FIA are operated normally to determine the performance of the system with respect to the VOC compounds present in the synthetic sample.
10.0 Calibration and Standardization
10.1 Calibration. The FIA equipment is able to be calibrated for almost any range of total organic concentrations. For high concentrations of organics (>1.0 percent by volume as propane), modifications to most commonly available analyzers are necessary. One accepted method of equipment modification is to decrease the size of the sample to the analyzer through the use of a smaller diameter sample capillary. Direct and continuous measurement of organic concentration is a necessary consideration when determining any modification design.
11.0 Procedure
11.1 Analytical Procedure. Upon completion of the pre-survey sampling, the sample fractions are to be analyzed by an appropriate chromatographic technique. (Ref: Method 18) The resulting chromatograms must be reviewed to ensure that the ratio of known peak area to total peak area is 95% or greater. It should be noted that if formaldehyde is a suspected analyte, it must be quantitated separately using a different analytical technique.
12.0 Data Analysis and Calculations
Chromatogram peaks will be ranked from greatest area to least area using peak integrator output. The area of all peaks will then be totaled, and the proportion of each peak area to the total area will be calculated. Beginning with the highest ranked area, each peak will be identified and the area added to previous areas until the cumulative area comprises at least 95% of the total area. The VOC compounds generating those identified peaks will comprise the compound list to be used in Method 18 testing of the subject source.
13.0 Method Performance [Reserved]
14.0 Pollution Prevention [Reserved]
15.0 Waste Management [Reserved]
16.0 References
16.1 CFR 40 Part 60, Appendix A, Method 18, Measurement of Gaseous Organic Compound Emissions by Gas Chromatography.
16.2 CFR 40 Part 60, Appendix A, Method 25A, Determination of Total Gaseous Organic Concentration Using a Flame Ionization Analyzer.
16.2 CFR 40 Part 60, Appendix A, Method 6, Determination of Sulfur Dioxide Emissions from Stationary Sources.
16.3 National Council for Air and Stream Improvement (NCASI), Method CI/WP-98.01 "Chilled Impinger Method for Use at Wood Products Mills to Measure Formaldehyde, Methanol, and Phenol.
17. Tables, Diagrams, Flowcharts, and Validation Data
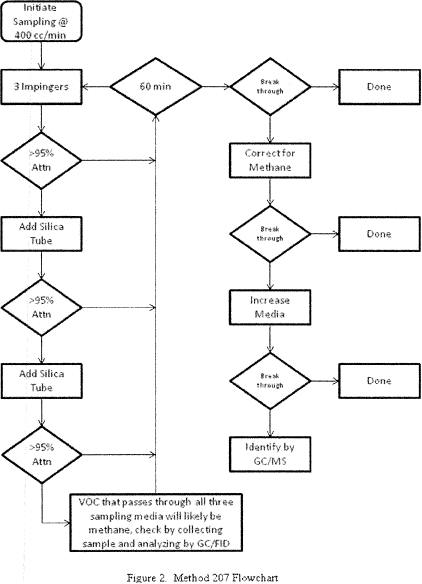
[55 FR 14249, Apr. 17, 1990; 55 FR 24687, June 18, 1990, as amended at 55 FR 37606, Sept. 12, 1990; 56 FR 6278, Feb. 15, 1991; 56 FR 65435, Dec. 17, 1991; 60 FR 28054, May 30, 1995; 62 FR 32502, June 16, 1997; 71 FR 55123, Sept. 21, 2006; 73 FR 30779, May 29, 2008; 75 FR 55644, Sept. 13, 2010; 75 FR 80134, Dec. 21, 2010; 79 FR 11235, Feb. 27, 2014; 79 FR 18453, Apr. 2, 2014; 81 FR 59806, Aug. 30, 2016; 83 FR 56720, Nov. 14, 2018; 85 FR 63401, Oct. 7, 2020; 88 FR 18401, March 29, 2023]
['Air Programs']
['Air Quality']
UPGRADE TO CONTINUE READING