Compliance Just Got Easier: Stay ahead of regulatory changes with instant notifications on updates that matter.
['Air Programs']
['Air Quality']
11/20/2023

Copyright 2026 J. J. Keller & Associate, Inc. For re-use options please contact copyright@jjkeller.com or call 800-558-5011.
Method 26 - Determination of Hydrogen Chloride Emissions From Stationary Sources
Method 26A - Determination of hydrogen halide and halogen emissions from stationary sources - isokinetic method
Method 27 - Determination of vapor tightness of gasoline delivery tank using pressure-vacuum test
Method 28 - Certification and auditing of wood heaters
Method 28A - Measurement of air to fuel ratio and minimum achievable burn rates for wood-fired appliances
Method 29 - Determination of metals emissions from stationary sources
The test methods in this appendix are referred to in §60.8 (Performance Tests) and §60.11 (Compliance With Standards and Maintenance Requirements) of 40 CFR part 60, subpart A (General Provisions). Specific uses of these test methods are described in the standards of performance contained in the subparts, beginning with Subpart D.
Within each standard of performance, a section title “Test Methods and Procedures” is provided to: (1) Identify the test methods to be used as reference methods to the facility subject to the respective standard and (2) identify any special instructions or conditions to be followed when applying a method to the respective facility. Such instructions (for example, establish sampling rates, volumes, or temperatures) are to be used either in addition to, or as a substitute for procedures in a test method. Similarly, for sources subject to emission monitoring requirements, specific instructions pertaining to any use of a test method as a reference method are provided in the subpart or in Appendix B.
Inclusion of methods in this appendix is not intended as an endorsement or denial of their applicability to sources that are not subject to standards of performance. The methods are potentially applicable to other sources; however, applicability should be confirmed by careful and appropriate evaluation of the conditions prevalent at such sources.
The approach followed in the formulation of the test methods involves specifications for equipment, procedures, and performance. In concept, a performance specification approach would be preferable in all methods because this allows the greatest flexibility to the user. In practice, however, this approach is impractical in most cases because performance specifications cannot be established. Most of the methods described herein, therefore, involve specific equipment specifications and procedures, and only a few methods in this appendix rely on performance criteria.
Minor changes in the test methods should not necessarily affect the validity of the results and it is recognized that alternative and equivalent methods exist. section 60.8 provides authority for the Administrator to specify or approve (1) equivalent methods, (2) alternative methods, and (3) minor changes in the methodology of the test methods. It should be clearly understood that unless otherwise identified all such methods and changes must have prior approval of the Administrator. An owner employing such methods or deviations from the test methods without obtaining prior approval does so at the risk of subsequent disapproval and retesting with approved methods.
Within the test methods, certain specific equipment or procedures are recognized as being acceptable or potentially acceptable and are specifically identified in the methods. The items identified as acceptable options may be used without approval but must be identified in the test report. The potentially approvable options are cited as “subject to the approval of the Administrator” or as “or equivalent.” Such potentially approvable techniques or alternatives may be used at the discretion of the owner without prior approval. However, detailed descriptions for applying these potentially approvable techniques or alternatives are not provided in the test methods. Also, the potentially approvable options are not necessarily acceptable in all applications. Therefore, an owner electing to use such potentially approvable techniques or alternatives is responsible for: (1) assuring that the techniques or alternatives are in fact applicable and are properly executed; (2) including a written description of the alternative method in the test report (the written method must be clear and must be capable of being performed without additional instruction, and the degree of detail should be similar to the detail contained in the test methods); and (3) providing any rationale or supporting data necessary to show the validity of the alternative in the particular application. Failure to meet these requirements can result in the Administrator's disapproval of the alternative.
Method 26 - Determination of Hydrogen Halide and Halogen Emissions From Stationary Sources Non-Isokinetic Method
1.0 Scope and Application
1.1 Analytes.
| Analytes | CAS No. |
|---|---|
| Hydrogen Chloride (HCl) | 7647-01-0 |
| Hydrogen Bromide (HBr) | 10035-10-6 |
| Hydrogen Fluoride (HF) | 7664-39-3 |
| Chlorine (Cl 2 ) | 7882-50-5 |
| Bromine (Br 2 ) | 7726-95-6 |
1.2 Applicability. This method is applicable for determining emissions of hydrogen halides (HX) (HCl, HBr, and HF) and halogens (X 2 ) (Cl 2 and Br 2 ) from stationary sources when specified by the applicable subpart. Sources, such as those controlled by wet scrubbers, that emit acid particulate matter must be sampled using Method 26A.
1.3 Data Quality Objectives. Adherence to the requirements of this method will enhance the quality of the data obtained from air pollutant sampling methods.
2.0 Summary of Method
2.1 An integrated sample is extracted from the source and passed through a prepurged heated probe and filter into dilute sulfuric acid and dilute sodium hydroxide solutions which collect the gaseous hydrogen halides and halogens, respectively. The filter collects particulate matter including halide salts but is not routinely recovered and analyzed. The hydrogen halides are solubilized in the acidic solution and form chloride (Cl − ), bromide (Br − ), and fluoride (F − ) ions. The halogens have a very low solubility in the acidic solution and pass through to the alkaline solution where they are hydrolyzed to form a proton (H ), the halide ion, and the hypohalous acid (HClO or HBrO). Sodium thiosulfate is added in excess to the alkaline solution to assure reaction with the hypohalous acid to form a second halide ion such that 2 halide ions are formed for each molecule of halogen gas. The halide ions in the separate solutions are measured by ion chromatography (IC).
3.0 Definitions [Reserved]
4.0 Interferences
4.1 Volatile materials, such as chlorine dioxide (ClO 2 ) and ammonium chloride (NH 4 Cl), which produce halide ions upon dissolution during sampling are potential interferents. Interferents for the halide measurements are the halogen gases which disproportionate to a hydrogen halide and a hydrohalous acid upon dissolution in water. However, the use of acidic rather than neutral or basic solutions for collection of the hydrogen halides greatly reduces the dissolution of any halogens passing through this solution.
4.2 The simultaneous presence of HBr and CL 2 may cause a positive bias in the HCL result with a corresponding negative bias in the Cl 2 result as well as affecting the HBr/Br 2 split.
4.3 High concentrations of nitrogen oxides (NO X ) may produce sufficient nitrate (NO 3 − to interfere with measurements of very low Br − levels.
4.4 A glass wool plug should not be used to remove particulate matter since a negative bias in the data could result.
4.5 There is anecdotal evidence that HF may be outgassed from new teflon components. If HF is a target analyte, then preconditioning of new teflon components, by heating should be considered.
5.0 Safety
5.1 Disclaimer. This method may involve hazardous materials, operations, and equipment. This test method may not address all of the safety problems associated with its use. It is the responsibility of the user to establish appropriate safety and health practices and determine the applicability of regulatory limitations before performing this test method.
5.2 Corrosive Reagents. The following reagents are hazardous. Personal protective equipment and safe procedures are useful in preventing chemical splashes. If contact occurs, immediately flush with copious amounts of water for at least 15 minutes. Remove clothing under shower and decontaminate. Treat residual chemical burns as thermal burns.
5.2.1 Sodium Hydroxide (NaOH). Causes severe damage to eyes and skin. Inhalation causes irritation to nose, throat, and lungs. Reacts exothermically with limited amounts of water.
5.2.2 Sulfuric Acid (H 2 SO 4 ). Rapidly destructive to body tissue. Will cause third degree burns. Eye damage may result in blindness. Inhalation may be fatal from spasm of the larynx, usually within 30 minutes. May cause lung tissue damage with edema. 1 mg/m 3 for 8 hours will cause lung damage or, in higher concentrations, death. Provide ventilation to limit inhalation. Reacts violently with metals and organics.
6.0 Equipment and Supplies
Note:
Mention of trade names or specific products does not constitute endorsement by the Environmental Protection Agency.
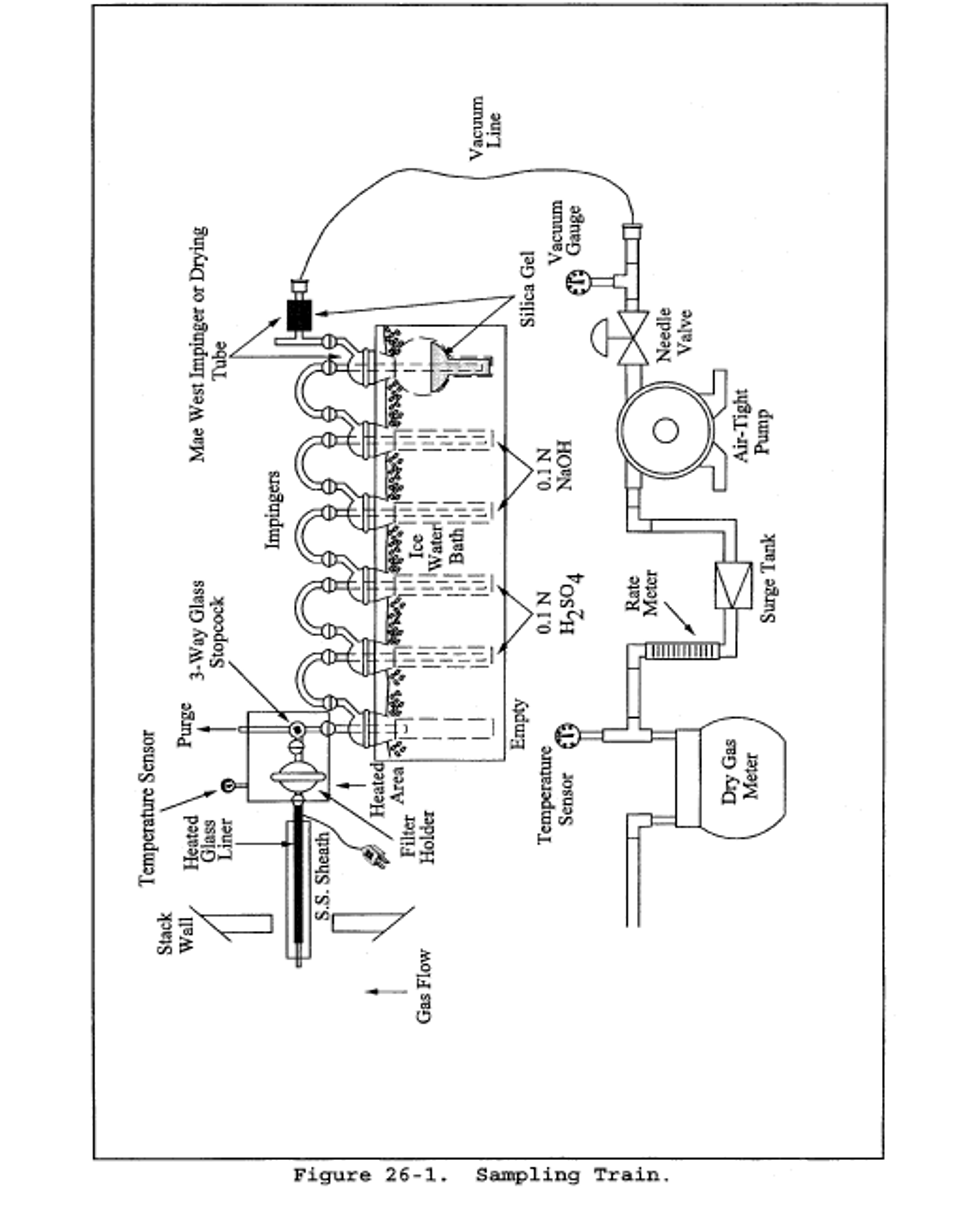
6.1 Sampling. The sampling train is shown in Figure 26-1, and component parts are discussed below.
6.1.1 Probe. Borosilicate glass, approximately 3/8-in. (9-mm) I.D. with a heating system capable of maintaining a probe gas temperature during sampling between 120 and 134°C (248 and 273°F) to prevent moisture condensation; or Teflon where stack probes are below 210°C. If HF is a target analyte, then preconditioning of new teflon components by heating should be considered to prevent potential HF outgassing. A Teflon-glass filter in a mat configuration should be installed to remove particulate matter from the gas stream.
6.1.2 Three-way Stopcock. A borosilicate-glass three-way stopcock with a heating system to prevent moisture condensation. The heated stopcock should connect to the outlet of the heated filter and the inlet of the first impinger. The heating system should be capable of preventing condensation up to the inlet of the first impinger. Silicone grease may be used, if necessary, to prevent leakage.
6.1.3 Impingers. Four 30-ml midget impingers with leak-free glass connectors. Silicone grease may be used, if necessary, to prevent leakage. For sampling at high moisture sources or for sampling times greater than one hour, a midget impinger with a shortened stem (such that the gas sample does not bubble through the collected condensate) should be used in front of the first impinger.
6.1.4 Drying Tube or Impinger. Tube or impinger, of Mae West design, filled with 6- to 16-mesh indicating type silica gel, or equivalent, to dry the gas sample and to protect the dry gas meter and pump. If the silica gel has been used previously, dry at 175°C (350°F) for 2 hours. New silica gel may be used as received. Alternatively, other types of desiccants (equivalent or better) may be used.
6.1.5 Heating System. Any heating system capable of maintaining a temperature around the probe and filter holder between 120 and 134°C (248 and 273°F) during sampling, or such other temperature as specified by an applicable subpart of the standards or approved by the Administrator for a particular application.
6.1.6 Filter Holder and Support. The filter holder shall be made of Teflon or quartz. The filter support shall be made of Teflon. All Teflon filter holders and supports are available from Savillex Corp., 5325 Hwy 101, Minnetonka, MN 55345.
6.1.7 Sample Line. Leak-free, with compatible fittings to connect the last impinger to the needle valve.
6.1.8 Rate Meter. Rotameter, or equivalent, capable of measuring flow rate to within 2 percent of the selected flow rate of 2 liters/min (0.07 ft 3 /min).
6.1.9 Purge Pump, Purge Line, Drying Tube, Needle Valve, and Rate Meter. Pump capable of purging the sampling probe at 2 liters/min, with drying tube, filled with silica gel or equivalent, to protect pump, and a rate meter capable of measuring 0 to 5 liters/min (0.2 ft 3 /min).
6.1.10 Stopcock Grease, Valve, Pump, Volume Meter, Barometer, and Vacuum Gauge. Same as in Method 6, sections 6.1.1.4, 6.1.1.7, 6.1.1.8, 6.1.1.10, 6.1.2, and 6.1.3.
6.1.11 Temperature Measuring Devices. Temperature sensors to monitor the temperature of the probe and to monitor the temperature of the sampling system from the outlet of the probe to the inlet of the first impinger.
6.1.12 Ice Water Bath. To minimize loss of absorbing solution.
6.2 Sample Recovery.
6.2.1 Wash Bottles. Polyethylene or glass, 500-ml or larger, two.
6.2.2 Storage Containers. 100- or 250-ml, high-density polyethylene or glass sample storage containers with Teflon screw cap liners to store impinger samples.
6.3 Sample Preparation and Analysis. The materials required for volumetric dilution and chromatographic analysis of samples are described below.
6.3.1 Volumetric Flasks. Class A, 100-ml size.
6.3.2 Volumetric Pipets. Class A, assortment. To dilute samples to the calibration range of the ion chromatograph.
6.3.3 Ion Chromatograph (IC). Suppressed or non-suppressed, with a conductivity detector and electronic integrator operating in the peak area mode. Other detectors, strip chart recorders, and peak height measurements may be used.
7.0 Reagents and Standards
Note:
Unless otherwise indicated, all reagents must conform to the specifications established by the Committee on Analytical Reagents of the American Chemical Society (ACS reagent grade). When such specifications are not available, the best available grade shall be used.
7.1 Sampling.
7.1.1 Filter. A 25-mm (1 in) (or other size) Teflon glass mat, Pallflex TX40HI75 (Pallflex Inc., 125 Kennedy Drive, Putnam, CT 06260). This filter is in a mat configuration to prevent fine particulate matter from entering the sampling train. Its composition is 75% Teflon/25% borosilicate glass. Other filters may be used, but they must be in a mat (as opposed to a laminate) configuration and contain at least 75% Teflon. For practical rather than scientific reasons, when the stack gas temperature exceeds 210°C (410°F) and the HCl concentration is greater than 20 ppm, a quartz-fiber filter may be used since Teflon becomes unstable above this temperature.
7.1.2 Water. Deionized, distilled water that conforms to American Society of Testing and Materials (ASTM) Specification D 1193-77 or 91, Type 3 (incorporated by reference - see §60.17 ).
7.1.3 Acidic Absorbing Solution, 0.1 N Sulfuric Acid (H 2 SO 4 ). To prepare 100 ml of the absorbing solution for the front impinger pair, slowly add 0.28 ml of concentrated H 2 SO 4 to about 90 ml of water while stirring, and adjust the final volume to 100 ml using additional water. Shake well to mix the solution.
7.1.4 Silica Gel. Indicating type, 6 to 16 mesh. If previously used, dry at 180°C (350°F) for 2 hours. New silica gel may be used as received. Alternatively, other types of desiccants may be used, subject to the approval of the Administrator.
7.1.5 Alkaline Adsorbing Solution, 0.1 N Sodium Hydroxide (NaOH). To prepare 100 ml of the scrubber solution for the third and fourth impinger, dissolve 0.40 g of solid NaOH in about 90 ml of water, and adjust the final solution volume to 100 ml using additional water. Shake well to mix the solution.
7.1.6 Sodium Thiosulfate (Na 2 S 2 O 3 5 H 2 O)
7.2 Sample Preparation and Analysis.
7.2.1 Water. Same as in section 7.1.2.
7.2.2 Absorbing Solution Blanks. A separate blank solution of each absorbing reagent should be prepared for analysis with the field samples. Dilute 30 ml of each absorbing solution to approximately the same final volume as the field samples using the blank sample of rinse water.
7.2.3 Halide Salt Stock Standard Solutions. Prepare concentrated stock solutions from reagent grade sodium chloride (NaCl), sodium bromide (NaBr), and sodium fluoride (NaF). Each must be dried at 110°C (230°F) for two or more hours and then cooled to room temperature in a desiccator immediately before weighing. Accurately weigh 1.6 to 1.7 g of the dried NaCl to within 0.1 mg, dissolve in water, and dilute to 1 liter. Calculate the exact Cl − concentration using Equation 26-1 in section 12.2. In a similar manner, accurately weigh and solubilize 1.2 to 1.3 g of dried NaBr and 2.2 to 2.3 g of NaF to make 1-liter solutions. Use Equations 26-2 and 26-3 in section 12.2, to calculate the Br − and F − concentrations. Alternately, solutions containing a nominal certified concentration of 1000 mg/l NaCl are commercially available as convenient stock solutions from which standards can be made by appropriate volumetric dilution. Refrigerate the stock standard solutions and store no longer than one month.
7.2.4 Chromatographic Eluent. Effective eluents for nonsuppressed IC using a resin-or silica-based weak ion exchange column are a 4 mM potassium hydrogen phthalate solution, adjusted to pH 4.0 using a saturated sodium borate solution, and a 4 mM 4-hydroxy benzoate solution, adjusted to pH 8.6 using 1 N NaOH. An effective eluent for suppressed ion chromatography is a solution containing 3 mM sodium bicarbonate and 2.4 mM sodium carbonate. Other dilute solutions buffered to a similar pH and containing no interfering ions may be used. When using suppressed ion chromatography, if the “water dip” resulting from sample injection interferes with the chloride peak, use a 2 mM NaOH/2.4 mM sodium bicarbonate eluent.
8.0 Sample Collection, Preservation, Storage, and Transport
Note:
Because of the complexity of this method, testers and analyst should be trained and experienced with the procedure to ensure reliable results.
8.1 Sampling.
8.1.1 Preparation of Collection Train. Prepare the sampling train as follows: Pour 15 ml of the acidic absorbing solution into each one of the first pair of impingers, and 15 ml of the alkaline absorbing solution into each one of the second pair of impingers. Connect the impingers in series with the knockout impinger first, if used, followed by the two impingers containing the acidic absorbing solution and the two impingers containing the alkaline absorbing solution. Place a fresh charge of silica gel, or equivalent, in the drying tube or impinger at the end of the impinger train.
8.1.2 Adjust the probe temperature and the temperature of the filter and the stopcock ( i.e., the heated area in Figure 26-1) to a temperature sufficient to prevent water condensation. This temperature must be maintained between 120 and 134 °C (248 and 273 °F). The temperature should be monitored throughout a sampling run to ensure that the desired temperature is maintained. It is important to maintain a temperature around the probe and filter in this range since it is extremely difficult to purge acid gases off these components. (These components are not quantitatively recovered and, hence, any collection of acid gases on these components would result in potential under reporting of these emissions. The applicable subparts may specify alternative higher temperatures.)
8.1.3 Leak-Check Procedure.
8.1.3.1 Sampling Train. A leak-check prior to the sampling run is optional; however, a leak-check after the sampling run is mandatory. The leak-check procedure is as follows: Temporarily attach a suitable [ e.g., 0-40 cc/min (0-2.4 in 3 /min)] rotameter to the outlet of the dry gas meter and place a vacuum gauge at or near the probe inlet. Plug the probe inlet, pull a vacuum of at least 250 mm Hg (10 in. Hg), and note the flow rate as indicated by the rotameter. A leakage rate not in excess of 2 percent of the average sampling rate is acceptable.
Note:
Carefully release the probe inlet plug before turning off the pump.
8.1.3.2 Pump. It is suggested (not mandatory) that the pump be leak-checked separately, either prior to or after the sampling run. If done prior to the sampling run, the pump leak-check shall precede the leak-check of the sampling train described immediately above; if done after the sampling run, the pump leak-check shall follow the train leak-check. To leak-check the pump, proceed as follows: Disconnect the drying tube from the probe-impinger assembly. Place a vacuum gauge at the inlet to either the drying tube or pump, pull a vacuum of 250 mm (10 in) Hg, plug or pinch off the outlet of the flow meter, and then turn off the pump. The vacuum should remain stable for at least 30 sec. Other leak-check procedures may be used, subject to the approval of the Administrator, U.S. Environmental Protection Agency.
8.1.4 Purge Procedure. Immediately before sampling, connect the purge line to the stopcock, and turn the stopcock to permit the purge pump to purge the probe (see Figure 1A of Figure 26-1). Turn on the purge pump, and adjust the purge rate to 2 liters/min (0.07 ft 3 /min). Purge for at least 5 minutes before sampling.
8.1.5 Sample Collection. Turn on the sampling pump, pull a slight vacuum of approximately 25 mm Hg (1 in Hg) on the impinger train, and turn the stopcock to permit stack gas to be pulled through the impinger train (see Figure 1C of Figure 26-1). Adjust the sampling rate to 2 liters/min, as indicated by the rate meter, and maintain this rate to within 10 percent during the entire sampling run. Take readings of the dry gas meter volume and temperature, rate meter, and vacuum gauge at least once every five minutes during the run. A sampling time of one hour is recommended. Shorter sampling times may introduce a significant negative bias in the HCl concentration. At the conclusion of the sampling run, remove the train from the stack, cool, and perform a leak-check as described in section 8.1.3.1.
8.2 Sample Recovery.
8.2.1 Disconnect the impingers after sampling. Quantitatively transfer the contents of the acid impingers and the knockout impinger, if used, to a leak-free storage bottle. Add the water rinses of each of these impingers and connecting glassware to the storage bottle.
8.2.2 Repeat this procedure for the alkaline impingers and connecting glassware using a separate storage bottle. Add 25 mg of sodium thiosulfate per the product of ppm of halogen anticipated to be in the stack gas times the volume (dscm) of stack gas sampled (0.7 mg per ppm-dscf).
Note:
This amount of sodium thiosulfate includes a safety factor of approximately 5 to assure complete reaction with the hypohalous acid to form a second Cl − ion in the alkaline solution.
8.2.3 Save portions of the absorbing reagents (0.1 N H 2 SO 4 and 0.1 N NaOH) equivalent to the amount used in the sampling train (these are the absorbing solution blanks described in section 7.2.2); dilute to the approximate volume of the corresponding samples using rinse water directly from the wash bottle being used. Add the same amount of sodium thiosulfate solution to the 0.1 N NaOH absorbing solution blank. Also, save a portion of the rinse water used to rinse the sampling train. Place each in a separate, prelabeled storage bottle. The sample storage bottles should be sealed, shaken to mix, and labeled. Mark the fluid level.
8.3 Sample Preparation for Analysis. Note the liquid levels in the storage bottles and confirm on the analysis sheet whether or not leakage occurred during transport. If a noticeable leakage has occurred, either void the sample or use methods, subject to the approval of the Administrator, to correct the final results. Quantitatively transfer the sample solutions to 100-ml volumetric flasks, and dilute to 100 ml with water.
9.0 Quality Control [Reserved]
10.0 Calibration and Standardization
Note:
Maintain a laboratory log of all calibrations.
10.1 Volume Metering System, Temperature Sensors, Rate Meter, and Barometer. Same as in Method 6, sections 10.1, 10.2, 10.3, and 10.4.
10.2 Ion Chromatograph.
10.2.1 To prepare the calibration standards, dilute given amounts (1.0 ml or greater) of the stock standard solutions to convenient volumes, using 0.1 N H 2 SO 4 or 0.1 N NaOH, as appropriate. Prepare at least four calibration standards for each absorbing reagent containing the appropriate stock solutions such that they are within the linear range of the field samples.
10.2.2 Using one of the standards in each series, ensure adequate baseline separation for the peaks of interest.
10.2.3 Inject the appropriate series of calibration standards, starting with the lowest concentration standard first both before and after injection of the quality control check sample, reagent blanks, and field samples. This allows compensation for any instrument drift occurring during sample analysis. The values from duplicate injections of these calibration samples should agree within 5 percent of their mean for the analysis to be valid.
10.2.4 Determine the peak areas, or heights, for the standards and plot individual values versus halide ion concentrations in µg/ml.
10.2.5 Draw a smooth curve through the points. Use linear regression to calculate a formula describing the resulting linear curve.
11.0 Analytical Procedures
11.1 Sample Analysis.
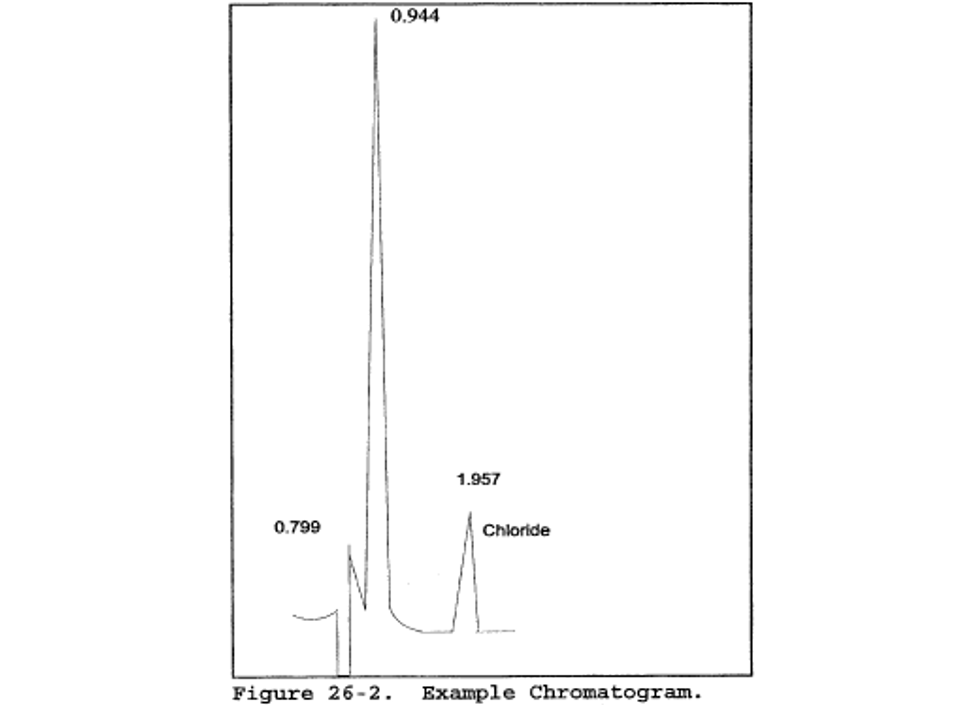
11.1.1 The IC conditions will depend upon analytical column type and whether suppressed or non-suppressed IC is used. An example chromatogram from a non-suppressed system using a 150-mm Hamilton PRP-X100 anion column, a 2 ml/min flow rate of a 4 mM 4-hydroxy benzoate solution adjusted to a pH of 8.6 using 1 N NaOH, a 50 µl sample loop, and a conductivity detector set on 1.0 µS full scale is shown in Figure 26-2.
11.1.2 Before sample analysis, establish a stable baseline. Next, inject a sample of water, and determine if any Cl − , Br − , or F − appears in the chromatogram. If any of these ions are present, repeat the load/injection procedure until they are no longer present. Analysis of the acid and alkaline absorbing solution samples requires separate standard calibration curves; prepare each according to section 10.2. Ensure adequate baseline separation of the analyses.
11.1.3 Between injections of the appropriate series of calibration standards, inject in duplicate the reagent blanks, quality control sample, and the field samples. Measure the areas or heights of the Cl − , Br − , and F − peaks. Use the mean response of the duplicate injections to determine the concentrations of the field samples and reagent blanks using the linear calibration curve. The values from duplicate injections should agree within 5 percent of their mean for the analysis to be valid. If the values of duplicate injections are not within 5 percent of the mean, the duplicate injections shall be repeated and all four values used to determine the average response. Dilute any sample and the blank with equal volumes of water if the concentration exceeds that of the highest standard.
12.0 Data Analysis and Calculations
Note:
Retain at least one extra decimal figure beyond those contained in the available data in intermediate calculations, and round off only the final answer appropriately.
12.1 Nomenclature.
B X − = Mass concentration of applicable absorbing solution blank, µg halide ion (Cl − , Br − , F − ) /ml, not to exceed 1 µg/ml which is 10 times the published analytical detection limit of 0.1 µg/ml.
C = Concentration of hydrogen halide (HX) or halogen (X 2 ), dry basis, mg/dscm.
K = 10 −3 mg/µg.
K HCl = 1.028 (µg HCl/µg-mole)/(µg Cl − /µg-mole).
K HBr = 1.013 (µg HBr/µg-mole)/(µg Br − /µg-mole).
K HF = 1.053 (µg HF/µg-mole)/(µg F − /µg-mole).
m HX = Mass of HCl, HBr, or HF in sample, µg.
m X2 = Mass of Cl 2 or Br 2 in sample, µg.
S X − = Analysis of sample, µg halide ion (Cl − , Br − , F − )/ml.
V m(std) = Dry gas volume measured by the dry gas meter, corrected to standard conditions, dscm.
V s = Volume of filtered and diluted sample, ml.
12.2 Calculate the exact Cl − , Br − , and F − concentration in the halide salt stock standard solutions using the following equations.
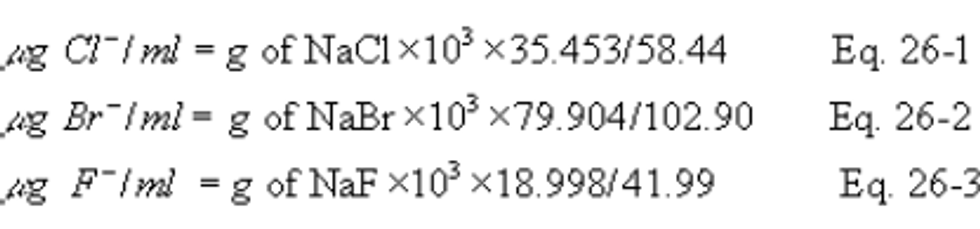
12.3 Sample Volume, Dry Basis, Corrected to Standard Conditions. Calculate the sample volume using Eq. 6-1 of Method 6.
12.4 Total µg HCl, HBr, or HF Per Sample.
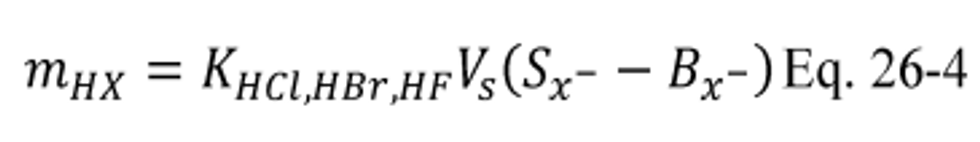
12.5 Total µg Cl 2 or Br 2 Per Sample.
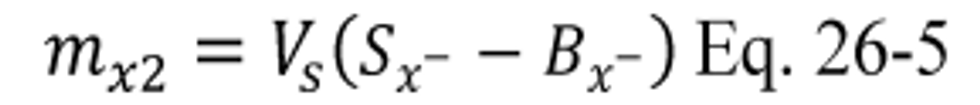
12.6 Concentration of Hydrogen Halide or Halogen in Flue Gas.
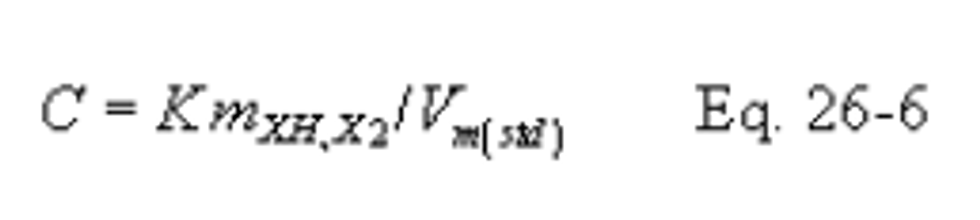
13.0 Method Performance
13.1 Precision and Bias. The within-laboratory relative standard deviations are 6.2 and 3.2 percent at HCl concentrations of 3.9 and 15.3 ppm, respectively. The method does not exhibit a bias to Cl 2 when sampling at concentrations less than 50 ppm.
13.2 Sample Stability. The collected Cl − samples can be stored for up to 4 weeks.
13.3 Detection Limit. A typical IC instrumental detection limit for Cl − is 0.2 µg/ml. Detection limits for the other analyses should be similar. Assuming 50 ml liquid recovered from both the acidified impingers, and the basic impingers, and 0.12 dscm (4.24 dscf) of stack gas sampled, then the analytical detection limit in the stack gas would be about 0.05 ppm for HCl and Cl 2 , respectively.
14.0 Pollution Prevention [Reserved]
15.0 Waste Management [Reserved]
16.0 Alternative Procedures
Method 26A. Method 26A, which uses isokinetic sampling equipment, is an acceptable alternative to Method 26.
17.0 References
1. Steinsberger, S. C. and J. H. Margeson, “Laboratory and Field Evaluation of a Methodology for Determination of Hydrogen Chloride Emissions from Municipal and Hazardous Waste Incinerators,” U.S. Environmental Protection Agency, Office of Research and Development, Report No. 600/3-89/064, April 1989. Available from the National Technical Information Service, Springfield, VA 22161 as PB89220586/AS.
2. State of California, Air Resources Board, Method 421, “Determination of Hydrochloric Acid Emissions from Stationary Sources,” March 18, 1987.
3. Cheney, J.L. and C.R. Fortune. Improvements in the Methodology for Measuring Hydrochloric Acid in Combustion Source Emissions. J. Environ. Sci. Health. A19 (3): 337-350. 1984.
4. Stern, D. A., B. M. Myatt, J. F. Lachowski, and K. T. McGregor. Speciation of Halogen and Hydrogen Halide Compounds in Gaseous Emissions. In: Incineration and Treatment of Hazardous Waste: Proceedings of the 9th Annual Research Symposium, Cincinnati, Ohio, May 2-4, 1983. Publication No. 600/9-84-015. July 1984. Available from National Technical Information Service, Springfield, VA 22161 as PB84-234525.
5. Holm, R. D. and S. A. Barksdale. Analysis of Anions in Combustion Products. In: Ion Chromatographic Analysis of Environmental Pollutants. E. Sawicki, J. D. Mulik, and E. Wittgenstein (eds.). Ann Arbor, Michigan, Ann Arbor Science Publishers. 1978. pp. 99-110.
18.0 Tables, Diagrams, Flowcharts, and Validation Data
Method 26A - Determination of Hydrogen Halide and Halogen Emissions From Stationary Sources Isokinetic Method
Note:
This method does not include all of the specifications ( e.g., equipment and supplies) and procedures ( e.g., sampling and analytical) essential to its performance. Some material is incorporated by reference from other methods in this part. Therefore, to obtain reliable results, persons using this method should have a thorough knowledge of at least the following additional test methods: Method 2, Method 5, and Method 26.
1.0 Scope and Application
1.1 Analytes.
| Analytes | CAS No. |
|---|---|
| Hydrogen Chloride (HCl) | 7647-01-0 |
| Hydrogen Bromide (HBr) | 10035-10-6 |
| Hydrogen Fluoride (HF) | 7664-39-3 |
| Chlorine (Cl 2 ) | 7882-50-5 |
| Bromine (Br 2 ) | 7726-95-6 |
1.2 This method is applicable for determining emissions of hydrogen halides (HX) [HCl, HBr, and HF] and halogens (X 2 ) [Cl 2 and Br 2 ] from stationary sources when specified by the applicable subpart. This method collects the emission sample isokinetically and is therefore particularly suited for sampling at sources, such as those controlled by wet scrubbers, emitting acid particulate matter ( e.g., hydrogen halides dissolved in water droplets).
1.3 Data Quality Objectives. Adherence to the requirements of this method will enhance the quality of the data obtained from air pollutant sampling methods.
2.0 Summary of Method
2.1 Principle. Gaseous and particulate pollutants are withdrawn isokinetically from the source and collected in an optional cyclone, on a filter, and in absorbing solutions. The cyclone collects any liquid droplets and is not necessary if the source emissions do not contain them; however, it is preferable to include the cyclone in the sampling train to protect the filter from any liquid present. The filter collects particulate matter including halide salts but is not routinely recovered or analyzed. Acidic and alkaline absorbing solutions collect the gaseous hydrogen halides and halogens, respectively. Following sampling of emissions containing liquid droplets, any halides/halogens dissolved in the liquid in the cyclone and on the filter are vaporized to gas and collected in the impingers by pulling conditioned ambient air through the sampling train. The hydrogen halides are solubilized in the acidic solution and form chloride (Cl − ), bromide (Br − ), and fluoride (F − ) ions. The halogens have a very low solubility in the acidic solution and pass through to the alkaline solution where they are hydrolyzed to form a proton (H ), the halide ion, and the hypohalous acid (HClO or HBrO). Sodium thiosulfate is added to the alkaline solution to assure reaction with the hypohalous acid to form a second halide ion such that 2 halide ions are formed for each molecule of halogen gas. The halide ions in the separate solutions are measured by ion chromatography (IC). If desired, the particulate matter recovered from the filter and the probe is analyzed following the procedures in Method 5.
Note:
If the tester intends to use this sampling arrangement to sample concurrently for particulate matter, the alternative Teflon probe liner, cyclone, and filter holder should not be used. The Teflon filter support must be used. The tester must also meet the probe and filter temperature requirements of both sampling trains.
3.0 Definitions [Reserved]
4.0 Interferences
4.1 Volatile materials, such as chlorine dioxide (ClO 2 ) and ammonium chloride (NH 4 Cl), which produce halide ions upon dissolution during sampling are potential interferents. Interferents for the halide measurements are the halogen gases which disproportionate to a hydrogen halide and a hypohalous acid upon dissolution in water. The use of acidic rather than neutral or basic solutions for collection of the hydrogen halides greatly reduces the dissolution of any halogens passing through this solution.
4.2 The simultaneous presence of both HBr and Cl 2 may cause a positive bias in the HCl result with a corresponding negative bias in the Cl 2 result as well as affecting the HBr/Br 2 split.
4.3 High concentrations of nitrogen oxides (NO X ) may produce sufficient nitrate (NO 3 − ) to interfere with measurements of very low Br − levels. Dissociating chloride salts ( e.g., ammonium chloride) at elevated temperatures interfere with halogen acid measurement in this method. Maintaining particulate probe/filter temperatures between 120°C and 134°C (248°F and 273°F) minimizes this interference.
4.4 There is anecdotal evidence that HF may be outgassed from new Teflon components. If HF is a target analyte then preconditioning of new Teflon components, by heating, should be considered.
5.0 Safety
5.1 Disclaimer. This method may involve hazardous materials, operations, and equipment. This test method may not address all of the safety problems associated with its use. It is the responsibility of the user to establish appropriate safety and health practices and determine the applicability of regulatory limitations before performing this test method.
5.2 Corrosive Reagents. The following reagents are hazardous. Personal protective equipment and safe procedures are useful in preventing chemical splashes. If contact occurs, immediately flush with copious amounts of water for at least 15 minutes. Remove clothing under shower and decontaminate. Treat residual chemical burns as thermal burns.
5.2.1 Sodium Hydroxide (NaOH). Causes severe damage to eyes and skin. Inhalation causes irritation to nose, throat, and lungs. Reacts exothermically with limited amounts of water.
5.2.2 Sulfuric Acid (H 2 SO 4 ). Rapidly destructive to body tissue. Will cause third degree burns. Eye damage may result in blindness. Inhalation may be fatal from spasm of the larynx, usually within 30 minutes. May cause lung tissue damage with edema. 1 mg/m 3 for 8 hours will cause lung damage or, in higher concentrations, death. Provide ventilation to limit inhalation. Reacts violently with metals and organics.
6.0. Equipment and Supplies
Note:
Mention of trade names or specific products does not constitute endorsement by the Environmental Protection Agency.
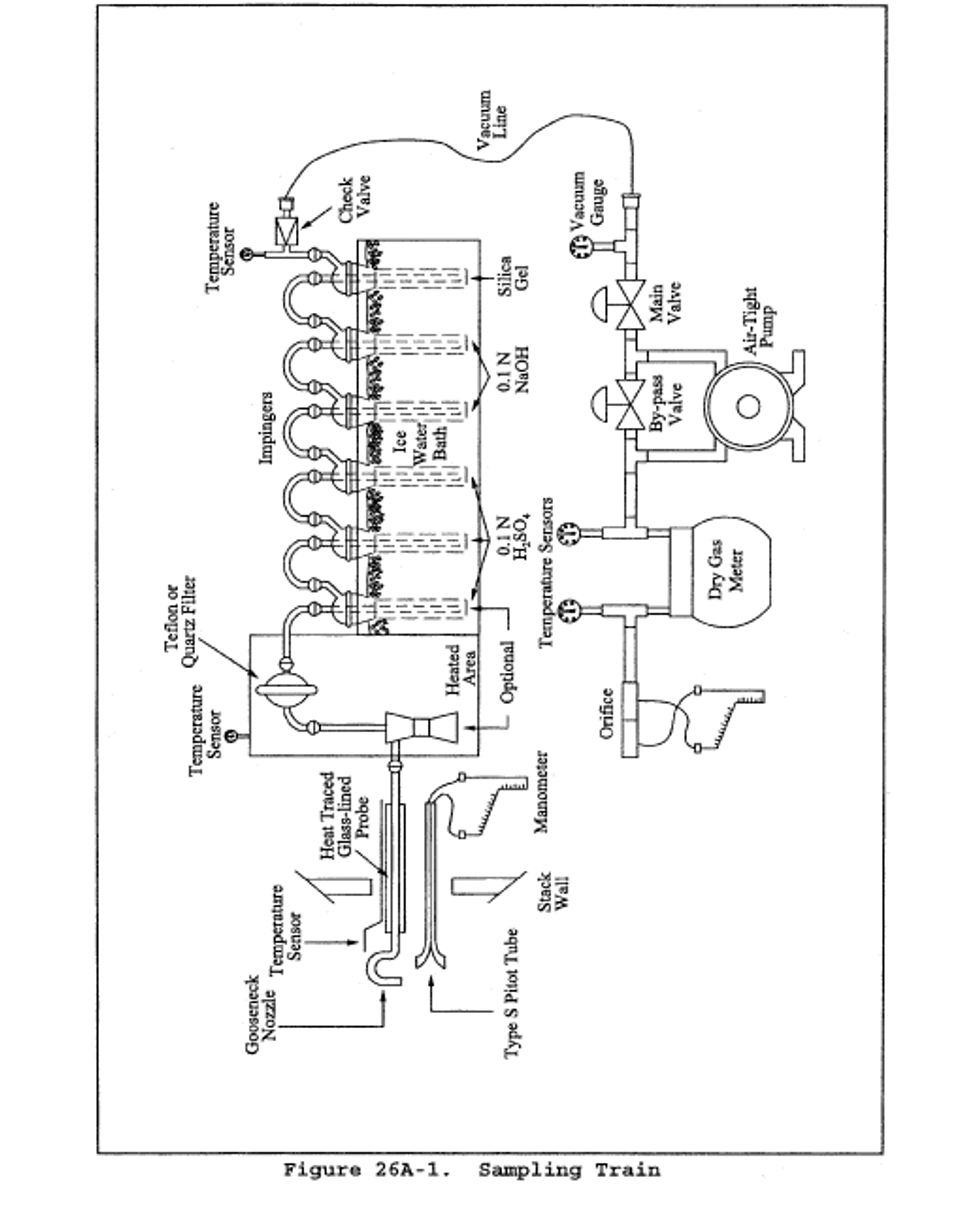
6.1 Sampling. The sampling train is shown in Figure 26A-1; the apparatus is similar to the Method 5 train where noted as follows:
6.1.1 Probe Nozzle. Borosilicate or quartz glass; constructed and calibrated according to Method 5, sections 6.1.1.1 and 10.1, and coupled to the probe liner using a Teflon union; a stainless steel nut is recommended for this union. When the stack temperature exceeds 210°C (410°F), a one-piece glass nozzle/liner assembly must be used.
6.1.2 Probe Liner. Same as Method 5, section 6.1.1.2, except metal liners shall not be used. Water-cooling of the stainless steel sheath is recommended at temperatures exceeding 500°C (932°F). Teflon may be used in limited applications where the minimum stack temperature exceeds 120°C (250°F) but never exceeds the temperature where Teflon is estimated to become unstable [approximately 210°C (410°F)].
6.1.3 Pitot Tube, Differential Pressure Gauge, Filter Heating System, Filter Temperature Sensor with a glass or Teflon encasement, Metering System, Barometer, Gas Density Determination Equipment. Same as Method 5, sections 6.1.1.3, 6.1.1.4, 6.1.1.6, 6.1.1.7, 6.1.1.9, 6.1.2, and 6.1.3.
6.1.4 Cyclone (Optional). Glass or Teflon. Use of the cyclone is required only when the sample gas stream is saturated with moisture; however, the cyclone is recommended to protect the filter from any liquid droplets present.
6.1.5 Filter Holder. Borosilicate or quartz glass, or Teflon filter holder, with a Teflon filter support and a sealing gasket. The sealing gasket shall be constructed of Teflon or equivalent materials. The holder design shall provide a positive seal against leakage at any point along the filter circumference. The holder shall be attached immediately to the outlet of the cyclone.
6.1.6 Impinger Train. The following system shall be used to determine the stack gas moisture content and to collect the hydrogen halides and halogens: five or six impingers connected in series with leak-free ground glass fittings or any similar leak-free noncontaminating fittings. The first impinger shown in Figure 26A-1 (knockout or condensate impinger) is optional and is recommended as a water knockout trap for use under high moisture conditions. If used, this impinger should be constructed as described below for the alkaline impingers, but with a shortened stem, and should contain 50 ml of 0.1 N H 2 SO 4 . The following two impingers (acid impingers which each contain 100 ml of 0.1 N H 2 SO 4 ) shall be of the Greenburg-Smith design with the standard tip (Method 5, section 6.1.1.8). The next two impingers (alkaline impingers which each contain 100 ml of 0.1 N NaOH) and the last impinger (containing silica gel) shall be of the modified Greenburg-Smith design (Method 5, section 6.1.1.8). The condensate, acid, and alkaline impingers shall contain known quantities of the appropriate absorbing reagents. The last impinger shall contain a known weight of silica gel or equivalent desiccant. Teflon impingers are an acceptable alternative.
6.1.7 Heating System. Any heating system capable of maintaining a temperature around the probe and filter holder between 120 and 134°C (248 to 273°F) during sampling, or such other temperature as specified by an applicable subpart of the standards or approved by the Administrator for a particular application.
6.1.8 Ambient Air Conditioning Tube (Optional). Tube tightly packed with approximately 150 g of fresh 8 to 20 mesh sodium hydroxide-coated silica, or equivalent, (Ascarite II has been found suitable) to dry and remove acid gases from the ambient air used to remove moisture from the filter and cyclone, when the cyclone is used. The inlet and outlet ends of the tube should be packed with at least 1-cm thickness of glass wool or filter material suitable to prevent escape of fines. Fit one end with flexible tubing, etc. to allow connection to probe nozzle following the test run.
6.2 Sample Recovery.
6.2.1 Probe-Liner and Probe-Nozzle Brushes, Wash Bottles, Petri Dishes, Graduated Cylinder and/or Balance, and Rubber Policeman. Same as Method 5, sections 6.2.1, 6.2.2, 6.2.4, 6.2.5, and 6.2.7.
6.2.2 Plastic Storage Containers. Screw-cap polypropylene or polyethylene containers to store silica gel. High-density polyethylene bottles with Teflon screw cap liners to store impinger reagents, 1-liter.
6.2.3 Funnels. Glass or high-density polyethylene, to aid in sample recovery.
6.2.4 Sample Storage Containers. High-density polyethylene or glass sample storage containers with Teflon screw cap liners to store impinger samples.
6.3 Sample Preparation and Analysis.
6.3.1 Volumetric Flasks. Class A, various sizes.
6.3.2 Volumetric Pipettes. Class A, assortment. To dilute samples to calibration range of the ion chromatograph (IC).
6.3.3 Ion Chromatograph (IC). Suppressed or nonsuppressed, with a conductivity detector and electronic integrator operating in the peak area mode. Other detectors, a strip chart recorder, and peak heights may be used.
7.0 Reagents and Standards
Note:
Unless otherwise indicated, all reagents must conform to the specifications established by the Committee on Analytical Reagents of the American Chemical Society (ACS reagent grade). When such specifications are not available, the best available grade shall be used.
7.1 Sampling.
7.1.1 Filter. Teflon mat ( e.g., Pallflex TX40HI45) filter. When the stack gas temperature exceeds 210°C (410°F) a quartz fiber filter may be used.
7.1.2 Water. Deionized, distilled water that conforms to American Society of Testing and Materials (ASTM) Specification D 1193-77 or 91, Type 3 (incorporated by reference - see §60.17 ).
7.1.3 Acidic Absorbing Solution, 0.1 N Sulfuric Acid (H 2 SO 4 ). To prepare 1 L, slowly add 2.80 ml of concentrated 17.9 M H2SO4 to about 900 ml of water while stirring, and adjust the final volume to 1 L using additional water. Shake well to mix the solution.
7.1.4 Silica Gel, Crushed Ice, and Stopcock Grease. Same as Method 5, sections 7.1.2, 7.1.4, and 7.1.5, respectively.
7.1.5 Alkaline Absorbing Solution, 0.1 N Sodium Hydroxide (NaOH). To prepare 1 L, dissolve 4.00 g of solid NaOH in about 900 ml of water and adjust the final volume to 1 L using additional water. Shake well to mix the solution.
7.1.6 Sodium Thiosulfate, (Na 2 S 2 O 3 3.5 H 2 O).
7.2 Sample Preparation and Analysis.
7.2.1 Water. Same as in section 7.1.2.
7.2.2 Absorbing Solution Blanks. A separate blank solution of each absorbing reagent should be prepared for analysis with the field samples. Dilute 200 ml of each absorbing solution (250 ml of the acidic absorbing solution, if a condensate impinger is used) to the same final volume as the field samples using the blank sample of rinse water. If a particulate determination is conducted, collect a blank sample of acetone.
7.2.3 Halide Salt Stock Standard Solutions. Prepare concentrated stock solutions from reagent grade sodium chloride (NaCl), sodium bromide (NaBr), and sodium fluoride (NaF). Each must be dried at 110°C (230°F) for two or more hours and then cooled to room temperature in a desiccator immediately before weighing. Accurately weigh 1.6 to 1.7 g of the dried NaCl to within 0.1 mg, dissolve in water, and dilute to 1 liter. Calculate the exact Cl − concentration using Equation 26A-1 in section 12.2. In a similar manner, accurately weigh and solubilize 1.2 to 1.3 g of dried NaBr and 2.2 to 2.3 g of NaF to make 1-liter solutions. Use Equations 26A-2 and 26A-3 in section 12.2, to calculate the Br − and F − concentrations. Alternately, solutions containing a nominal certified concentration of 1000 mg/L NaCl are commercially available as convenient stock solutions from which standards can be made by appropriate volumetric dilution. Refrigerate the stock standard solutions and store no longer than one month.
7.2.4 Chromatographic Eluent. Same as Method 26, section 7.2.4.
7.2.5 Water. Same as section 7.1.1.
7.2.6 Acetone. Same as Method 5, section 7.2.
8.0 Sample Collection, Preservation, Storage, and Transport
Note:
Because of the complexity of this method, testers and analysts should be trained and experienced with the procedures to ensure reliable results.
8.1 Sampling.
8.1.1 Pretest Preparation. Follow the general procedure given in Method 5, section 8.1, except the filter need only be desiccated and weighed if a particulate determination will be conducted.
8.1.2 Preliminary Determinations. Same as Method 5, section 8.2.
8.1.3 Preparation of Sampling Train. Follow the general procedure given in Method 5, section 8.1.3, except for the following variations: Add 50 ml of 0.1 N H 2 SO 4 to the condensate impinger, if used. Place 100 ml of 0.1 N H 2 SO 4 in each of the next two impingers. Place 100 ml of 0.1 N NaOH in each of the following two impingers. Finally, transfer approximately 200-300 g of preweighed silica gel from its container to the last impinger. Set up the train as in Figure 26A-1. When used, the optional cyclone is inserted between the probe liner and filter holder and located in the heated filter box.
8.1.4 Leak-Check Procedures. Follow the leak-check procedures given in Method 5, sections 8.4.2 (Pretest Leak-Check), 8.4.3 (Leak-Checks During the Sample Run), and 8.4.4 (Post-Test Leak-Check).
8.1.5 Sampling Train Operation. Follow the general procedure given in Method 5, Section 8.5. It is important to maintain a temperature around the probe, filter (and cyclone, if used) between 120 and 134 °C (248 and 273 °F) since it is extremely difficult to purge acid gases off these components. (These components are not quantitatively recovered and hence any collection of acid gases on these components would result in potential under reporting these emissions. The applicable subparts may specify alternative higher temperatures.) For each run, record the data required on a data sheet such as the one shown in Method 5, Figure 5-3. If the condensate impinger becomes too full, it may be emptied, recharged with 50 ml of 0.1 N H2SO4, and replaced during the sample run. The condensate emptied must be saved and included in the measurement of the volume of moisture collected and included in the sample for analysis. The additional 50 ml of absorbing reagent must also be considered in calculating the moisture. Before the sampling train integrity is compromised by removing the impinger, conduct a leak-check as described in Method 5, section 8.4.2.
8.1.6 Post-Test Moisture Removal (Optional). When the optional cyclone is included in the sampling train or when liquid is visible on the filter at the end of a sample run even in the absence of a cyclone, perform the following procedure. Upon completion of the test run, connect the ambient air conditioning tube at the probe inlet and operate the train with the filter heating system between 120 and 134°C (248 and 273°F) at a low flow rate ( e.g., ΔH = 1 in. H 2 O) to vaporize any liquid and hydrogen halides in the cyclone or on the filter and pull them through the train into the impingers. After 30 minutes, turn off the flow, remove the conditioning tube, and examine the cyclone and filter for any visible liquid. If liquid is visible, repeat this step for 15 minutes and observe again. Keep repeating until the cyclone is dry.
Note:
It is critical that this procedure is repeated until the cyclone is completely dry.
8.2 Sample Recovery. Allow the probe to cool. When the probe can be handled safely, wipe off all the external surfaces of the tip of the probe nozzle and place a cap loosely over the tip to prevent gaining or losing particulate matter. Do not cap the probe tip tightly while the sampling train is cooling down because this will create a vacuum in the filter holder, drawing water from the impingers into the holder. Before moving the sampling train to the cleanup site, remove the probe from the sample train, wipe off any silicone grease, and cap the open outlet of the impinger train, being careful not to lose any condensate that might be present. Wipe off any silicone grease and cap the filter or cyclone inlet. Remove the umbilical cord from the last impinger and cap the impinger. If a flexible line is used between the first impinger and the filter holder, disconnect it at the filter holder and let any condensed water drain into the first impinger. Wipe off any silicone grease and cap the filter holder outlet and the impinger inlet. Ground glass stoppers, plastic caps, serum caps, Teflon tape, Parafilm, or aluminum foil may be used to close these openings. Transfer the probe and filter/impinger assembly to the cleanup area. This area should be clean and protected from the weather to minimize sample contamination or loss. Inspect the train prior to and during disassembly and note any abnormal conditions. Treat samples as follows:
8.2.1 Container No. 1 (Optional; Filter Catch for Particulate Determination). Same as Method 5, section 8.7.6.1, Container No. 1.
8.2.2 Container No. 2 (Optional; Front-Half Rinse for Particulate Determination). Same as Method 5, section 8.7.6.2, Container No. 2.
8.2.3 Container No. 3 (Knockout and Acid Impinger Catch for Moisture and Hydrogen Halide Determination). Disconnect the impingers. Measure the liquid in the acid and knockout impingers to ±1 ml by using a graduated cylinder or by weighing it to ±0.5 g by using a balance. Record the volume or weight of liquid present. This information is required to calculate the moisture content of the effluent gas. Quantitatively transfer this liquid to a leak-free sample storage container. Rinse these impingers and connecting glassware including the back portion of the filter holder (and flexible tubing, if used) with water and add these rinses to the storage container. Seal the container, shake to mix, and label. The fluid level should be marked so that if any sample is lost during transport, a correction proportional to the lost volume can be applied. Retain rinse water and acidic absorbing solution blanks to be analyzed with the samples.
8.2.4 Container No. 4 (Alkaline Impinger Catch for Halogen and Moisture Determination). Measure and record the liquid in the alkaline impingers as described in section 8.2.3. Quantitatively transfer this liquid to a leak-free sample storage container. Rinse these two impingers and connecting glassware with water and add these rinses to the container. Add 25 mg of sodium thiosulfate per ppm halogen anticipated to be in the stack gas multiplied by the volume (dscm) of stack gas sampled (0.7 mg/ppm-dscf). Seal the container, shake to mix, and label; mark the fluid level. Retain alkaline absorbing solution blank to be analyzed with the samples.
Note:
25 mg per sodium thiosulfate per ppm halogen anticipated to be in the stack includes a safety factor of approximately 5 to assure complete reaction with the hypohalous acid to form a second Cl − ion in the alkaline solution.
8.2.5 Container No. 5 (Silica Gel for Moisture Determination). Same as Method 5, section 8.7.6.3, Container No. 3.
8.2.6 Container Nos. 6 through 9 (Reagent Blanks). Save portions of the absorbing reagents (0.1 N H 2 SO 4 and 0.1 N NaOH) equivalent to the amount used in the sampling train; dilute to the approximate volume of the corresponding samples using rinse water directly from the wash bottle being used. Add the same ratio of sodium thiosulfate solution used in container No. 4 to the 0.1 N NaOH absorbing reagent blank. Also, save a portion of the rinse water alone and a portion of the acetone equivalent to the amount used to rinse the front half of the sampling train. Place each in a separate, prelabeled sample container.
8.2.7 Prior to shipment, recheck all sample containers to ensure that the caps are well-secured. Seal the lids of all containers around the circumference with Teflon tape. Ship all liquid samples upright and all particulate filters with the particulate catch facing upward.
9.0 Quality Control
9.1 Miscellaneous Quality Control Measures.
| Section | Quality control measure | Effect |
|---|---|---|
| 8.1.4, 10.1 | Sampling equipment leak-check and calibration | Ensure accurate measurement of stack gas flow rate, sample volume. |
9.2 Volume Metering System Checks. Same as Method 5, section 9.2.
10.0 Calibration and Standardization
Note:
Maintain a laboratory log of all calibrations.
10.1 Probe Nozzle, Pitot Tube Assembly, Dry Gas Metering System, Probe Heater, Temperature Sensors, Leak-Check of Metering System, and Barometer. Same as Method 5, sections 10.1, 10.2, 10.3, 10.4, 10.5, 8.4.1, and 10.6, respectively.
10.2 Ion Chromatograph.
10.2.1 To prepare the calibration standards, dilute given amounts (1.0 ml or greater) of the stock standard solutions to convenient volumes, using 0.1 N H 2 SO 4 or 0.1 N NaOH, as appropriate. Prepare at least four calibration standards for each absorbing reagent containing the three stock solutions such that they are within the linear range of the field samples.
10.2.2 Using one of the standards in each series, ensure adequate baseline separation for the peaks of interest.
10.2.3 Inject the appropriate series of calibration standards, starting with the lowest concentration standard first both before and after injection of the quality control check sample, reagent blanks, and field samples. This allows compensation for any instrument drift occurring during sample analysis. The values from duplicate injections of these calibration samples should agree within 5 percent of their mean for the analysis to be valid.
10.2.4 Determine the peak areas, or height, of the standards and plot individual values versus halide ion concentrations in µg/ml.
10.2.5 Draw a smooth curve through the points. Use linear regression to calculate a formula describing the resulting linear curve.
11.0 Analytical Procedures
Note:
The liquid levels in the sample containers and confirm on the analysis sheet whether or not leakage occurred during transport. If a noticeable leakage has occurred, either void the sample or use methods, subject to the approval of the Administrator, to correct the final results.
11.1 Sample Analysis.
11.1.1 The IC conditions will depend upon analytical column type and whether suppressed or non-suppressed IC is used. An example chromatogram from a non-suppressed system using a 150-mm Hamilton PRP-X100 anion column, a 2 ml/min flow rate of a 4 mM 4-hydroxy benzoate solution adjusted to a pH of 8.6 using 1 N NaOH, a 50 µl sample loop, and a conductivity detector set on 1.0 µS full scale is shown in Figure 26-2.
11.1.2 Before sample analysis, establish a stable baseline. Next, inject a sample of water, and determine if any Cl − , Br − , or F − appears in the chromatogram. If any of these ions are present, repeat the load/injection procedure until they are no longer present. Analysis of the acid and alkaline absorbing solution samples requires separate standard calibration curves; prepare each according to section 10.2. Ensure adequate baseline separation of the analyses.
11.1.3 Between injections of the appropriate series of calibration standards, inject in duplicate the reagent blanks, quality control sample, and the field samples. Measure the areas or heights of the Cl − , Br − , and F − peaks. Use the mean response of the duplicate injections to determine the concentrations of the field samples and reagent blanks using the linear calibration curve. The values from duplicate injections should agree within 5 percent of their mean for the analysis to be valid. If the values of duplicate injections are not within 5 percent of the mean, the duplicator injections shall be repeated and all four values used to determine the average response. Dilute any sample and the blank with equal volumes of water if the concentration exceeds that of the highest standard.
11.2 Container Nos. 1 and 2 and Acetone Blank (Optional; Particulate Determination). Same as Method 5, sections 11.2.1 and 11.2.2, respectively.
11.3 Container No. 5. Same as Method 5, section 11.2.3 for silica gel.
12.0 Data Analysis and Calculations
Note:
Retain at least one extra decimal figure beyond those contained in the available data in intermediate calculations, and round off only the final answer appropriately.
12.1 Nomenclature. Same as Method 5, section 12.1. In addition:
B X− = Mass concentration of applicable absorbing solution blank, µg halide ion (Cl − , Br − , F − )/ml, not to exceed 1 µg/ml which is 10 times the published analytical detection limit of 0.1 µg/ml. (It is also approximately 5 percent of the mass concentration anticipated to result from a one hour sample at 10 ppmv HCl.)
C = Concentration of hydrogen halide (HX) or halogen (X 2 ), dry basis, mg/dscm.
K = 10 −3 mg/µg.
K HCl = 1.028 (µg HCl/µg-mole)/(µg Cl − /µg-mole).
K HBr = 1.013 (µg HBr/µg-mole)/(µg Br − /µg-mole).
K HF = 1.053 (µg HF/µg-mole)/(µg F − /µg-mole).
m HX = Mass of HCl, HBr, or HF in sample, ug.
m X2 = Mass of Cl 2 or Br 2 in sample, ug.
S X− = Analysis of sample, ug halide ion (Cl − , Br − , F − )/ml.
V s = Volume of filtered and diluted sample, ml.
12.2 Calculate the exact Cl − , Br − , and F − concentration in the halide salt stock standard solutions using the following equations.
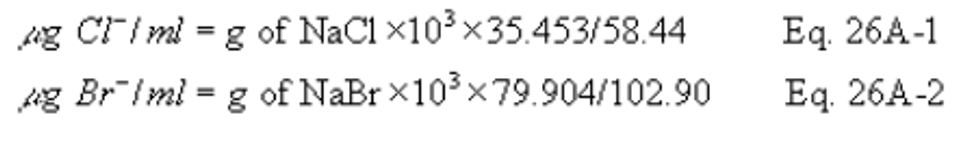
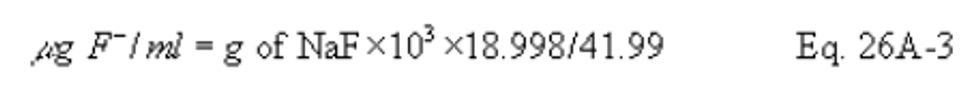
12.3 Average Dry Gas Meter Temperature and Average Orifice Pressure Drop. See data sheet (Figure 5-3 of Method 5).
12.4 Dry Gas Volume. Calculate V m(std) and adjust for leakage, if necessary, using the equation in section 12.3 of Method 5.
12.5 Volume of Water Vapor and Moisture Content. Calculate the volume of water vapor V w(std) and moisture content B ws from the data obtained in this method (Figure 5-3 of Method 5); use Equations 5-2 and 5-3 of Method 5.
12.6 Isokinetic Variation and Acceptable Results. Use Method 5, section 12.11.
12.7 Acetone Blank Concentration, Acetone Wash Blank Residue Weight, Particulate Weight, and Particulate Concentration. For particulate determination.
12.8 Total µg HCl, HBr, or HF Per Sample.
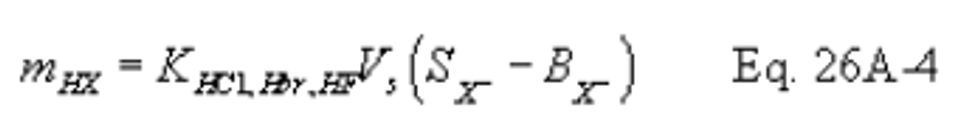
12.9 Total µg Cl 2 or Br 2 Per Sample.
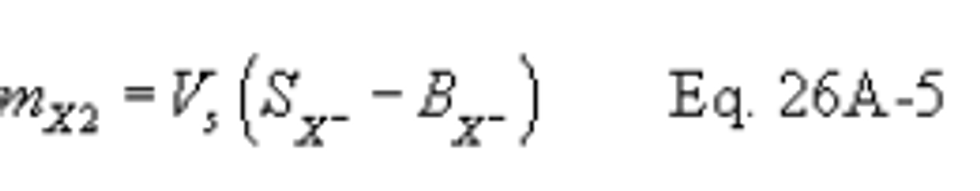
12.10 Concentration of Hydrogen Halide or Halogen in Flue Gas.
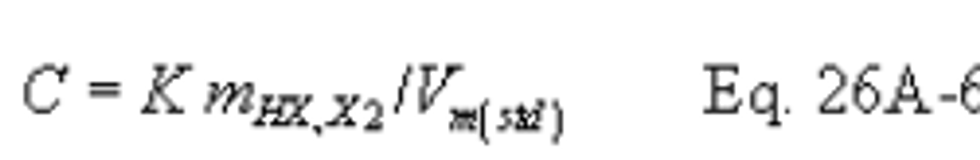
12.11 Stack Gas Velocity and Volumetric Flow Rate. Calculate the average stack gas velocity and volumetric flow rate, if needed, using data obtained in this method and the equations in sections 12.3 and 12.4 of Method 2.
13.0 Method Performance
13.1 Precision and Bias. The method has a possible measurable negative bias below 20 ppm HCl perhaps due to reaction with small amounts of moisture in the probe and filter. Similar bias for the other hydrogen halides is possible.
13.2 Sample Stability. The collected Cl-samples can be stored for up to 4 weeks for analysis for HCl and Cl2.
13.3 Detection Limit. A typical analytical detection limit for HCl is 0.2 µg/ml. Detection limits for the other analyses should be similar. Assuming 300 ml of liquid recovered for the acidified impingers and a similar amounts recovered from the basic impingers, and 1 dscm of stack gas sampled, the analytical detection limits in the stack gas would be about 0.04 ppm for HCl and Cl2, respectively.
14.0 Pollution Prevention [Reserved]
15.0 Waste Management [Reserved]
16.0 References
1. Steinsberger, S. C. and J. H. Margeson. Laboratory and Field Evaluation of a Methodology for Determination of Hydrogen Chloride Emissions from Municipal and Hazardous Waste Incinerators. U.S. Environmental Protection Agency, Office of Research and Development. Publication No. 600/3-89/064. April 1989. Available from National Technical Information Service, Springfield, VA 22161 as PB89220586/AS.
2. State of California Air Resources Board. Method 421 - Determination of Hydrochloric Acid Emissions from Stationary Sources. March 18, 1987.
3. Cheney, J.L. and C.R. Fortune. Improvements in the Methodology for Measuring Hydrochloric Acid in Combustion Source Emissions. J. Environ. Sci. Health. A19 (3): 337-350. 1984.
4. Stern, D.A., B.M. Myatt, J.F. Lachowski, and K.T. McGregor. Speciation of Halogen and Hydrogen Halide Compounds in Gaseous Emissions. In: Incineration and Treatment of Hazardous Waste: Proceedings of the 9th Annual Research Symposium, Cincinnati, Ohio, May 2-4, 1983. Publication No. 600/9-84-015. July 1984. Available from National Technical Information Service, Springfield, VA 22161 as PB84-234525.
5. Holm, R.D. and S.A. Barksdale. Analysis of Anions in Combustion Products. In: Ion Chromatographic Analysis of Environmental Pollutants, E. Sawicki, J.D. Mulik, and E. Wittgenstein (eds.). Ann Arbor, Michigan, Ann Arbor Science Publishers. 1978. pp. 99-110.
17.0 Tables, Diagrams, Flowcharts, and Validation Data
Method 27 - Determination of Vapor Tightness of Gasoline Delivery Tank Using Pressure Vacuum Test
1.0 Scope and Application
1.1 Applicability. This method is applicable for the determination of vapor tightness of a gasoline delivery collection equipment.
2.0 Summary of Method
2.1 Pressure and vacuum are applied alternately to the compartments of a gasoline delivery tank and the change in pressure or vacuum is recorded after a specified period of time.
3.0 Definitions
3.1 Allowable pressure change (Δp) means the allowable amount of decrease in pressure during the static pressure test, within the time period t, as specified in the appropriate regulation, in mm H 2 O.
3.2 Allowable vacuum change (Δv) means the allowable amount of decrease in vacuum during the static vacuum test, within the time period t, as specified in the appropriate regulation, in mm H 2 O.
3.3 Compartment means a liquid-tight division of a delivery tank.
3.4 Delivery tank means a container, including associated pipes and fittings, that is attached to or forms a part of any truck, trailer, or railcar used for the transport of gasoline.
3.5 Delivery tank vapor collection equipment means any piping, hoses, and devices on the delivery tank used to collect and route gasoline vapors either from the tank to a bulk terminal vapor control system or from a bulk plant or service station into the tank.
3.6 Gasoline means a petroleum distillate or petroleum distillate/alcohol blend having a Reid vapor pressure of 27.6 kilopascals or greater which is used as a fuel for internal combustion engines.
3.7 Initial pressure (P i ) means the pressure applied to the delivery tank at the beginning of the static pressure test, as specified in the appropriate regulation, in mm H 2 O.
3.8 Initial vacuum (V i ) means the vacuum applied to the delivery tank at the beginning of the static vacuum test, as specified in the appropriate regulation, in mm H 3 .
3.9 Time period of the pressure or vacuum test (t) means the time period of the test, as specified in the appropriate regulation, during which the change in pressure or vacuum is monitored, in minutes.
4.0 Interferences [Reserved]
5.0 Safety
5.1 Gasoline contains several volatile organic compounds ( e.g., benzene and hexane) which presents a potential for fire and/or explosions. It is advisable to take appropriate precautions when testing a gasoline vessel's vapor tightness, such as refraining from smoking and using explosion-proof equipment.
5.2 This method may involve hazardous materials, operations, and equipment. This test method may not address all of the safety problems associated with its use. It is the responsibility of the user of this test method to establish appropriate safety and health practices and determine the applicability of regulatory limitations prior to performing this test method
6.0 Equipment and Supplies
The following equipment and supplies are required for testing:
6.1 Pressure Source. Pump or compressed gas cylinder of air or inert gas sufficient to pressurize the delivery tank to 500 mm (20 in.) H 2 O above atmospheric pressure.
6.2 Regulator. Low pressure regulator for controlling pressurization of the delivery tank.
6.3 Vacuum Source. Vacuum pump capable of evacuating the delivery tank to 250 mm (10 in.) H 2 O below atmospheric pressure.
6.4 Pressure-Vacuum Supply Hose.
6.5 Manometer. Liquid manometer, or equivalent instrument, capable of measuring up to 500 mm (20 in.) H 2 O gauge pressure with ±2.5 mm (0.1 in.) H 2 O precision.
6.6 Pressure-Vacuum Relief Valves. The test apparatus shall be equipped with an inline pressure-vacuum relief valve set to activate at 675 mm (26.6 in.) H 2 O above atmospheric pressure or 250 mm (10 in.) H2O below atmospheric pressure, with a capacity equal to the pressurizing or evacuating pumps.
6.7 Test Cap for Vapor Recovery Hose. This cap shall have a tap for manometer connection and a fitting with shut-off valve for connection to the pressure-vacuum supply hose.
6.8 Caps for Liquid Delivery Hoses.
7.0 Reagents and Standards [Reserved]
8.0 Sample Collection, Preservation, Storage, and Transport
8.1 Pretest Preparations.
8.1.1 Summary. Testing problems may occur due to the presence of volatile vapors and/or temperature fluctuations inside the delivery tank. Under these conditions, it is often difficult to obtain a stable initial pressure at the beginning of a test, and erroneous test results may occur. To help prevent this, it is recommended that prior to testing, volatile vapors be removed from the tank and the temperature inside the tank be allowed to stabilize. Because it is not always possible to completely attain these pretest conditions, a provision to ensure reproducible results is included. The difference in results for two consecutive runs must meet the criteria in sections 8.2.2.5 and 8.2.3.5.
8.1.2 Emptying of Tank. The delivery tank shall be emptied of all liquid.
8.1.3 Purging of Vapor. As much as possible the delivery tank shall be purged of all volatile vapors by any safe, acceptable method. One method is to carry a load of non-volatile liquid fuel, such as diesel or heating oil, immediately prior to the test, thus flushing out all the volatile gasoline vapors. A second method is to remove the volatile vapors by blowing ambient air into each tank compartment for at least 20 minutes. This second method is usually not as effective and often causes stabilization problems, requiring a much longer time for stabilization during the testing.
8.1.4 Temperature Stabilization. As much as possible, the test shall be conducted under isothermal conditions. The temperature of the delivery tank should be allowed to equilibrate in the test environment. During the test, the tank should be protected from extreme environmental and temperature variability, such as direct sunlight.
8.2 Test Procedure.
8.2.1 Preparations.
8.2.1.1 Open and close each dome cover.
8.2.1.2 Connect static electrical ground connections to the tank. Attach the liquid delivery and vapor return hoses, remove the liquid delivery elbows, and plug the liquid delivery fittings.
Note:
The purpose of testing the liquid delivery hoses is to detect tears or holes that would allow liquid leakage during a delivery. Liquid delivery hoses are not considered to be possible sources of vapor leakage, and thus, do not have to be attached for a vapor leakage test. Instead, a liquid delivery hose could be either visually inspected, or filled with water to detect any liquid leakage.
8.2.1.3 Attach the test cap to the end of the vapor recovery hose.
8.2.1.4 Connect the pressure-vacuum supply hose and the pressure-vacuum relief valve to the shut-off valve. Attach a manometer to the pressure tap.
8.2.1.5 Connect compartments of the tank internally to each other if possible. If not possible, each compartment must be tested separately, as if it were an individual delivery tank.
8.2.2 Pressure Test.
8.2.2.1 Connect the pressure source to the pressure-vacuum supply hose.
8.2.2.2 Open the shut-off valve in the vapor recovery hose cap. Apply air pressure slowly, pressurize the tank to P i , the initial pressure specified in the regulation.
8.2.2.3 Close the shut-off and allow the pressure in the tank to stabilize, adjusting the pressure if necessary to maintain pressure of P i . When the pressure stabilizes, record the time and initial pressure.
8.2.2.4 At the end of the time period (t) specified in the regulation, record the time and final pressure.
8.2.2.5 Repeat steps 8.2.2.2 through 8.2.2.4 until the change in pressure for two consecutive runs agrees within 12.5 mm (0.5 in.) H 2 O. Calculate the arithmetic average of the two results.
8.2.2.6 Compare the average measured change in pressure to the allowable pressure change, Δp, specified in the regulation. If the delivery tank does not satisfy the vapor tightness criterion specified in the regulation, repair the sources of leakage, and repeat the pressure test until the criterion is met.
8.2.2.7 Disconnect the pressure source from the pressure-vacuum supply hose, and slowly open the shut-off valve to bring the tank to atmospheric pressure.
8.2.3 Vacuum Test.
8.2.3.1 Connect the vacuum source to the pressure-vacuum supply hose.
8.2.3.2 Open the shut-off valve in the vapor recovery hose cap. Slowly evacuate the tank to V i , the initial vacuum specified in the regulation.
8.2.3.3 Close the shut-off valve and allow the pressure in the tank to stabilize, adjusting the pressure if necessary to maintain a vacuum of V i . When the pressure stabilizes, record the time and initial vacuum.
8.2.3.4 At the end of the time period specified in the regulation (t), record the time and final vacuum.
8.2.3.5 Repeat steps 8.2.3.2 through 8.2.3.4 until the change in vacuum for two consecutive runs agrees within 12.5 mm (0.5 in.) H 2 O. Calculate the arithmetic average of the two results.
8.2.3.6 Compare the average measured change in vacuum to the allowable vacuum change, Δv, as specified in the regulation. If the delivery tank does not satisfy the vapor tightness criterion specified in the regulation, repair the sources of leakage, and repeat the vacuum test until the criterion is met.
8.2.3.7 Disconnect the vacuum source from the pressure-vacuum supply hose, and slowly open the shut-off valve to bring the tank to atmospheric pressure.
8.2.4 Post-Test Clean-up. Disconnect all test equipment and return the delivery tank to its pretest condition.
9.0 Quality Control
| Section(s) | Quality control measure | Effect |
|---|---|---|
| 8.2.2.5, 8.3.3.5 | Repeat test procedures until change in pressure or vacuum for two consecutive runs agrees within ±12.5 mm (0.5 in.) H 2 O | Ensures data precision. |
10.0 Calibration and Standardization [Reserved]
11.0 Analytical Procedures [Reserved]
12.0 Data Analysis and Calculations [Reserved]
13.0 Method Performance
13.1 Precision. The vapor tightness of a gasoline delivery tank under positive or negative pressure, as measured by this method, is precise within 12.5 mm (0.5 in.) H 2 O
13.2 Bias. No bias has been identified.
14.0 Pollution Prevention [Reserved]
15.0 Waste Management [Reserved]
16.0 Alternative Procedures
16.1 The pumping of water into the bottom of a delivery tank is an acceptable alternative to the pressure source described above. Likewise, the draining of water out of the bottom of a delivery tank may be substituted for the vacuum source. Note that some of the specific step-by-step procedures in the method must be altered slightly to accommodate these different pressure and vacuum sources.
16.2 Techniques other than specified above may be used for purging and pressurizing a delivery tank, if prior approval is obtained from the Administrator. Such approval will be based upon demonstrated equivalency with the above method.
17.0 References [Reserved]
18.0 Tables, Diagrams, Flowcharts, and Validation Data [Reserved]
Method 28 - Certification and Auditing of Wood Heaters
Note:
This method does not include all of the specifications ( e.g., equipment and supplies) and procedures ( e.g., sampling and analytical) essential to its performance. Some material is incorporated by reference from other methods in this part. Therefore, to obtain reliable results, persons using this method should have a thorough knowledge of at least the following additional test methods: Method 1, Method 2, Method 3, Method 4, Method 5, Method 5G, Method 5H, Method 6, Method 6C, and Method 16A.
1.0 Scope and Application
1.1 Analyte. Particulate matter (PM). No CAS number assigned.
1.2 Applicability. This method is applicable for the certification and auditing of wood heaters, including pellet burning wood heaters.
1.3 Data Quality Objectives. Adherence to the requirements of this method will enhance the quality of the data obtained from air pollutant sampling methods.
2.0 Summary of Method
2.1 Particulate matter emissions are measured from a wood heater burning a prepared test fuel crib in a test facility maintained at a set of prescribed conditions. Procedures for determining burn rates and particulate emission rates and for reducing data are provided.
3.0 Definitions
3.1 2 × 4 or 4 × 4 means two inches by four inches or four inches by four inches (50 mm by 100 mm or 100 mm by 100 mm), as nominal dimensions for lumber.
3.2 Burn rate means the rate at which test fuel is consumed in a wood heater. Measured in kilograms or lbs of wood (dry basis) per hour (kg/hr or lb/hr).
3.3 Certification or audit test means a series of at least four test runs conducted for certification or audit purposes that meets the burn rate specifications in section 8.4.
3.4 Firebox means the chamber in the wood heater in which the test fuel charge is placed and combusted.
3.5 Height means the vertical distance extending above the loading door, if fuel could reasonably occupy that space, but not more than 2 inches above the top (peak height) of the loading door, to the floor of the firebox ( i.e., below a permanent grate) if the grate allows a 1-inch diameter piece of wood to pass through the grate, or, if not, to the top of the grate. Firebox height is not necessarily uniform but must account for variations caused by internal baffles, air channels, or other permanent obstructions.
3.6 Length means the longest horizontal fire chamber dimension that is parallel to a wall of the chamber.
3.7 Pellet burning wood heater means a wood heater which meets the following criteria: (1) The manufacturer makes no reference to burning cord wood in advertising or other literature, (2) the unit is safety listed for pellet fuel only, (3) the unit operating and instruction manual must state that the use of cordwood is prohibited by law, and (4) the unit must be manufactured and sold including the hopper and auger combination as integral parts.
3.8 Secondary air supply means an air supply that introduces air to the wood heater such that the burn rate is not altered by more than 25 percent when the secondary air supply is adjusted during the test run. The wood heater manufacturer can document this through design drawings that show the secondary air is introduced only into a mixing chamber or secondary chamber outside the firebox.
3.9 Test facility means the area in which the wood heater is installed, operated, and sampled for emissions.
3.10 Test fuel charge means the collection of test fuel pieces placed in the wood heater at the start of the emission test run.
3.11 Test fuel crib means the arrangement of the test fuel charge with the proper spacing requirements between adjacent fuel pieces.
3.12 Test fuel loading density means the weight of the as-fired test fuel charge per unit volume of usable firebox.
3.13 Test fuel piece means the 2 × 4 or 4 × 4 wood piece cut to the length required for the test fuel charge and used to construct the test fuel crib.
3.14 Test run means an individual emission test which encompasses the time required to consume the mass of the test fuel charge.
3.15 Usable firebox volume means the volume of the firebox determined using its height, length, and width as defined in this section.
3.16 Width means the shortest horizontal fire chamber dimension that is parallel to a wall of the chamber.
3.17 Wood heater means an enclosed, woodburning appliance capable of and intended for space heating or domestic water heating, as defined in the applicable regulation.
4.0 Interferences [Reserved]
5.0 Safety
5.1 Disclaimer. This method may involve hazardous materials, operations, and equipment. This test method may not address all of the safety problems associated with its use. It is the responsibility of the user of this test method to establish appropriate safety and health practices and to determine the applicability of regulatory limitations prior to performing this test method.
6.0 Equipment and Supplies
Same as section 6.0 of either Method 5G or Method 5H, with the addition of the following:
6.1 Insulated Solid Pack Chimney. For installation of wood heaters. Solid pack insulated chimneys shall have a minimum of 2.5 cm (1 in.) solid pack insulating material surrounding the entire flue and possess a label demonstrating conformance to U.L. 103 (incorporated by reference - see §60.17 ).
6.2 Platform Scale and Monitor. For monitoring of fuel load weight change. The scale shall be capable of measuring weight to within 0.05 kg (0.1 lb) or 1 percent of the initial test fuel charge weight, whichever is greater.
6.3 Wood Heater Temperature Monitors. Seven, each capable of measuring temperature to within 1.5 percent of expected absolute temperatures.
6.4 Test Facility Temperature Monitor. A thermocouple located centrally in a vertically oriented 150 mm (6 in.) long, 50 mm (2 in.) diameter pipe shield that is open at both ends, capable of measuring temperature to within 1.5 percent of expected temperatures.
6.5 Balance (optional). Balance capable of weighing the test fuel charge to within 0.05 kg (0.1 lb).
6.6 Moisture Meter. Calibrated electrical resistance meter for measuring test fuel moisture to within 1 percent moisture content.
6.7 Anemometer. Device capable of detecting air velocities less than 0.10 m/sec (20 ft/min), for measuring air velocities near the test appliance.
6.8 Barometer. Mercury, aneroid or other barometer capable of measuring atmospheric pressure to within 2.5 mm Hg (0.1 in. Hg).
6.9 Draft Gauge. Electromanometer or other device for the determination of flue draft or static pressure readable to within 0.50 Pa (0.002 in. H 2 O).
6.10 Humidity Gauge. Psychrometer or hygrometer for measuring room humidity.
6.11 Wood Heater Flue.
6.11.1 Steel flue pipe extending to 2.6 ±0.15 m (8.5 ±0.5 ft) above the top of the platform scale, and above this level, insulated solid pack type chimney extending to 4.6 ±0.3 m (15 ±1 ft) above the platform scale, and of the size specified by the wood heater manufacturer. This applies to both freestanding and insert type wood heaters.
6.11.2 Other chimney types ( e.g., solid pack insulated pipe) may be used in place of the steel flue pipe if the wood heater manufacturer's written appliance specifications require such chimney for home installation ( e.g., zero clearance wood heater inserts). Such alternative chimney or flue pipe must remain and be sealed with the wood heater following the certification test.
6.12 Test Facility. The test facility shall meet the following requirements during testing:
6.12.1 The test facility temperature shall be maintained between 18 and 32°C (65 and 90°F) during each test run.
6.12.2 Air velocities within 0.6 m (2 ft) of the test appliance and exhaust system shall be less than 0.25 m/sec (50 ft/min) without fire in the unit.
6.12.3 The flue shall discharge into the same space or into a space freely communicating with the test facility. Any hood or similar device used to vent combustion products shall not induce a draft greater than 1.25 Pa (0.005 in. H 2 O) on the wood heater measured when the wood heater is not operating.
6.12.4 For test facilities with artificially induced barometric pressures ( e.g., pressurized chambers), the barometric pressure in the test facility shall not exceed 775 mm Hg (30.5 in. Hg) during any test run.
7.0 Reagents and Standards
Same as section 6.0 of either Method 5G or Method 5H, with the addition of the following:
7.1 Test Fuel. The test fuel shall conform to the following requirements:
7.1.1 Fuel Species. Untreated, air-dried, Douglas fir lumber. Kiln-dried lumber is not permitted. The lumber shall be certified C grade (standard) or better Douglas fir by a lumber grader at the mill of origin as specified in the West Coast Lumber Inspection Bureau Standard No. 16 (incorporated by reference - see §60.17 ).
7.1.2 Fuel Moisture. The test fuel shall have a moisture content range between 16 to 20 percent on a wet basis (19 to 25 percent dry basis). Addition of moisture to previously dried wood is not allowed. It is recommended that the test fuel be stored in a temperature and humidity-controlled room.
7.1.3 Fuel Temperature. The test fuel shall be at the test facility temperature of 18 to 32°C (65 to 90°F).
7.1.4 Fuel Dimensions. The dimensions of each test fuel piece shall conform to the nominal measurements of 2 × 4 and 4 × 4 lumber. Each piece of test fuel (not including spacers) shall be of equal length, except as necessary to meet requirements in section 8.8, and shall closely approximate 5/6 the dimensions of the length of the usable firebox. The fuel piece dimensions shall be determined in relation to the appliance's firebox volume according to guidelines listed below:
7.1.4.1 If the usable firebox volume is less than or equal to 0.043 m 3 (1.5 ft 3 ), use 2 × 4 lumber.
7.1.4.2 If the usable firebox volume is greater than 0.043 m 3 (1.5 ft 3 ) and less than or equal to 0.085 m 3 (3.0 ft 3 ), use 2 × 4 and 4 × 4 lumber. About half the weight of the test fuel charge shall be 2 × 4 lumber, and the remainder shall be 4 × 4 lumber.
7.1.4.3 If the usable firebox volume is greater than 0.085 m 3 (3.0 ft 3 ), use 4 × 4 lumber.
7.2 Test Fuel Spacers. Air-dried, Douglas fir lumber meeting the requirements outlined in sections 7.1.1 through 7.1.3. The spacers shall be 130 × 40 × 20 mm (5 × 1.5 × 0.75 in.).
8.0 Sample Collection, Preservation, Storage, and Transport
8.1 Test Run Requirements.
8.1.1 Burn Rate Categories. One emission test run is required in each of the following burn rate categories:
| Category 1 | Category 2 | Category 3 | Category 4 |
|---|---|---|---|
| <0.80 | 0.80 to 1.25 | 1.25 to 1.90 | Maximum. |
| (<1.76) | (1.76 to 2.76) | (2.76 to 4.19) | burn rate. |
8.1.1.1 Maximum Burn Rate. For Category 4, the wood heater shall be operated with the primary air supply inlet controls fully open (or, if thermostatically controlled, the thermostat shall be set at maximum heat output) during the entire test run, or the maximum burn rate setting specified by the manufacturer's written instructions.
8.1.1.2 Other Burn Rate Categories. For burn rates in Categories 1 through 3, the wood heater shall be operated with the primary air supply inlet control, or other mechanical control device, set at a predetermined position necessary to obtain the average burn rate required for the category.
8.1.1.3 Alternative Burn Rates for Burn Rate Categories 1 and 2.
8.1.1.3.1 If a wood heater cannot be operated at a burn rate below 0.80 kg/hr (1.76 lb/hr), two test runs shall be conducted with burn rates within Category 2. If a wood heater cannot be operated at a burn rate below 1.25 kg/hr (2.76 lb/hr), the flue shall be dampered or the air supply otherwise controlled in order to achieve two test runs within Category 2.
8.1.1.3.2 Evidence that a wood heater cannot be operated at a burn rate less than 0.80 kg/hr shall include documentation of two or more attempts to operate the wood heater in burn rate Category 1 and fuel combustion has stopped, or results of two or more test runs demonstrating that the burn rates were greater than 0.80 kg/hr when the air supply controls were adjusted to the lowest possible position or settings. Stopped fuel combustion is evidenced when an elapsed time of 30 minutes or more has occurred without a measurable (<0.05 kg (0.1 lb) or 1.0 percent, whichever is greater) weight change in the test fuel charge. See also section 8.8.3. Report the evidence and the reasoning used to determine that a test in burn rate Category 1 cannot be achieved; for example, two unsuccessful attempts to operate at a burn rate of 0.4 kg/hr are not sufficient evidence that burn rate Category 1 cannot be achieved.
Note:
After July 1, 1990, if a wood heater cannot be operated at a burn rate less than 0.80 kg/hr, at least one test run with an average burn rate of 1.00 kg/hr or less shall be conducted. Additionally, if flue dampering must be used to achieve burn rates below 1.25 kg/hr (or 1.0 kg/hr), results from a test run conducted at burn rates below 0.90 kg/hr need not be reported or included in the test run average provided that such results are replaced with results from a test run meeting the criteria above.
8.2 Catalytic Combustor and Wood Heater Aging. The catalyst-equipped wood heater or a wood heater of any type shall be aged before the certification test begins. The aging procedure shall be conducted and documented by a testing laboratory accredited according to procedures in §60.535 of 40 CFR part 60.
8.2.1 Catalyst-equipped Wood Heater. Operate the catalyst-equipped wood heater using fuel meeting the specifications outlined in sections 7.1.1 through 7.1.3, or cordwood with a moisture content between 15 and 25 percent on a wet basis. Operate the wood heater at a medium burn rate (Category 2 or 3) with a new catalytic combustor in place and in operation for at least 50 hours. Record and report hourly catalyst exit temperature data (Section 8.6.2) and the hours of operation.
8.2.2 Non-Catalyst Wood Heater. Operate the wood heater using the fuel described in section 8.4.1 at a medium burn rate for at least 10 hours. Record and report the hours of operation.
8.3 Pretest Recordkeeping. Record the test fuel charge dimensions and weights, and wood heater and catalyst descriptions as shown in the example in Figure 28-1.
8.4 Wood Heater Installation. Assemble the wood heater appliance and parts in conformance with the manufacturer's written installation instructions. Place the wood heater centrally on the platform scale and connect the wood heater to the flue described in section 6.11. Clean the flue with an appropriately sized, wire chimney brush before each certification test.
8.5 Wood Heater Temperature Monitors.
8.5.1 For catalyst-equipped wood heaters, locate a temperature monitor (optional) about 25 mm (1 in.) upstream of the catalyst at the centroid of the catalyst face area, and locate a temperature monitor (mandatory) that will indicate the catalyst exhaust temperature. This temperature monitor is centrally located within 25 mm (1 in.) downstream at the centroid of catalyst face area. Record these locations.
8.5.2 Locate wood heater surface temperature monitors at five locations on the wood heater firebox exterior surface. Position the temperature monitors centrally on the top surface, on two sidewall surfaces, and on the bottom and back surfaces. Position the monitor sensing tip on the firebox exterior surface inside of any heat shield, air circulation walls, or other wall or shield separated from the firebox exterior surface. Surface temperature locations for unusual design shapes ( e.g., spherical, etc.) shall be positioned so that there are four surface temperature monitors in both the vertical and horizontal planes passing at right angles through the centroid of the firebox, not including the fuel loading door (total of five temperature monitors).
8.6 Test Facility Conditions.
8.6.1 Locate the test facility temperature monitor on the horizontal plane that includes the primary air intake opening for the wood heater. Locate the temperature monitor 1 to 2 m (3 to 6 ft) from the front of the wood heater in the 90° sector in front of the wood heater.
8.6.2 Use an anemometer to measure the air velocity. Measure and record the room air velocity before the pretest ignition period (Section 8.7) and once immediately following the test run completion.
8.6.3 Measure and record the test facility's ambient relative humidity, barometric pressure, and temperature before and after each test run.
8.6.4 Measure and record the flue draft or static pressure in the flue at a location no greater than 0.3 m (1 ft) above the flue connector at the wood heater exhaust during the test run at the recording intervals (Section 8.8.2).
8.7 Wood Heater Firebox Volume.
8.7.1 Determine the firebox volume using the definitions for height, width, and length in section 3. Volume adjustments due to presence of firebrick and other permanent fixtures may be necessary. Adjust width and length dimensions to extend to the metal wall of the wood heater above the firebrick or permanent obstruction if the firebrick or obstruction extending the length of the side(s) or back wall extends less than one-third of the usable firebox height. Use the width or length dimensions inside the firebrick if the firebrick extends more than one-third of the usable firebox height. If a log retainer or grate is a permanent fixture and the manufacturer recommends that no fuel be placed outside the retainer, the area outside of the retainer is excluded from the firebox volume calculations.
8.7.2 In general, exclude the area above the ash lip if that area is less than 10 percent of the usable firebox volume. Otherwise, take into account consumer loading practices. For instance, if fuel is to be loaded front-to-back, an ash lip may be considered usable firebox volume.
8.7.3 Include areas adjacent to and above a baffle (up to two inches above the fuel loading opening) if four inches or more horizontal space exist between the edge of the baffle and a vertical obstruction ( e.g., sidewalls or air channels).
8.8 Test Fuel Charge.
8.8.1 Prepare the test fuel pieces in accordance with the specifications outlined in sections 7.1 and 7.2. Determine the test fuel moisture content with a calibrated electrical resistance meter or other equivalent performance meter. If necessary, convert fuel moisture content values from dry basis (%M d ) to wet basis (%M w ) in section 12.2 using Equation 28-1. Determine fuel moisture for each fuel piece (not including spacers) by averaging at least three moisture meter readings, one from each of three sides, measured parallel to the wood grain. Average all the readings for all the fuel pieces in the test fuel charge. If an electrical resistance type meter is used, penetration of insulated electrodes shall be one-fourth the thickness of the test fuel piece or 19 mm (0.75 in.), whichever is greater. Measure the moisture content within a 4-hour period prior to the test run. Determine the fuel temperature by measuring the temperature of the room where the wood has been stored for at least 24 hours prior to the moisture determination.
8.8.2 Attach the spacers to the test fuel pieces with uncoated, ungalvanized nails or staples as illustrated in Figure 28-2. Attachment of spacers to the top of the test fuel piece(s) on top of the test fuel charge is optional.
8.8.3 To avoid stacking difficulties, or when a whole number of test fuel pieces does not result, all piece lengths shall be adjusted uniformly to remain within the specified loading density. The shape of the test fuel crib shall be geometrically similar to the shape of the firebox volume without resorting to special angular or round cuts on the individual fuel pieces.
8.8.4 The test fuel loading density shall be 112 ±11.2 kg/m 3 (7 ±0.7 lb/ft 3 ) of usable firebox volume on a wet basis.
8.9 Sampling Equipment. Prepare the sampling equipment as defined by the selected method ( i.e., either Method 5G or Method 5H). Collect one particulate emission sample for each test run.
8.10 Secondary Air Adjustment Validation.
8.10.1 If design drawings do not show the introduction of secondary air into a chamber outside the firebox (see “secondary air supply” under section 3.0, Definitions), conduct a separate test of the wood heater's secondary air supply. Operate the wood heater at a burn rate in Category 1 (Section 8.1.1) with the secondary air supply operated following the manufacturer's written instructions. Start the secondary air validation test run as described in section 8.8.1, except no emission sampling is necessary and burn rate data shall be recorded at 5-minute intervals.
8.10.2 After the start of the test run, operate the wood heater with the secondary air supply set as per the manufacturer's instructions, but with no adjustments to this setting. After 25 percent of the test fuel has been consumed, adjust the secondary air supply controls to another setting, as per the manufacturer's instructions. Record the burn rate data (5-minute intervals) for 20 minutes following the air supply adjustment.
8.10.3 Adjust the air supply control(s) to the original position(s), operate at this condition for at least 20 minutes, and repeat the air supply adjustment procedure above. Repeat the procedure three times at equal intervals over the entire burn period as defined in section 8.8. If the secondary air adjustment results in a burn rate change of more than an average of 25 percent between the 20-minute periods before and after the secondary adjustments, the secondary air supply shall be considered a primary air supply, and no adjustment to this air supply is allowed during the test run.
8.10.4 The example sequence below describes a typical secondary air adjustment validation check. The first cycle begins after at least 25 percent of the test fuel charge has been consumed.
Cycle 1
Part 1, sec air adjusted to final position - 20 min
Part 2, sec air adjusted to final position - 20 min
Part 3, sec air adjusted to final position - 20 min
Cycle 2
Part 1, sec air adjusted to final position - 20 min
Part 2, sec air adjusted to final position - 20 min
Part 3, sec air adjusted to final position - 20 min
Cycle 3
Part 1, sec air adjusted to final position - 20 min
Part 2, sec air adjusted to final position - 20 min
Part 3, sec air adjusted to final position - 20 min
Note that the cycles may overlap; that is, Part 3 of Cycle 1 may coincide in part or in total with Part 1 of Cycle 2. The calculation of the secondary air percent effect for this example is as follows:
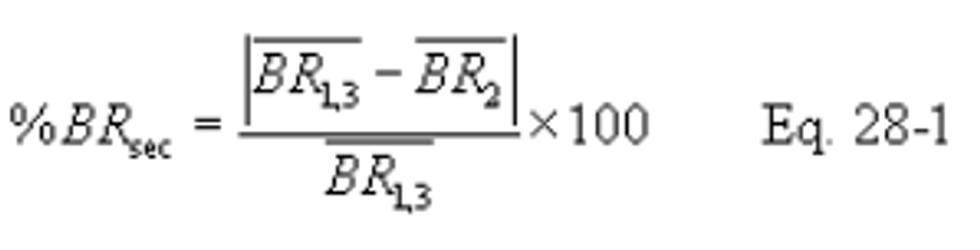
8.11 Pretest Ignition. Build a fire in the wood heater in accordance with the manufacturer's written instructions.
8.11.1 Pretest Fuel Charge. Crumpled newspaper loaded with kindling may be used to help ignite the pretest fuel. The pretest fuel, used to sustain the fire, shall meet the same fuel requirements prescribed in section 7.1. The pretest fuel charge shall consist of whole 2 × 4's that are no less than 1/3 the length of the test fuel pieces. Pieces of 4 × 4 lumber in approximately the same weight ratio as for the test fuel charge may be added to the pretest fuel charge.
8.11.2 Wood Heater Operation and Adjustments. Set the air inlet supply controls at any position that will maintain combustion of the pretest fuel load. At least one hour before the start of the test run, set the air supply controls at the approximate positions necessary to achieve the burn rate desired for the test run. Adjustment of the air supply controls, fuel addition or subtractions, and coalbed raking shall be kept to a minimum but are allowed up to 15 minutes prior to the start of the test run. For the purposes of this method, coalbed raking is the use of a metal tool (poker) to stir coals, break burning fuel into smaller pieces, dislodge fuel pieces from positions of poor combustion, and check for the condition of uniform charcoalization. Record all adjustments made to the air supply controls, adjustments to and additions or subtractions of fuel, and any other changes to wood heater operations that occur during pretest ignition period. Record fuel weight data and wood heater temperature measurements at 10-minute intervals during the hour of the pretest ignition period preceding the start of the test run. During the 15-minute period prior to the start of the test run, the wood heater loading door shall not be open more than a total of 1 minute. Coalbed raking is the only adjustment allowed during this period.
Note:
One purpose of the pretest ignition period is to achieve uniform charcoalization of the test fuel bed prior to loading the test fuel charge. Uniform charcoalization is a general condition of the test fuel bed evidenced by an absence of large pieces of burning wood in the coal bed and the remaining fuel pieces being brittle enough to be broken into smaller charcoal pieces with a metal poker. Manipulations to the fuel bed prior to the start of the test run should be done to achieve uniform charcoalization while maintaining the desired burn rate. In addition, some wood heaters ( e.g., high mass units) may require extended pretest burn time and fuel additions to reach an initial average surface temperature sufficient to meet the thermal equilibrium criteria in section 8.3.
8.11.3 The weight of pretest fuel remaining at the start of the test run is determined as the difference between the weight of the wood heater with the remaining pretest fuel and the tare weight of the cleaned, dry wood heater with or without dry ash or sand added consistent with the manufacturer's instructions and the owner's manual. The tare weight of the wood heater must be determined with the wood heater (and ash, if added) in a dry condition.
8.12 Test Run. Complete a test run in each burn rate category, as follows:
8.12.1 Test Run Start.
8.12.1.1 When the kindling and pretest fuel have been consumed to leave a fuel weight between 20 and 25 percent of the weight of the test fuel charge, record the weight of the fuel remaining and start the test run. Record and report any other criteria, in addition to those specified in this section, used to determine the moment of the test run start ( e.g., firebox or catalyst temperature), whether such criteria are specified by the wood heater manufacturer or the testing laboratory. Record all wood heater individual surface temperatures, catalyst temperatures, any initial sampling method measurement values, and begin the particulate emission sampling. Within 1 minute following the start of the test run, open the wood heater door, load the test fuel charge, and record the test fuel charge weight. Recording of average, rather than individual, surface temperatures is acceptable for tests conducted in accordance with §60.533(o)(3)(i) of 40 CFR part 60.
8.12.1.2 Position the fuel charge so that the spacers are parallel to the floor of the firebox, with the spacer edges abutting each other. If loading difficulties result, some fuel pieces may be placed on edge. If the usable firebox volume is between 0.043 and 0.085 m 3 (1.5 and 3.0 ft 3 ), alternate the piece sizes in vertical stacking layers to the extent possible. For example, place 2 × 4's on the bottom layer in direct contact with the coal bed and 4 × 4's on the next layer, etc. (See Figure 28-3). Position the fuel pieces parallel to each other and parallel to the longest wall of the firebox to the extent possible within the specifications in section 8.8.
8.12.1.3 Load the test fuel in appliances having unusual or unconventional firebox design maintaining air space intervals between the test fuel pieces and in conformance with the manufacturer's written instructions. For any appliance that will not accommodate the loading arrangement specified in the paragraph above, the test facility personnel shall contact the Administrator for an alternative loading arrangement.
8.12.1.4 The wood heater door may remain open and the air supply controls adjusted up to five minutes after the start of the test run in order to make adjustments to the test fuel charge and to ensure ignition of the test fuel charge has occurred. Within the five minutes after the start of the test run, close the wood heater door and adjust the air supply controls to the position determined to produce the desired burn rate. No other adjustments to the air supply controls or the test fuel charge are allowed (except as specified in sections 8.12.3 and 8.12.4) after the first five minutes of the test run. Record the length of time the wood heater door remains open, the adjustments to the air supply controls, and any other operational adjustments.
8.12.2 Data Recording. Record on a data sheet similar to that shown in Figure 28-4, at intervals no greater than 10 minutes, fuel weight data, wood heater individual surface and catalyst temperature measurements, other wood heater operational data ( e.g., draft), test facility temperature and sampling method data.
8.12.3 Test Fuel Charge Adjustment. The test fuel charge may be adjusted ( i.e., repositioned) once during a test run if more than 60 percent of the initial test fuel charge weight has been consumed and more than 10 minutes have elapsed without a measurable (<0.05 kg (0.1 lb) or 1.0 percent, whichever is greater) weight change. The time used to make this adjustment shall be less than 15 seconds.
8.12.4 Air Supply Adjustment. Secondary air supply controls may be adjusted once during the test run following the manufacturer's written instructions (see section 8.10). No other air supply adjustments are allowed during the test run. Recording of wood heater flue draft during the test run is optional for tests conducted in accordance with §60.533(o)(3)(i) of 40 CFR part 60.
8.12.5 Auxiliary Wood Heater Equipment Operation. Heat exchange blowers sold with the wood heater shall be operated during the test run following the manufacturer's written instructions. If no manufacturer's written instructions are available, operate the heat exchange blower in the “high” position. (Automatically operated blowers shall be operated as designed.) Shaker grates, by-pass controls, or other auxiliary equipment may be adjusted only one time during the test run following the manufacturer's written instructions.
Record all adjustments on a wood heater operational written record.
Note:
If the wood heater is sold with a heat exchange blower as an option, test the wood heater with the heat exchange blower operating as described in sections 8.1 through 8.12 and report the results. As an alternative to repeating all test runs without the heat exchange blower operating, one additional test run may be without the blower operating as described in section 8.12.5 at a burn rate in Category 2 (Section 8.1.1). If the emission rate resulting from this test run without the blower operating is equal to or less than the emission rate plus 1.0 g/hr (0.0022 lb/hr) for the test run in burn rate Category 2 with the blower operating, the wood heater may be considered to have the same average emission rate with or without the blower operating. Additional test runs without the blower operating are unnecessary.
8.13 Test Run Completion. Continue emission sampling and wood heater operation for 2 hours. The test run is completed when the remaining weight of the test fuel charge is 0.00 kg (0.0 lb). End the test run when the scale has indicated a test fuel charge weight of 0.00 kg (0.0 lb) or less for 30 seconds. At the end of the test run, stop the particulate sampling, and record the final fuel weight, the run time, and all final measurement values.
8.14 Wood Heater Thermal Equilibrium. The average of the wood heater surface temperatures at the end of the test run shall agree with the average surface temperature at the start of the test run to within 70°C (126°F).
8.15 Consecutive Test Runs. Test runs on a wood heater may be conducted consecutively provided that a minimum one-hour interval occurs between test runs.
8.16 Additional Test Runs. The testing laboratory may conduct more than one test run in each of the burn rate categories specified in section 8.1.1. If more than one test run is conducted at a specified burn rate, the results from at least two-thirds of the test runs in that burn rate category shall be used in calculating the weighted average emission rate (see section 12.2). The measurement data and results of all test runs shall be reported regardless of which values are used in calculating the weighted average emission rate (see note in section 8.1).
9.0 Quality Control
Same as section 9.0 of either Method 5G or Method 5H.
10.0 Calibration and Standardizations
Same as section 10.0 of either Method 5G or Method 5H, with the addition of the following:
10.1 Platform Scale. Perform a multi-point calibration (at least five points spanning the operational range) of the platform scale before its initial use. The scale manufacturer's calibration results are sufficient for this purpose. Before each certification test, audit the scale with the wood heater in place by weighing at least one calibration weight (Class F) that corresponds to between 20 percent and 80 percent of the expected test fuel charge weight. If the scale cannot reproduce the value of the calibration weight within 0.05 kg (0.1 lb) or 1 percent of the expected test fuel charge weight, whichever is greater, recalibrate the scale before use with at least five calibration weights spanning the operational range of the scale.
10.2 Balance (optional). Calibrate as described in section 10.1.
10.3 Temperature Monitor. Calibrate as in Method 2, section 4.3, before the first certification test and semiannually thereafter.
10.4 Moisture Meter. Calibrate as per the manufacturer's instructions before each certification test.
10.5 Anemometer. Calibrate the anemometer as specified by the manufacturer's instructions before the first certification test and semiannually thereafter.
10.6 Barometer. Calibrate against a mercury barometer before the first certification test and semiannually thereafter.
10.7 Draft Gauge. Calibrate as per the manufacturer's instructions; a liquid manometer does not require calibration.
10.8 Humidity Gauge. Calibrate as per the manufacturer's instructions before the first certification test and semiannually thereafter.
11.0 Analytical Procedures
Same as section 11.0 of either Method 5G or Method 5H.
12.0 Data Analysis and Calculations
Same as section 12.0 of either Method 5G or Method 5H, with the addition of the following:
12.1 Nomenclature.
BR = Dry wood burn rate, kg/hr (lb/hr)
E i = Emission rate for test run, i, from Method 5G or 5H, g/hr (lb/hr)
E w = Weighted average emission rate, g/hr (lb/hr)
k i = Test run weighting factor = P i 1 − P i−1
%M d = Fuel moisture content, dry basis, percent.
%M w = Average moisture in test fuel charge, wet basis, percent.
n = Total number of test runs.
P i = Probability for burn rate during test run, i, obtained from Table 28-1. Use linear interpolation to determine probability values for burn rates between those listed on the table.
W wd = Total mass of wood burned during the test run, kg (lb).
12.2 Wet Basis Fuel Moisture Content.
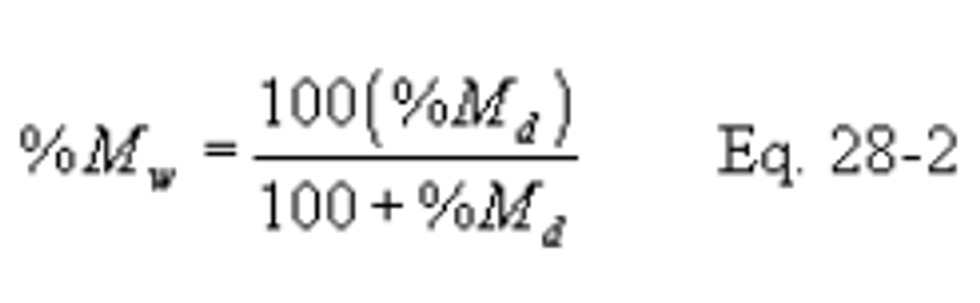
12.3 Weighted Average Emission Rate. Calculate the weighted average emission rate (E w ) using Equation 28-1:
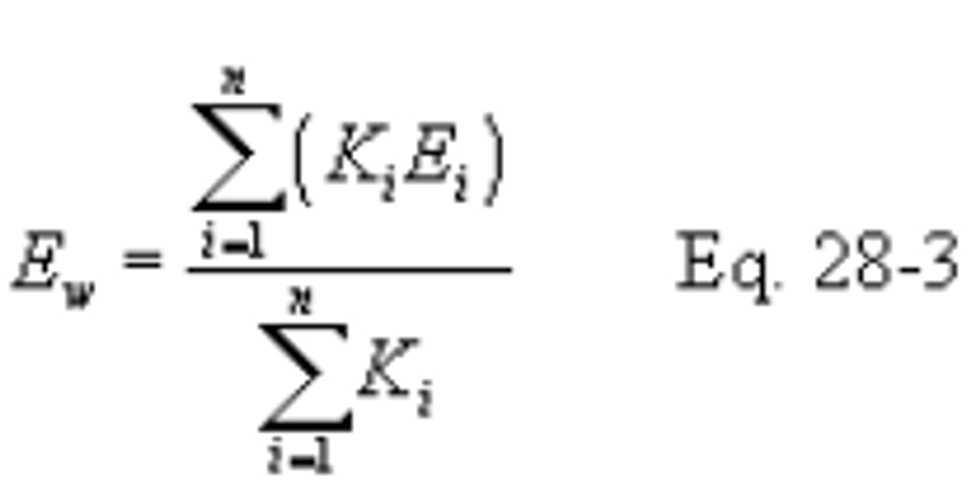
Note:
P o always equals 0, P (n 1) always equals 1, P 1 corresponds to the probability of the lowest recorded burn rate, P 2 corresponds to the probability of the next lowest burn rate, etc. An example calculation is in section 12.3.1.
12.3.1 Example Calculation of Weighted Average Emission Rate.
| Burn rate category | Test No. | Burn rate (Dry-kg/hr) | Emissions (g/hr) |
|---|---|---|---|
| 1 | 1 | 0.65 | 5.0 |
| 2 1 | 2 | 0.85 | 6.7 |
| 2 | 3 | 0.90 | 4.7 |
| 2 | 4 | 1.00 | 5.3 |
| 3 | 5 | 1.45 | 3.8 |
| 4 | 6 | 2.00 | 5.1 |
| 1 As permitted in section 6.6, this test run may be omitted from the calculation of the weighted average emission rate because three runs were conducted for this burn rate category. | |||
| Test No. | Burn rate | P i | E i | K i |
|---|---|---|---|---|
| 0 | 0.000 | |||
| 1 | 0.65 | 0.121 | 5.0 | 0.300 |
| 2 | 0.90 | 0.300 | 4.7 | 0.259 |
| 3 | 1.00 | 0.380 | 5.3 | 0.422 |
| 4 | 1.45 | 0.722 | 3.8 | 0.532 |
| 5 | 2.00 | 0.912 | 5.1 | 0.278 |
| 6 | 1.000 | |||
| K 1 = P 2 − P 0 = 0.300 − 0 = 0.300 K 2 = P 3 − P 1 = 0.381 − 0.121 = 0.259 K 3 = P 4 − P 2 = 0.722 − 0.300 = 0.422 K 4 = P 5 − P 3 = 0.912 − 0.380 = 0.532 K 5 = P 6 − P 4 = 1.000 − 0.722 = 0.278 | ||||
Weighted Average Emission Rate, E w , Calculation
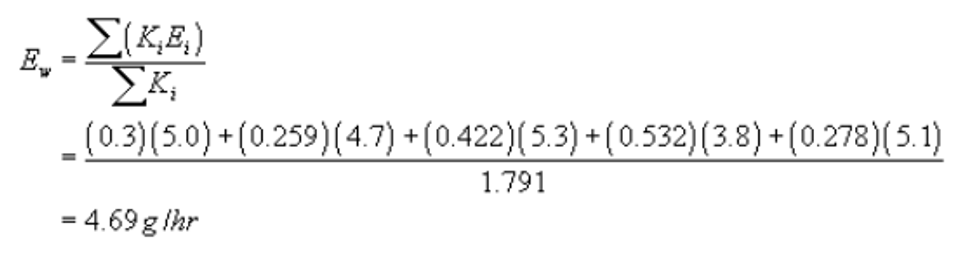
12.4 Average Wood Heater Surface Temperatures. Calculate the average of the wood heater surface temperatures for the start of the test run (Section 8.12.1) and for the test run completion (Section 8.13). If the two average temperatures do not agree within 70°C (125°F), report the test run results, but do not include the test run results in the test average. Replace such test run results with results from another test run in the same burn rate category.
12.5 Burn Rate. Calculate the burn rate (BR) using Equation 28-3:
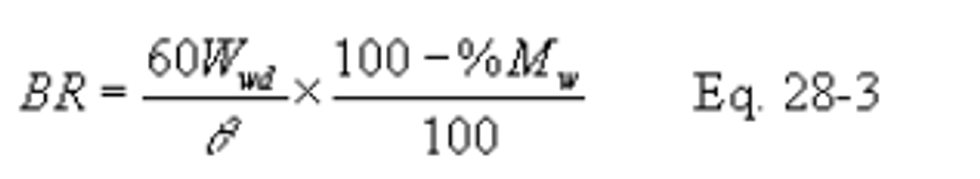
12.6 Reporting Criteria. Submit both raw and reduced test data for wood heater tests.
12.6.1 Suggested Test Report Format.
12.6.1.1 Introduction.
12.6.1.1.1 Purpose of test-certification, audit, efficiency, research and development.
12.6.1.1.2 Wood heater identification-manufacturer, model number, catalytic/noncatalytic, options.
12.6.1.1.3 Laboratory-name, location (altitude), participants.
12.6.1.1.4 Test information-date wood heater received, date of tests, sampling methods used, number of test runs.
12.6.1.2 Summary and Discussion of Results
12.6.1.2.1 Table of results (in order of increasing burn rate)-test run number, burn rate, particulate emission rate, efficiency (if determined), averages (indicate which test runs are used).
12.6.1.2.2 Summary of other data-test facility conditions, surface temperature averages, catalyst temperature averages, pretest fuel weights, test fuel charge weights, run times.
12.6.1.2.3 Discussion-Burn rate categories achieved, test run result selection, specific test run problems and solutions.
12.6.1.3 Process Description.
12.6.1.3.1 Wood heater dimensions-volume, height, width, lengths (or other linear dimensions), weight, volume adjustments.
12.6.1.3.2 Firebox configuration-air supply locations and operation, air supply introduction location, refractory location and dimensions, catalyst location, baffle and by-pass location and operation (include line drawings or photographs).
12.6.1.3.3 Process operation during test-air supply settings and adjustments, fuel bed adjustments, draft.
12.6.1.3.4 Test fuel-test fuel properties (moisture and temperature), test fuel crib description (include line drawing or photograph), test fuel loading density.
12.6.1.4 Sampling Locations.
12.6.1.4.1 Describe sampling location relative to wood heater. Include drawing or photograph.
12.6.1.5 Sampling and Analytical Procedures
12.6.1.5.1 Sampling methods-brief reference to operational and sampling procedures and optional and alternative procedures used.
12.6.1.5.2 Analytical methods-brief description of sample recovery and analysis procedures.
12.6.1.6 Quality Control and Assurance Procedures and Results
12.6.1.6.1 Calibration procedures and results-certification procedures, sampling and analysis procedures.
12.6.1.6.2 Test method quality control procedures-leak-checks, volume meter checks, stratification (velocity) checks, proportionality results.
12.6.1.7 Appendices
12.6.1.7.1 Results and Example Calculations. Complete summary tables and accompanying examples of all calculations.
12.6.1.7.2 Raw Data. Copies of all uncorrected data sheets for sampling measurements, temperature records and sample recovery data. Copies of all pretest burn rate and wood heater temperature data.
12.6.1.7.3 Sampling and Analytical Procedures. Detailed description of procedures followed by laboratory personnel in conducting the certification test, emphasizing particular parts of the procedures differing from the methods ( e.g., approved alternatives).
12.6.1.7.4 Calibration Results. Summary of all calibrations, checks, and audits pertinent to certification test results with dates.
12.6.1.7.5 Participants. Test personnel, manufacturer representatives, and regulatory observers.
12.6.1.7.6 Sampling and Operation Records. Copies of uncorrected records of activities not included on raw data sheets ( e.g., wood heater door open times and durations).
12.6.1.7.7 Additional Information. Wood heater manufacturer's written instructions for operation during the certification test.
12.6.2.1 Wood Heater Identification. Report wood heater identification information. An example data form is shown in Figure 28-4.
12.6.2.2 Test Facility Information. Report test facility temperature, air velocity, and humidity information. An example data form is shown on Figure 28-4.
12.6.2.3 Test Equipment Calibration and Audit Information. Report calibration and audit results for the platform scale, test fuel balance, test fuel moisture meter, and sampling equipment including volume metering systems and gaseous analyzers.
12.6.2.4 Pretest Procedure Description. Report all pretest procedures including pretest fuel weight, burn rates, wood heater temperatures, and air supply settings. An example data form is shown on Figure 28-4.
12.6.2.5 Particulate Emission Data. Report a summary of test results for all test runs and the weighted average emission rate. Submit copies of all data sheets and other records collected during the testing. Submit examples of all calculations.
13.0 Method Performance [Reserved]
14.0 Pollution Prevention [Reserved]
15.0 Waste Management [Reserved]
16.0 Alternative Procedures
16.1 Pellet Burning Heaters. Certification testing requirements and procedures for pellet burning wood heaters are identical to those for other wood heaters, with the following exceptions:
16.1.1 Test Fuel Properties. The test fuel shall be all wood pellets with a moisture content no greater than 20 percent on a wet basis (25 percent on a dry basis). Determine the wood moisture content with either ASTM D 2016-74 or 83, (Method A), ASTM D 4444-92, or ASTM D 4442-84 or 92 (all noted ASTM standards are incorporated by reference - see §60.17 ).
16.1.2 Test Fuel Charge Specifications. The test fuel charge size shall be as per the manufacturer's written instructions for maintaining the desired burn rate.
16.1.3 Wood Heater Firebox Volume. The firebox volume need not be measured or determined for establishing the test fuel charge size. The firebox dimensions and other heater specifications needed to identify the heater for certification purposes shall be reported.
16.1.4 Heater Installation. Arrange the heater with the fuel supply hopper on the platform scale as described in section 8.6.1.
16.1.5 Pretest Ignition. Start a fire in the heater as directed by the manufacturer's written instructions, and adjust the heater controls to achieve the desired burn rate. Operate the heater at the desired burn rate for at least 1 hour before the start of the test run.
16.1.6 Test Run. Complete a test run in each burn rate category as follows:
16.1.6.1 Test Run Start. When the wood heater has operated for at least 1 hour at the desired burn rate, add fuel to the supply hopper as necessary to complete the test run, record the weight of the fuel in the supply hopper (the wood heater weight), and start the test run. Add no additional fuel to the hopper during the test run.
Record all the wood heater surface temperatures, the initial sampling method measurement values, the time at the start of the test, and begin the emission sampling. Make no adjustments to the wood heater air supply or wood supply rate during the test run.
16.1.6.2 Data Recording. Record the fuel (wood heater) weight data, wood heater temperature and operational data, and emission sampling data as described in section 8.12.2.
16.1.6.3 Test Run Completion. Continue emission sampling and wood heater operation for 2 hours. At the end of the test run, stop the particulate sampling, and record the final fuel weight, the run time, and all final measurement values, including all wood heater individual surface temperatures.
16.1.7 Calculations. Determine the burn rate using the difference between the initial and final fuel (wood heater) weights and the procedures described in section 12.4. Complete the other calculations as described in section 12.0.
17.0 References
Same as Method 5G, with the addition of the following:
1. Radian Corporation. OMNI Environmental Services, Inc., Cumulative Probability for a Given Burn Rate Based on Data Generated in the CONEG and BPA Studies. Package of materials submitted to the Fifth Session of the Regulatory Negotiation Committee, July 16-17, 1986.
18.0 Tables, Diagrams, Flowcharts, and Validation Data
| Burn rate (kg/hr-dry) | Cumulative probability (P) | Burn rate (kg/hr-dry) | Cumulative probability (P) | Burn rate (kg/hr-dry) | Cumulative probability (P) |
|---|---|---|---|---|---|
| 0.00 | 0.000 | 1.70 | 0.840 | 3.40 | 0.989 |
| 0.05 | 0.002 | 1.75 | 0.857 | 3.45 | 0.989 |
| 0.10 | 0.007 | 1.80 | 0.875 | 3.50 | 0.990 |
| 0.15 | 0.012 | 1.85 | 0.882 | 3.55 | 0.991 |
| 0.20 | 0.016 | 1.90 | 0.895 | 3.60 | 0.991 |
| 0.25 | 0.021 | 1.95 | 0.906 | 3.65 | 0.992 |
| 0.30 | 0.028 | 2.00 | 0.912 | 3.70 | 0.992 |
| 0.35 | 0.033 | 2.05 | 0.920 | 3.75 | 0.992 |
| 0.40 | 0.041 | 2.10 | 0.925 | 3.80 | 0.993 |
| 0.45 | 0.054 | 2.15 | 0.932 | 3.85 | 0.994 |
| 0.50 | 0.065 | 2.20 | 0.936 | 3.90 | 0.994 |
| 0.55 | 0.086 | 2.25 | 0.940 | 3.95 | 0.994 |
| 0.60 | 0.100 | 2.30 | 0.945 | 4.00 | 0.994 |
| 0.65 | 0.121 | 2.35 | 0.951 | 4.05 | 0.995 |
| 0.70 | 0.150 | 2.40 | 0.956 | 4.10 | 0.995 |
| 0.75 | 0.185 | 2.45 | 0.959 | 4.15 | 0.995 |
| 0.80 | 0.220 | 2.50 | 0.964 | 4.20 | 0.995 |
| 0.85 | 0.254 | 2.55 | 0.968 | 4.25 | 0.995 |
| 0.90 | 0.300 | 2.60 | 0.972 | 4.30 | 0.996 |
| 0.95 | 0.328 | 2.65 | 0.975 | 4.35 | 0.996 |
| 1.00 | 0.380 | 2.70 | 0.977 | 4.40 | 0.996 |
| 1.05 | 0.407 | 2.75 | 0.979 | 4.45 | 0.996 |
| 1.10 | 0.460 | 2.80 | 0.980 | 4.50 | 0.996 |
| 1.15 | 0.490 | 2.85 | 0.981 | 4.55 | 0.996 |
| 1.20 | 0.550 | 2.90 | 0.982 | 4.60 | 0.996 |
| 1.25 | 0.572 | 2.95 | 0.984 | 4.65 | 0.996 |
| 1.30 | 0.620 | 3.00 | 0.984 | 4.70 | 0.996 |
| 1.35 | 0.654 | 3.05 | 0.985 | 4.75 | 0.997 |
| 1.40 | 0.695 | 3.10 | 0.986 | 4.80 | 0.997 |
| 1.45 | 0.722 | 3.15 | 0.987 | 4.85 | 0.997 |
| 1.50 | 0.750 | 3.20 | 0.987 | 4.90 | 0.997 |
| 1.55 | 0.779 | 3.25 | 0.988 | 4.95 | 0.997 |
| 1.60 | 0.800 | 3.30 | 0.988 | ≥5.00 | 1.000 |
| 1.65 | 0.825 | 3.35 | 0.989 |

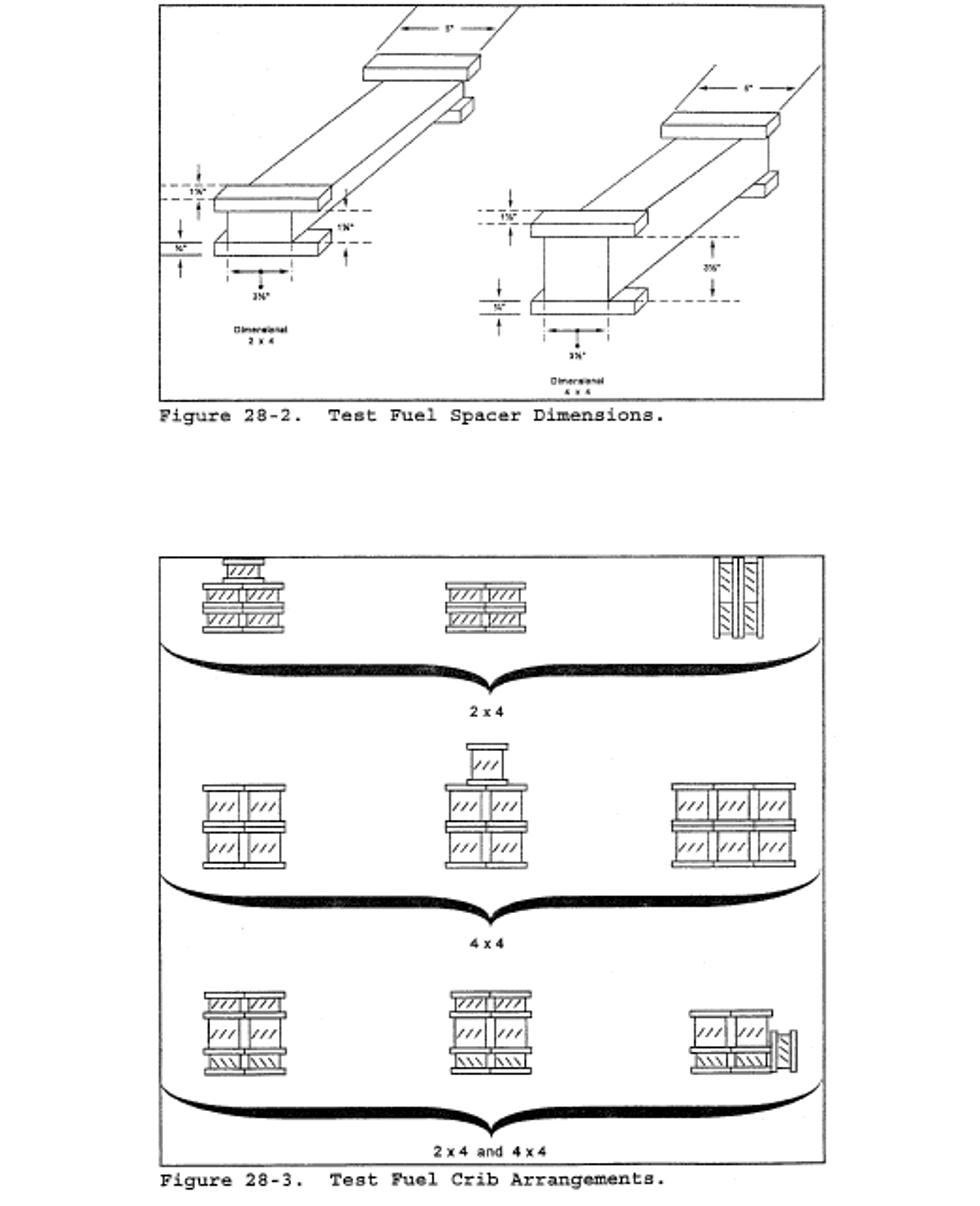

Method 28A - Measurement of Air- to-Fuel Ratio and Minimum Achievable Burn Rates for Wood-Fired Appliances
Note:
This method does not include all or the specifications ( e.g., equipment and supplies) and procedures ( e.g., sampling and analytical) essential to its performance. Some material is incorporated by reference from other methods in this part. Therefore, to obtain reliable results, persons using this method should also have a thorough knowledge of at least the following additional test methods: Method 3, Method 3A, Method 5H, Method 6C, and Method 28.
1.0 Scope and Application
1.1 Analyte. Particulate matter (PM). No CAS number assigned.
1.2 Applicability. This method is applicable for the measurement of air-to-fuel ratios and minimum achievable burn rates, for determining whether a wood-fired appliance is an affected facility, as specified in 40 CFR 60.530 .
1.3 Data Quality Objectives. Adherence to the requirements of this method will enhance the quality of the data obtained from air pollutant sampling methods.
2.0 Summary of Method
2.1 A gas sample is extracted from a location in the stack of a wood-fired appliance while the appliance is operating at a prescribed set of conditions. The gas sample is analyzed for carbon dioxide (CO 2 ), oxygen (O 2 ), and carbon monoxide (CO). These stack gas components are measured for determining the dry molecular weight of the exhaust gas. Total moles of exhaust gas are determined stoichiometrically. Air-to-fuel ratio is determined by relating the mass of dry combustion air to the mass of dry fuel consumed.
3.0 Definitions
Same as Method 28, section 3.0, with the addition of the following:
3.1 Air-to-fuel ratio means the ratio of the mass of dry combustion air introduced into the firebox to the mass of dry fuel consumed (grams of dry air per gram of dry wood burned).
4.0 Interferences [Reserved]
5.0 Safety
5.1 Disclaimer. This method may involve hazardous materials, operations, and equipment. This test method may not address all of the safety problems associated with its use. It is the responsibility of the user of this test method to establish appropriate safety and health practices and to determine the applicability of regulatory limitations prior to performing this test method.
6.0 Equipment and Supplies
6.1 Test Facility. Insulated Solid Pack Chimney, Platform Scale and Monitor, Test Facility Temperature Monitor, Balance, Moisture Meter, Anemometer, Barometer, Draft Gauge, Humidity Gauge, Wood Heater Flue, and Test Facility. Same as Method 28, sections 6.1, 6.2, and 6.4 to 6.12, respectively.
6.2 Sampling System. Probe, Condenser, Valve, Pump, Rate Meter, Flexible Bag, Pressure Gauge, and Vacuum Gauge. Same as Method 3, sections 6.2.1 to 6.2.8, respectively. Alternatively, the sampling system described in Method 5H, section 6.1 may be used.
6.3 Exhaust Gas Analysis. Use one or both of the following:
6.3.1 Orsat Analyzer. Same as Method 3, section 6.1.3
6.3.2 Instrumental Analyzers. Same as Method 5H, sections 6.1.3.4 and 6.1.3.5, for CO 2 and CO analyzers, except use a CO analyzer with a range of 0 to 5 percent and use a CO 2 analyzer with a range of 0 to 5 percent. Use an O 2 analyzer capable of providing a measure of O 2 in the range of 0 to 25 percent by volume at least once every 10 minutes.
7.0 Reagents and Standards
7.1 Test Fuel and Test Fuel Spacers. Same as Method 28, sections 7.1 and 7.2, respectively.
7.2 Cylinder Gases. For each of the three analyzers, use the same concentration as specified in sections 7.2.1, 7.2.2, and 7.2.3 of Method 6C.
8.0 Sample Collection, Preservation, Storage, and Transport
8.1 Wood Heater Air Supply Adjustments.
8.1.1 This section describes how dampers are to be set or adjusted and air inlet ports closed or sealed during Method 28A tests. The specifications in this section are intended to ensure that affected facility determinations are made on the facility configurations that could reasonably be expected to be employed by the user. They are also intended to prevent circumvention of the standard through the addition of an air port that would often be blocked off in actual use. These specifications are based on the assumption that consumers will remove such items as dampers or other closure mechanism stops if this can be done readily with household tools; that consumers will block air inlet passages not visible during normal operation of the appliance using aluminum tape or parts generally available at retail stores; and that consumers will cap off any threaded or flanged air inlets. They also assume that air leakage around glass doors, sheet metal joints or through inlet grilles visible during normal operation of the appliance would not be further blocked or taped off by a consumer.
8.1.2 It is not the intention of this section to cause an appliance that is clearly designed, intended, and, in most normal installations, used as a fireplace to be converted into a wood heater for purposes of applicability testing. Such a fireplace would be identifiable by such features as large or multiple glass doors or panels that are not gasketed, relatively unrestricted air inlets intended, in large part, to limit smoking and fogging of glass surfaces, and other aesthetic features not normally included in wood heaters.
8.1.3 Adjustable Air Supply Mechanisms. Any commercially available flue damper, other adjustment mechanism or other air inlet port that is designed, intended or otherwise reasonably expected to be adjusted or closed by consumers, installers, or dealers and which could restrict air into the firebox shall be set so as to achieve minimum air into the firebox ( i.e., closed off or set in the most closed position).
8.1.3.1 Flue dampers, mechanisms and air inlet ports which could reasonably be expected to be adjusted or closed would include:
8.1.3.1.1 All internal or externally adjustable mechanisms (including adjustments that affect the tightness of door fittings) that are accessible either before and/or after installation.
8.1.3.1.2 All mechanisms, other inlet ports, or inlet port stops that are identified in the owner's manual or in any dealer literature as being adjustable or alterable. For example, an inlet port that could be used to provide access to an outside air duct but which is identified as being closable through use of additional materials whether or not they are supplied with the facility.
8.1.3.1.3 Any combustion air inlet port or commercially available flue damper or mechanism stop, which would readily lend itself to closure by consumers who are handy with household tools by the removal of parts or the addition of parts generally available at retail stores ( e.g., addition of a pipe cap or plug, addition of a small metal plate to an inlet hole on a nondecorative sheet metal surface, or removal of riveted or screwed damper stops).
8.1.3.1.4 Any flue damper, other adjustment mechanisms or other air inlet ports that are found and documented in several ( e.g., a number sufficient to reasonably conclude that the practice is not unique or uncommon) actual installations as having been adjusted to a more closed position, or closed by consumers, installers, or dealers.
8.1.4 Air Supply Adjustments During Test. The test shall be performed with all air inlets identified under this section in the closed or most closed position or in the configuration which otherwise achieves the lowest air inlet ( i.e., greatest blockage).
Note:
For the purposes of this section, air flow shall not be minimized beyond the point necessary to maintain combustion or beyond the point that forces smoke into the room.
8.1.5 Notwithstanding section 8.1.1, any flue damper, adjustment mechanism, or air inlet port (whether or not equipped with flue dampers or adjusting mechanisms) that is visible during normal operation of the appliance and which could not reasonably be closed further or blocked except through means that would significantly degrade the aesthetics of the facility ( e.g., through use of duct tape) will not be closed further or blocked.
8.2 Sampling System.
8.2.1 Sampling Location. Same as Method 5H, section 8.1.2.
8.2.2 Sampling System Set Up. Set up the sampling equipment as described in Method 3, section 8.1.
8.3 Wood Heater Installation, Test Facility Conditions, Wood Heater Firebox Volume, and Test Fuel Charge. Same as Method 28, sections 8.4 and 8.6 to 8.8, respectively.
8.4 Pretest Ignition. Same as Method 28, section 8.11. Set the wood heater air supply settings to achieve a burn rate in Category 1 or the lowest achievable burn rate (see section 8.1).
8.5 Test Run. Same as Method 28, section 8.12. Begin sample collection at the start of the test run as defined in Method 28, section 8.12.1.
8.5.1 Gas Analysis.
8.5.1.1 If Method 3 is used, collect a minimum of two bag samples simultaneously at a constant sampling rate for the duration of the test run. A minimum sample volume of 30 liters (1.1 ft 3 ) per bag is recommended.
8.5.1.2 If instrumental gas concentration measurement procedures are used, conduct the gas measurement system performance tests, analyzer calibration, and analyzer calibration error check outlined in Method 6C, sections 8.2.3, 8.2.4, 8.5, and 10.0, respectively. Sample at a constant rate for the duration of the test run.
8.5.2 Data Recording. Record wood heater operational data, test facility temperature, sample train flow rate, and fuel weight data at intervals of no greater than 10 minutes.
8.5.3 Test Run Completion. Same as Method 28, section 8.13.
9.0 Quality Control
9.1 Data Validation. The following quality control procedure is suggested to provide a check on the quality of the data.
9.1.1 Calculate a fuel factor, F o , using Equation 28A-1 in section 12.2.
9.1.2 If CO is present in quantities measurable by this method, adjust the O 2 and CO 2 values before performing the calculation for F o as shown in section 12.3 and 12.4.
9.1.3 Compare the calculated F o factor with the expected F o range for wood (1.000-1.120). Calculated F o values beyond this acceptable range should be investigated before accepting the test results. For example, the strength of the solutions in the gas analyzer and the analyzing technique should be checked by sampling and analyzing a known concentration, such as air. If no detectable or correctable measurement error can be identified, the test should be repeated. Alternatively, determine a range of air-to-fuel ratio results that could include the correct value by using an F o value of 1.05 and calculating a potential range of CO 2 and O 2 values. Acceptance of such results will be based on whether the calculated range includes the exemption limit and the judgment of the Administrator.
9.2 Method 3 Analyses. Compare the results of the analyses of the two bag samples. If all the gas components (O 2 , CO, and CO 2 ) values for the two analyses agree within 0.5 percent ( e.g., 6.0 percent O 2 for bag 1 and 6.5 percent O 2 for bag 2, agree within 0.5 percent), the results of the bag analyses may be averaged for the calculations in section 12. If the analysis results do not agree within 0.5 percent for each component, calculate the air-to-fuel ratio using both sets of analyses and report the results.
10.0 Calibration and Standardization [Reserved]
11.0 Analytical Procedures
11.1 Method 3 Integrated Bag Samples. Within 4 hours after the sample collection, analyze each bag sample for percent CO 2 , O 2 , and CO using an Orsat analyzer as described in Method 3, section 11.0.
11.2 Instrumental Analyzers. Average the percent CO 2 , CO, and O 2 values for the test run.
12.0 Data Analyses and Calculations
Carry out calculations, retaining at least one extra significant figure beyond that of the acquired data. Round off figure after the final calculation. Other forms of the equations may be used as long as they give equivalent results.
12.1 Nomenclature.
M d = Dry molecular weight, g/g-mole (lb/lb-mole).
N T = Total gram-moles of dry exhaust gas per kg of wood burned (lb-moles/lb).
%CO 2 = Percent CO 2 by volume (dry basis).
%CO = Percent CO by volume (dry basis).
%N 2 = Percent N 2 by volume (dry basis).
%O 2 = Percent O2 by volume (dry basis).
Y HC = Assumed mole fraction of HC (dry as CH 4 ) = 0.0088 for catalytic wood heaters; = 0.0132 for noncatalytic wood heaters. = 0.0080 for pellet-fired wood heaters.
Y CO = Measured mole fraction of CO ( e.g., 1 percent CO = .01 mole fraction), g/g-mole (lb/lb-mole).
Y CO2 = Measured mole fraction of CO CO2 ( e.g., 10 percent CO 2 = .10 mole fraction), g/g-mole (lb/lb-mole).
0.280 = Molecular weight of N 2 or CO, divided by 100.
0.320 = Molecular weight of O 2 divided by 100.
0.440 = Molecular weight of CO 2 divided by 100.
20.9 = Percent O 2 by volume in ambient air.
42.5 = Gram-moles of carbon in 1 kg of dry wood assuming 51 percent carbon by weight dry basis (.0425 lb/lb-mole).
510 = Grams of carbon in exhaust gas per kg of wood burned.
1,000 = Grams in 1 kg.
12.2 Fuel Factor. Use Equation 28A-1 to calculate the fuel factor.
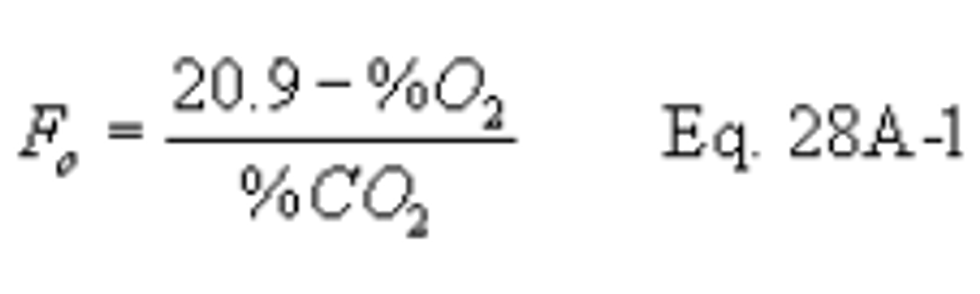
12. 3 Adjusted %CO 2 . Use Equation 28A-2 to adjust CO 2 values if measurable CO is present.
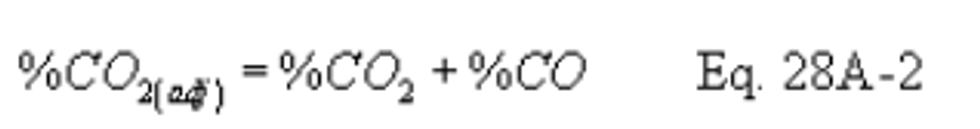
12.4 Adjusted %O 2 . Use Equation 28A-3 to adjust O 2 value if measurable CO is present.
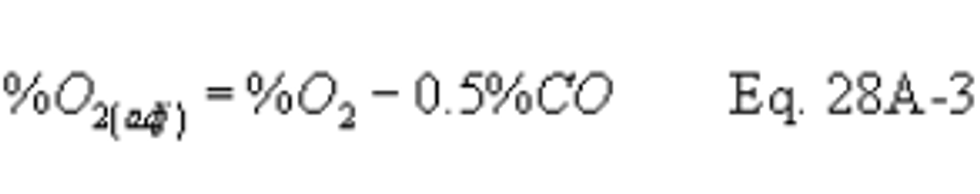
12.5 Dry Molecular Weight. Use Equation 28A-4 to calculate the dry molecular weight of the stack gas.
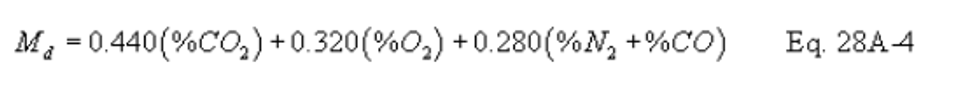
Note:
The above equation does not consider argon in air (about 0.9 percent, molecular weight of 39.9). A negative error of about 0.4 percent is introduced. Argon may be included in the analysis using procedures subject to approval of the Administrator.
12.6 Dry Moles of Exhaust Gas. Use Equation 28A-5 to calculate the total moles of dry exhaust gas produced per kilogram of dry wood burned.
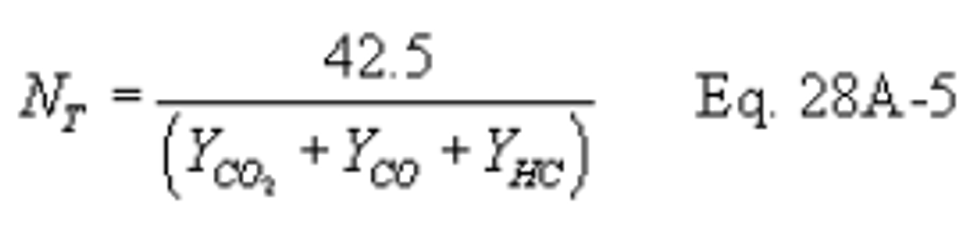
12.7 Air-to-Fuel Ratio. Use Equation 28A-6 to calculate the air-to-fuel ratio on a dry mass basis.
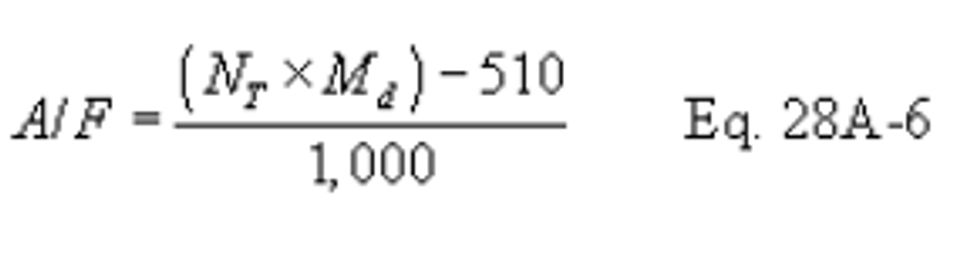
12.8 Burn Rate. Calculate the fuel burn rate as in Method 28, section 12.4.
13.0 Method Performance [Reserved]
14.0 Pollution Prevention [Reserved]
15.0 Waste Management [Reserved]
16.0 References
Same as section 16.0 of Method 3 and section 17 of Method 5G.
17.0 Tables, Diagrams, Flowcharts, and Validation Data [Reserved]
Test Method 28R for Certification and Auditing of Wood Heaters
1.0 Scope and Application
1.1 This test method applies to certification and auditing of wood-fired room heaters and fireplace inserts.
1.2 The test method covers the fueling and operating protocol for measuring particulate emissions, as well as determining burn rates, heat output and efficiency.
1.3 Particulate emissions are measured by the dilution tunnel method as specified in ASTM E2515-11 Standard Test Method for Determination of Particulate Matter Emissions Collected in a Dilution Tunnel (IBR, see §60.17 ). Upon request, four-inch filters may be used. Upon request, Teflon membrane filters or Teflon-coated glass fiber filters may be used.
2.0 Procedures
2.1 This method incorporates the provisions of ASTM E2780-10 (IBR, see §60.17 ) except as follows:
2.1.1 The burn rate categories, low burn rate requirement, and weightings in Method 28 shall be used.
2.1.2 The startup procedures shall be the same as in Method 28.
2.1.3 Manufacturers shall not specify a smaller volume of the firebox for testing than the full usable firebox.
2.1.4 Prior to testing, the heater must be operated for a minimum of 50 hours using a medium burn rate. The conditioning may be at the manufacturer's facility prior to the certification test. If the conditioning is at the certification test laboratory, the pre-burn for the first test can be included as part of the conditioning requirement.
2.2 Manufacturers may use ASTM E871-82 (reapproved 2013) (IBR, see §60.17 ) as an alternative to the procedures in Method 5H or Method 28 for determining total weight basis moisture in the analysis sample of particulate wood fuel.
Test Method 28WHH for Measurement of Particulate Emissions and Heating Efficiency of Wood-Fired Hydronic Heating Appliances
1.0 Scope and Application
1.1 This test method applies to wood-fired hydronic heating appliances. The units typically transfer heat through circulation of a liquid heat exchange media such as water or a water-antifreeze mixture.
1.2 The test method measures particulate emissions and delivered heating efficiency at specified heat output rates based on the appliance's rated heating capacity.
1.3 Particulate emissions are measured by the dilution tunnel method as specified in ASTM E2515-11 Standard Test Method for Determination of Particulate Matter Emissions Collected in a Dilution Tunnel (IBR, see §60.17 ). Upon request, four-inch filters may be used. Upon request, Teflon membrane filters or Teflon-coated glass fiber filters may be used. Delivered efficiency is measured by determining the heat output through measurement of the flow rate and temperature change of water circulated through a heat exchanger external to the appliance and determining the input from the mass of dry wood fuel and its higher heating value. Delivered efficiency does not attempt to account for pipeline loss.
1.4 Products covered by this test method include both pressurized and non-pressurized heating appliances intended to be fired with wood. These products are wood-fired hydronic heating appliances that the manufacturer specifies for indoor or outdoor installation. They are often connected to a heat exchanger by insulated pipes and normally include a pump to circulate heated liquid. They are used to heat structures such as homes, barns and greenhouses and can heat domestic hot water, spas or swimming pools.
1.5 Distinguishing features of products covered by this standard include:
1.5.1 Manufacturer specifies for indoor or outdoor installation.
1.5.2 A firebox with an access door for hand loading of fuel.
1.5.3 Typically, an aquastat that controls combustion air supply to maintain the liquid in the appliance within a predetermined temperature range provided sufficient fuel is available in the firebox.
1.5.4 A chimney or vent that exhausts combustion products from the appliance.
1.6 The values stated are to be regarded as the standard whether in I-P or SI units. The values given in parentheses are for information only.
2.0 Summary of Method and References
2.1 Particulate matter emissions are measured from a wood-fired hydronic heating appliance burning a prepared test fuel crib in a test facility maintained at a set of prescribed conditions. Procedures for determining burn rates, and particulate emissions rates and for reducing data are provided.
2.2 Referenced Documents
2.2.1 EPA Standards
2.2.1.1 Method 28 Certification and Auditing of Wood Heaters
2.2.2 Other Standards
2.2.2.1 ASTM E2515-11 - Standard Test Method for Determination of Particulate Matter Emissions Collected in a Dilution Tunnel (IBR, see §60.17 ).
2.2.2.2 CSA-B415.1-10 Performance Testing of Solid-Fuel-Burning Heating Appliances (IBR, see §60.17 ).
3.0 Terminology
3.1 Definitions.
3.1.1 Hydronic Heating - A heating system in which a heat source supplies energy to a liquid heat exchange media such as water that is circulated to a heating load and returned to the heat source through pipes.
3.1.2 Aquastat - A control device that opens or closes a circuit to control the rate of fuel consumption in response to the temperature of the heating media in the heating appliance.
3.1.3 Delivered Efficiency - The percentage of heat available in a test fuel charge that is delivered to a simulated heating load as specified in this test method.
3.1.4 Manufacturer's Rated Heat Output Capacity - The value in Btu/hr (MJ/hr) that the manufacturer specifies that a particular model of hydronic heating appliance is capable of supplying at its design capacity as verified by testing, in accordance with Section 13.
3.1.5 Burn Rate - The rate at which test fuel is consumed in an appliance. Measured in pounds (lbs) or kilograms of wood (dry basis) per hour (lb/hr or kg/hr).
3.1.6 Firebox - The chamber in the appliance in which the test fuel charge is placed and combusted.
3.1.7 Test Fuel Charge - The collection of test fuel layers placed in the appliance at the start of the emission test run.
3.1.8 Test Fuel Layer - Horizontal arrangement of test fuel units.
3.1.9 Test Fuel Unit - One or more test fuel pieces with 3/4 inch (19 mm) spacers attached to the bottom and to one side. If composed of multiple test fuel pieces, the bottom spacer may be one continuous piece.
3.1.10 Test Fuel Piece - A single 4 × 4 (4 ±0.25 inches by 4 ±0.25 inches) [100 ±6 mm by 100 ±6 mm] white or red oak wood piece cut to the length required.
3.1.11 Test Run - An individual emission test that encompasses the time required to consume the mass of the test fuel charge.
3.1.12 Overall Efficiency (SLM) - The efficiency for each test run as determined using the CSA B415.1-10 (IBR, see §60.17 ) stack loss method.
3.1.13 Thermopile - A device consisting of a number of thermocouples connected in series, used for measuring differential temperature.
4.0 Summary of Test Method
4.1 Dilution Tunnel. Emissions are determined using the “dilution tunnel” method specified in ASTM E2515-11 Standard Test Method for Determination of Particulate Matter Emissions Collected in a Dilution Tunnel (IBR, see §60.17 ). The flow rate in the dilution tunnel is maintained at a constant level throughout the test cycle and accurately measured. Samples of the dilution tunnel flow stream are extracted at a constant flow rate and drawn through high efficiency filters. The filters are dried and weighed before and after the test to determine the emissions catch and this value is multiplied by the ratio of tunnel flow to filter flow to determine the total particulate emissions produced in the test cycle.
4.2 Efficiency. The efficiency test procedure takes advantage of the fact that this type of appliance delivers heat through circulation of the heated liquid (water) from the appliance to a remote heat exchanger and back to the appliance. Measurements of the water temperature difference as it enters and exits the heat exchanger along with the measured flow rate allow for an accurate determination of the useful heat output of the appliance. The input is determined by weight of the test fuel charge, adjusted for moisture content, multiplied by the higher heating value. Additional measurements of the appliance weight and temperature at the beginning and end of a test cycle are used to correct for heat stored in the appliance. Overall efficiency (SLM) is determined using the CSA B415.1-10 (IBR, see §60.17 ) stack loss method for data quality assurance purposes.
4.3 Operation. Appliance operation is conducted on a hot-to-hot test cycle meaning that the appliance is brought to operating temperature and a coal bed is established prior to the addition of the test fuel charge and measurements are made for each test fuel charge cycle. The measurements are made under constant heat draw conditions within predetermined ranges. No attempt is made to modulate the heat demand to simulate an indoor thermostat cycling on and off in response to changes in the indoor environment. Four test categories are used. These are:
4.3.1 Category I: A heat output of 15 percent or less of manufacturer's rated heat output capacity.
4.3.2 Category II: A heat output of 16 percent to 24 percent of manufacturer's rated heat output capacity.
4.3.3 Category III: A heat output of 25 percent to 50 percent of manufacturer's rated heat output capacity.
4.3.4 Category IV: Manufacturer's rated heat output capacity.
5.0 Significance and Use
5.1 The measurement of particulate matter emission rates is an important test method widely used in the practice of air pollution control.
5.1.1 These measurements, when approved by state or federal agencies, are often required for the purpose of determining compliance with regulations and statutes.
5.1.2 The measurements made before and after design modifications are necessary to demonstrate the effectiveness of design changes in reducing emissions and make this standard an important tool in manufacturers' research and development programs.
5.2 Measurement of heating efficiency provides a uniform basis for comparison of product performance that is useful to the consumer. It is also required to relate emissions produced to the useful heat production.
5.3 This is a laboratory method and is not intended to be fully representative of all actual field use. It is recognized that users of hand-fired, wood-burning equipment have a great deal of influence over the performance of any wood-burning appliance. Some compromises in realism have been made in the interest of providing a reliable and repeatable test method.
6.0 Test Equipment
6.1 Scale. A platform scale capable of weighing the appliance under test and associated parts and accessories when completely filled with water to an accuracy of ±1.0 pound (±0.5 kg).
6.2 Heat Exchanger. A water-to-water heat exchanger capable of dissipating the expected heat output from the system under test.
6.3 Water Temperature Difference Measurement. A Type - T 'special limits' thermopile with a minimum of 5 pairs of junctions shall be used to measure the temperature difference in water entering and leaving the heat exchanger. The temperature difference measurement uncertainty of this type of thermopile is equal to or less than ±0.50°F (±0.25°C). Other temperature measurement methods may be used if the temperature difference measurement uncertainty is equal to or less than ±0.50°F (±0.25°C).
6.4 Water Flow Meter. A water flow meter shall be installed in the inlet to the load side of the heat exchanger. The flow meter shall have an accuracy of ±1 percent of measured flow.
6.4.1 Optional - Appliance Side Water Flow Meter. A water flow meter with an accuracy of ±1 percent of the flow rate is recommended to monitor supply side water flow rate.
6.5 Optional Recirculation Pump. Circulating pump used during test to prevent stratification of liquid being heated.
6.6 Water Temperature Measurement - Thermocouples or other temperature sensors to measure the water temperature at the inlet and outlet of the load side of the heat exchanger. Must meet the calibration requirements specified in section 10.1.
6.7 Wood Moisture Meter - Calibrated electrical resistance meter capable of measuring test fuel moisture to within 1 percent moisture content. Must meet the calibration requirements specified in section 10.4.
6.8 Flue Gas Temperature Measurement - Must meet the requirements of CSA B415.1-10 (IBR, see §60.17 ), clause 6.2.2.
6.9 Test Room Temperature Measurement - Must meet the requirements of CSA B415.1-10 (IBR, see §60.17 ), clause 6.2.1.
6.10 Flue Gas Composition Measurement - Must meet the requirements of CSA B415.1-10 (IBR, see §60.17 ), clauses 6.3.1 through 6.3.3.
7.0 Safety
7.1 These tests involve combustion of wood fuel and substantial release of heat and products of combustion. The heating system also produces large quantities of very hot water and the potential for steam production and system pressurization. Appropriate precautions must be taken to protect personnel from burn hazards and respiration of products of combustion.
8.0 Sampling, Test Specimens and Test Appliances
8.1 Test specimens shall be supplied as complete appliances including all controls and accessories necessary for installation in the test facility. A full set of specifications and design and assembly drawings shall be provided when the product is to be placed under certification of a third-party agency. The manufacturer's written installation and operating instructions are to be used as a guide in the set-up and testing of the appliance.
9.0 Preparation of Test Equipment
9.1 The appliance is to be placed on a scale capable of weighing the appliance fully loaded with a resolution of ±1.0 lb (0.5 kg).
9.2 The appliance shall be fitted with the type of chimney recommended or provided by the manufacturer and extending to 15 ±0.5 feet (4.6 ±0.15 m) from the upper surface of the scale. If no flue or chimney system is recommended or provided by the manufacturer, connect the appliance to a flue of a diameter equal to the flue outlet of the appliance. The flue section from the appliance flue collar to 8 ±0.5 feet above the scale shall be single wall stove pipe and the remainder of the flue shall be double wall insulated class A chimney.
9.3 Optional Equipment Use
9.3.1 A recirculation pump may be installed between connections at the top and bottom of the appliance to minimize thermal stratification if specified by the manufacturer. The pump shall not be installed in such a way as to change or affect the flow rate between the appliance and the heat exchanger.
9.3.2 If the manufacturer specifies that a thermal control valve or other device be installed and set to control the return water temperature to a specific set point, the valve or other device shall be installed and set per the manufacturer's written instructions.
9.4 Prior to filling the tank, weigh and record the appliance mass.
9.5 Heat Exchanger
9.5.1 Plumb the unit to a water-to-water heat exchanger with sufficient capacity to draw off heat at the maximum rate anticipated. Route hoses, electrical cables, and instrument wires in a manner that does not influence the weighing accuracy of the scale as indicated by placing dead weights on the platform and verifying the scale's accuracy.
9.5.2 Locate thermocouples to measure the water temperature at the inlet and outlet of the load side of the heat exchanger.
9.5.3 Install a thermopile meeting the requirements of section 6.3 to measure the water temperature difference between the inlet and outlet of the load side of the heat exchanger.
9.5.4 Install a calibrated water flow meter in the heat exchanger load side supply line. The water flow meter is to be installed on the cooling water inlet side of the heat exchanger so that it will operate at the temperature at which it is calibrated.
9.5.5 Place the heat exchanger in a box with 2 in. (50 mm) of expanded polystyrene (EPS) foam insulation surrounding it to minimize heat losses from the heat exchanger.
9.5.6 The reported efficiency and heat output rate shall be based on measurements made on the load side of the heat exchanger.
9.5.7 Temperature instrumentation per section 6.6 shall be installed in the appliance outlet and return lines. The average of the outlet and return water temperature on the supply side of the system shall be considered the average appliance temperature for calculation of heat storage in the appliance (TF avg and TI avg ). Installation of a water flow meter in the supply side of the system is optional.
9.6 Fill the system with water. Determine the total weight of the water in the appliance when the water is circulating. Verify that the scale indicates a stable weight under operating conditions. Make sure air is purged properly.
10.0 Calibration and Standardization
10.1 Water Temperature Sensors. Temperature measuring equipment shall be calibrated before initial use and at least semi-annually thereafter. Calibrations shall be in compliance with National Institute of Standards and Technology (NIST) Monograph 175, Standard Limits of Error.
10.2 Heat Exchanger Load Side Water Flow Meter.
10.2.1 The heat exchanger load side water flow meter shall be calibrated within the flow range used for the test run using NIST traceable methods. Verify the calibration of the water flow meter before and after each test run and at least once during each test run by comparing the water flow rate indicated by the flow meter to the mass of water collected from the outlet of the heat exchanger over a timed interval. Volume of the collected water shall be determined based on the water density calculated from section 13, Eq. 8, using the water temperature measured at the flow meter. The uncertainty in the verification procedure used shall be 1 percent or less. The water flow rate determined by the collection and weighing method shall be within 1 percent of the flow rate indicated by the water flow meter.
10.3 Scales. The scales used to weigh the appliance and test fuel charge shall be calibrated using NIST traceable methods at least once every 6 months.
10.4 Moisture Meter. The moisture meter shall be calibrated per the manufacturer's instructions and checked before each use.
10.5 Flue Gas Analyzers - In accordance with CSA B415.1-10 (IBR, see §60.17 ), clause 6.8.
11.0 Conditioning
11.1 Prior to testing, the appliance is to be operated for a minimum of 50 hours using a medium heat draw rate. The conditioning may be at the manufacturer's facility prior to the certification test. If the conditioning is at the certification test laboratory, the pre-burn for the first test can be included as part of the conditioning requirement. If conditioning is included in pre-burn, then the appliance shall be aged with fuel meeting the specifications outlined in sections 12.2 with a moisture content between 19 and 25 percent on a dry basis. Operate the appliance at a medium burn rate (Category II or III) for at least 10 hours for noncatalytic appliances and 50 hours for catalytic appliances. Record and report hourly flue gas exit temperature data and the hours of operation. The aging procedure shall be conducted and documented by a testing laboratory.
12.0 Procedure
12.1 Appliance Installation. Assemble the appliance and parts in conformance with the manufacturer's written installation instructions. Clean the flue with an appropriately sized, wire chimney brush before each certification test series.
12.2 Fuel. Test fuel charge fuel shall be red ( Quercus ruba L. ) or white ( Quercus alba ) oak 19 to 25 percent moisture content on a dry basis. Piece length shall be 80 percent of the firebox depth rounded down to the nearest 1 inch (25mm) increment. For example, if the firebox depth is 46 inches (1168mm) the 4 × 4 piece length would be 36 inches (46 inches × 0.8 = 36.8 inches rounded down to 36 inches). Pieces are to be placed in the firebox parallel to the longest firebox dimension. For fireboxes with sloped surfaces that create a non-uniform firebox length, the piece length shall be adjusted for each layer based on 80 percent of the length at the level where the layer is placed. Pieces are to be spaced 3/4 inches (19 mm) apart on all faces. The first fuel layer may be assembled using fuel units consisting of multiple 4 × 4s consisting of single pieces with bottom and side spacers of 3 or more pieces if needed for a stable layer. The second layer may consist of fuel units consisting of no more than two pieces with spacers attached on the bottom and side. The top two layers of the fuel charge must consist of single pieces unless the fuel charge is only three layers. In that instance only the top layer must consist of single units. Three-quarter inch (19 mm) by 1.5 inch (38 mm) spacers shall be attached to the bottom of piece to maintain a 3/4 inch (19 mm) separation. When a layer consists of two or more units of 4 × 4s an additional 3/4 inch (19 mm) thick by 1.5 inch (38 mm) wide spacer shall be attached to the vertical face of each end of one 4 × 4, such that the 3/4 inch (19 mm) space will be maintained when two 4 × 4 units or pieces are loaded side by side. In cases where a layer contains an odd number of 4 × 4s one piece shall not be attached, but shall have spacers attached in a manner that will provide for the 3/4 inch (19 mm) space to be maintained (See Figure 1). Spacers shall be attached perpendicular to the length of the 4 × 4s such that the edge of the spacer is 1 ± 0.25 inch from the end of the 4 × 4s in the previous layers. Spacers shall be red or white oak and will be attached with either nails (non-galvanized), brads or oak dowels. The use of kiln-dried wood is not allowed.
12.2.1 Using a fuel moisture meter as specified in section 6.7 of the test method, determine the fuel moisture for each test fuel piece used for the test fuel load by averaging at least five fuel moisture meter readings measured parallel to the wood grain. Penetration of the moisture meter insulated electrodes for all readings shall be 1/4 the thickness of the fuel piece or 19 mm ( 3/4 in.), whichever is lesser. One measurement from each of three sides shall be made at approximately 3 inches from each end and the center. Two additional measurements shall be made centered between the other three locations. Each individual moisture content reading shall be in the range of 18 to 28 percent on a dry basis. The average moisture content of each piece of test fuel shall be in the range of 19 to 25 percent. It is not required to measure the moisture content of the spacers. Moisture shall not be added to previously dried fuel pieces except by storage under high humidity conditions and temperature up to 100°F. Fuel moisture shall be measured within 4 hours of using the fuel for a test.
12.2.2 Firebox Volume. Determine the firebox volume in cubic feet. Firebox volume shall include all areas accessible through the fuel loading door where firewood could reasonably be placed up to the horizontal plane defined by the top of the loading door. A drawing of the firebox showing front, side and plan views or an isometric view with interior dimensions shall be provided by the manufacturer and verified by the laboratory. Calculations for firebox volume from computer aided design (CAD) software programs are acceptable and shall be included in the test report if used. If the firebox volume is calculated by the laboratory the firebox drawings and calculations shall be included in the test report.
12.2.3 Test Fuel Charge. Test fuel charges shall be determined by multiplying the firebox volume by 10 pounds (4.54 kg) per ft 3 (28L), or a higher load density as recommended by the manufacturer's printed operating instructions, of wood (as used wet weight). Select the number of pieces of standard fuel that most nearly match this target weight. This is the standard fuel charge for all tests. For example, if the firebox loading area volume is 10 ft 3 (280L) and the firebox depth is 46 inches (1168 mm), test fuel charge target is 100 lbs (45 kg) minimum and the piece length is 36 inches (914 mm). If eight 4 × 4s, 36 inches long weigh 105 lbs (48 kg), use 8 pieces for each test fuel charge. All test fuel charges will be of the same configuration.
12.3 Sampling Equipment. Prepare the particulate emission sampling equipment as defined by ASTM E2515-11 Standard Test Method for Determination of Particulate Matter Emissions Collected in a Dilution Tunnel (IBR, see §60.17 ). Upon request, four-inch filters may be used. Upon request, Teflon membrane filters or Teflon-coated glass fiber filters may be used.
12.4 Appliance Startup. The appliance shall be fired with wood fuel of any species, size and moisture content at the laboratories' discretion to bring it up to operating temperature. Operate the appliance until the water is heated to the upper operating control limit and has cycled at least two times. Then remove all unburned fuel, zero the scale and verify the scales accuracy using dead weights.
12.4.1 Pretest Burn Cycle. Reload appliance with oak wood and allow it to burn down to the specified coal bed weight. The pretest burn cycle fuel charge weight shall be within ±10 percent of the test fuel charge weight. Piece size and length shall be selected such that charcoalization is achieved by the time the fuel charge has burned down to the required coal bed weight. Pieces with a maximum thickness of approximately 2 inches have been found to be suitable. Charcoalization is a general condition of the test fuel bed evidenced by an absence of large pieces of burning wood in the coal bed and the remaining fuel pieces being brittle enough to be broken into smaller charcoal pieces with a metal poker. Manipulations to the fuel bed prior to the start of the test run are to be done to achieve charcoalization while maintaining the desired heat output rate. During the pre-test burn cycle and at least one hour prior to starting the test run, adjust water flow to the heat exchanger to establish the target heat draw for the test. For the first test run the heat draw rate shall be equal to the manufacturer's rated heat output capacity.
12.4.1.1 Allowable Adjustments. Fuel addition or subtractions, and coal bed raking shall be kept to a minimum but are allowed up to 15 minutes prior to the start of the test run. For the purposes of this method, coal bed raking is the use of a metal tool (poker) to stir coals, break burning fuel into smaller pieces, dislodge fuel pieces from positions of poor combustion, and check for the condition of charcoalization. Record all adjustments to and additions or subtractions of fuel, and any other changes to the appliance operations that occur during pretest ignition period. During the 15-minute period prior to the start of the test run, the wood heater loading door shall not be open more than a total of 1 minute. Coal bed raking is the only adjustment allowed during this period.
12.4.2 Coal Bed Weight. The appliance is to be loaded with the test fuel charge when the coal bed weight is between 10 percent and 20 percent of the test fuel charge weight. Coals may be raked as necessary to level the coal bed but may only be raked and stirred once between 15 to 20 minutes prior to the addition of the test fuel charge.
12.5 Test Runs. For all test runs, the return water temperature to the hydronic heater must be equal to or greater than 120°F. Aquastat or other heater output control device settings that are adjustable shall be set using manufacturer specifications, either as factory set or in accordance with the owner's manual, and shall remain the same for all burn categories.
Complete a test run in each heat output rate category, as follows:
12.5.1 Test Run Start. Once the appliance is operating normally and the pretest coal bed weight has reached the target value per section 12.4.2, tare the scale and load the full test charge into the appliance. Time for loading shall not exceed 5 minutes. The actual weight of the test fuel charge shall be measured and recorded within 30 minutes prior to loading. Start all sampling systems.
12.5.1.1 Record all water temperatures, differential water temperatures and water flow rates at time intervals of one minute or less.
12.5.1.2 Record particulate emissions data per the requirements of ASTM E2515 (IBR, see §60.17 ).
12.5.1.3 Record data needed to determine overall efficiency (SLM) per the requirements of CSA B415.1-10 (IBR, see §60.17 ), clauses 6.2.1, 6.2.2, 6.3, 8.5.7, 10.4.3 (a), 10.4.3(f), and 13.7.9.3
12.5.1.3.1 Measure and record the test room air temperature in accordance with the requirements of CSA B415.1-10 (IBR, see §60.17 ), clauses 6.2.1, 8.5.7 and 10.4.3 (g).
12.5.1.3.2 Measure and record the flue gas temperature in accordance with the requirements of CSA B415.1-10 (IBR, see §60.17 ), clauses 6.2.2, 8.5.7 and 10.4.3 (f).
12.5.1.3.3 Determine and record the carbon monoxide (CO) and carbon dioxide (CO 2 ) concentrations in the flue gas in accordance with CSA B415.1-10 (IBR, see §60.17 ), clauses 6.3, 8.5.7 and 10.4.3 (i) and (j).
12.5.1.3.4 Measure and record the test fuel weight per the requirements of CSA B415.1-10 (IBR, see §60.17 ), clauses 8.5.7 and 10.4.3 (h).
12.5.1.3.5 Record the test run time per the requirements of CSA B415.1-10 (IBR, see §60.17 ), clauses 10.4.3 (a).
12.5.1.4 Monitor the average heat output rate on the load side of the heat exchanger. If the heat output rate gets close to the upper or lower limit of the target range (±5 percent) adjust the water flow through the heat exchanger to compensate. Make changes as infrequently as possible while maintaining the target heat output rate. The first test run shall be conducted at the Category IV heat output rate to validate that the appliance is capable of producing the manufacturer's rated heat output capacity.
12.5.2 Test Fuel Charge Adjustment. It is acceptable to adjust the test fuel charge ( i.e., reposition) once during a test run if more than 60 percent of the initial test fuel charge weight has been consumed and more than 10 minutes have elapsed without a measurable (1 lb or 0. 5 kg) weight change while the operating control is in the demand mode. The time used to make this adjustment shall be less than 60 seconds.
12.5.3 Test Run Completion. The test run is completed when the remaining weight of the test fuel charge is 0.0 lb (0.0 kg). End the test run when the scale has indicated a test fuel charge weight of 0.0 lb (0.0 kg) or less for 30 seconds.
12.5.3.1 At the end of the test run, stop the particulate sampling train and overall efficiency (SLM) measurements, and record the run time, and all final measurement values.
12.5.4 Heat Output Capacity Validation. The first test run must produce a heat output rate that is within 10 percent of the manufacturer's rated heat output capacity (Category IV) throughout the test run and an average heat output rate within 5 percent of the manufacturer's rated heat output capacity. If the appliance is not capable of producing a heat output within these limits, the manufacturer's rated heat output capacity is considered not validated and testing is to be terminated. In such cases, the tests may be restarted using a lower heat output capacity if requested by the manufacturer.
12.5.5 Additional Test Runs. Using the manufacturer's rated heat output capacity as a basis, conduct a test for additional heat output categories as specified in section 4.3. It is not required to run these tests in any particular order.
12.5.6 Alternative Heat Output Rate for Category I. If an appliance cannot be operated in the Category I heat output range due to stopped combustion, two test runs shall be conducted at heat output rates within Category II, provided that the completed test run burn rate is no greater than the burn rate expected in home use. If this rate cannot be achieved, the test is not valid.
When the alternative heat output rate is used, the weightings for the weighted averages indicated in Table 2 shall be the average of the Category I and II weightings and shall be applied to both Category II results. The two completed runs in Category II will be deemed to meet the requirement for runs completed in both Category I and Category II. Appliances that are not capable of operation within Category II (<25 percent of maximum) cannot be evaluated by this test method. The test report must include full documentation and discussion of the attempted runs, completed rums and calculations.
12.5.6.1 Stopped Fuel Combustion. Evidence that an appliance cannot be operated at a Category I heat output rate due to stopped fuel combustion shall include documentation of two or more attempts to operate the appliance in burn rate Category I and fuel combustion has stopped prior to complete consumption of the test fuel charge. Stopped fuel combustion is evidenced when an elapsed time of 60 minutes or more has occurred without a measurable (1 lb or 0.5 kg) weight change in the test fuel charge while the appliance operating control is in the demand mode. Report the evidence and the reasoning used to determine that a test in burn rate Category I cannot be achieved. For example, two unsuccessful attempts to operate at an output rate of 10 percent of the rated output capacity are not sufficient evidence that burn rate Category I cannot be achieved. Note that section 12.5.6 requires that the completed test run burn rate can be no greater than the burn rate expected in home use. If this rate cannot be achieved, the test is not valid.
12.5.7 Appliance Overheating. Appliances shall be capable of operating in all heat output categories without overheating to be rated by this test method. Appliance overheating occurs when the rate of heat withdrawal from the appliance is lower than the rate of heat production when the unit control is in the idle mode. This condition results in the water in the appliance continuing to increase in temperature well above the upper limit setting of the operating control. Evidence of overheating includes: 1 hour or more of appliance water temperature increase above the upper temperature set-point of the operating control, exceeding the temperature limit of a safety control device (independent from the operating control), boiling water in a non-pressurized system or activation of a pressure or temperature relief valve in a pressurized system.
12.6 Additional Test Runs. The testing laboratory may conduct more than one test run in each of the heat output categories specified in section 4.3.1. If more than one test run is conducted at a specified heat output rate, the results from at least two-thirds of the test runs in that heat output rate category shall be used in calculating the weighted average emission rate (See section 14.1.14). The measurement data and results of all test runs shall be reported regardless of which values are used in calculating the weighted average emission rate.
13.0 Calculation of Results
13.1 Nomenclature
E T - Total particulate emissions for the full test run as determined per ASTM E2515-11 (IBR, see §60.17 ) in grams
E g/MJ - Emissions rate in grams per megajoule of heat output
E lb/mmBtu output - Emissions rate in pounds per million Btu of heat output
E g/kg - Emissions factor in grams per kilogram of dry fuel burned
E g/hr - Emissions factor in grams per hour
HHV - Higher heating value of fuel = 8600 Btu/lb (19.990 MJ/kg)
LHV - Lower heating value of fuel = 7988 Btu/lb (18.567 MJ/kg)
ΔT - Temperature difference between water entering and exiting the heat exchanger
Q out - Total heat output in BTU's (megajoules)
Q in - Total heat input available in test fuel charge in BTU (megajoules)
M - Mass flow rate of water in lb/min (kg/min)
V i - Volume of water indicated by a totalizing flow meter at the ith reading in gallons (liters)
V f - Volumetric flow rate of water in heat exchange system in gallons per minute (liters/min)
Θ - Total length of test run in hours
ti - Data sampling interval in minutes
η del - Delivered heating efficiency in percent
Fi - Weighting factor for heat output category i (See Table 2)
T1 - Temperature of water at the inlet on the supply side of the heat exchanger
T2 - Temperature of the water at the outlet on the supply side of the heat exchanger
T3 - Temperature of water at the inlet to the load side of the heat exchanger
TIavg - Average temperature of the appliance and water at start of the test

TF avg - Average temperature of the appliance and water at the end of the test
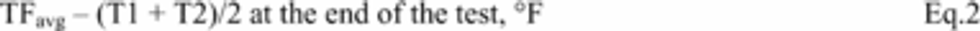
MC - Fuel moisture content in percent dry basis
MC i - Average moisture content of individual 4 × 4 fuel pieces in percent dry basis
MC sp - Moisture content of spacers assumed to be 10 percent dry basis
σ - Density of water in pounds per gallon
C p - Specific heat of water in Btu/lb,°F
C steel - Specific heat of steel (0.1 Btu/lb, ºF)
W fuel - Fuel charge weight in pounds (kg)
W i - Weight of individual fuel 4 × 4 pieces in pounds (kg)
W sp - Weight of all spacers used in a fuel load in pounds (kg)
W app - Weight of empty appliance in pounds
W wa - Weight of water in supply side of the system in pounds
13.2 After the test is completed, determine the particulate emissions E T in accordance with ASTM E2515-11 (IBR, see §60.17 ).
13.3 Determine Average Fuel Load Moisture Content
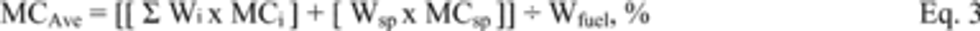
13.4 Determine heat input
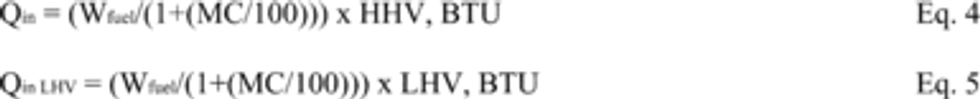
13.5 Determine Heat Output and Efficiency
13.5.1 Determine heat output as:
Q out = Σ [Heat output determined for each sampling time interval] Change in heat stored in the appliance.
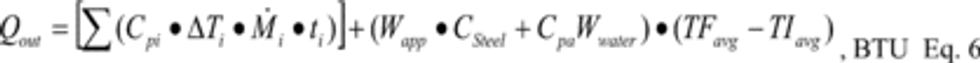
Note:
The subscript (i) indicates the parameter value for sampling time interval t i .
M i = Mass flow rate = gal/min × density of water (lb/gal) = lb/min

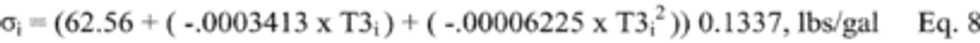
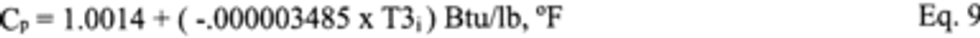
C steel = 0.1 Btu/lb,° F
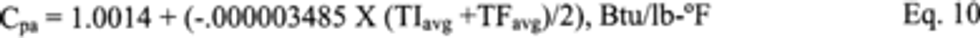

Note:
Vi is the total water volume at the end of interval i and Vi-1 is the total water volume at the beginning of the time interval. This calculation is necessary when a totalizing type water meter is used.
13.5.2 Determine heat output rate as:

13.5.3 Determine emission rates and emission factors as:

13.5.4 Determine delivered efficiency as:

13.5.5 Determine η SLM - Overall Efficiency (SLM) using Stack Loss
For determination of the average overall thermal efficiency (η SLM ) for the test run, use the data collected over the full test run and the calculations in accordance with CSA B415.1-10 (IBR, see §60.17 ), clause 13.7 except for 13.7.2 (e), (f), (g), and (h), use the following average fuel properties for oak: percent C = 50.0, percent H = 6.6, percent O = 43.2, percent ash = 0.2 percent. The averaging period for determination of efficiency by the stack loss method allows averaging over 10 minute time periods for flue gas temperature, flue gas CO 2 , and flue gas CO for the determination of the efficiency. However, under some cycling conditions the “on” period may be short relative to this 10 minute period. For this reason, during cycling operation the averaging period for these parameters may not be longer than the burner on period divided by 10. The averaging period need not be shorter than one minute. During the off period, under cycling operation, the averaging periods specified may be used. Where short averaging times are used, however, the averaging period for fuel consumption may still be at 10 minutes. This average wood consumption rate shall be applied to all of the smaller time intervals included.
13.5.5.1 Whenever the CSA B415.1-10 (IBR, see §60.17 ) overall efficiency is found to be lower than the overall efficiency based on load side measurements, as determined by Eq. 16 of this method, section 14.1.7 of the test report must include a discussion of the reasons for this result.
13.6 Weighted Average Emissions and Efficiency
13.6.1 Determine the weighted average emission rate and delivered efficiency from the individual tests in the specified heat output categories. The weighting factors (Fi) are derived from an analysis of ASHRAE bin data which provides details of normal building heating requirements in terms of percent of design capacity and time in a particular capacity range - or “bin” - over the course of a heating season. The values used in this method represent an average of data from several cities located in the northern United States.
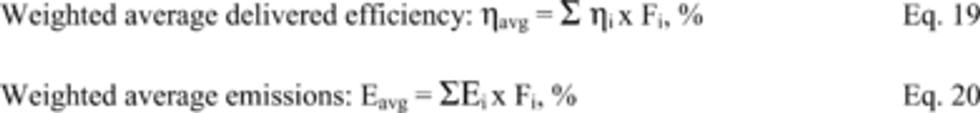
13.7 Average Heat Output (Q out-8hr ) and Efficiency ((η avg-8hr ) for 8 hour burn time.
13.7.1 Units tested under this standard typically require infrequent fuelling, 8 to 12 hours intervals being typical. Rating unit's based on an average output sustainable over an 8 hour duration will assist consumers in appropriately sizing units to match the theoretical heat demand of their application.
13.7.2 Calculations:
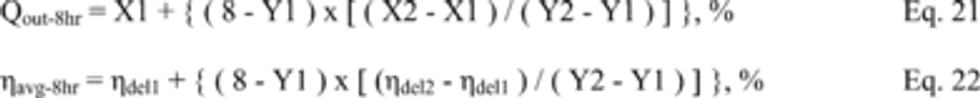
Where:
Y1 = Test duration just above 8 hrs
Y2 = Test duration just below 8 hrs
X1 = Actual load for duration Y1
X2 = Actual load for duration Y2
η del1 = Average delivered efficiency for duration Y1
η del2 = Average delivered efficiency for duration Y2
13.7.2.1 Determine the test durations and actual load for each category as recorded in Table 1A.
13.7.2.2 Determine the data point that has the nearest duration greater than 8 hrs.
X1 = Actual load,
Y1 = Test duration, and
η del1 = Average delivered efficiency for this data point
13.7.2.3 Determine the data point that has the nearest duration less than 8 hours.
X2 = Actual load,
Y2 = Test duration, and
η del2 = Average delivered efficiency for this data point
13.7.2.4 Example:
| (Btu/Hr) | (Hr) | (%) |
|---|---|---|
| 1 15,000 | 10.2 | 70.0 |
| 2 26,000 | 8.4 | 75.5 |
| 3 50,000 | 6.4 | 80.1 |
| 4 100,000 | 4.7 | 80.9 |
Category 2 duration is just above 8 hours, therefore: X1 = 26,000 Btu/hr, η del1 = 75.5% and Y1 = 8.4 hrs
Category 3 duration is just below 8 hours, therefore: X2 = 50,000 Btu/hr, η del2 = 80.1% and Y2 = 6.4 hrs
Q out-8hr = 26,000 {(8 - 8.4) × [(50,000 - 26,000)/(6.4 - 8.4)]} = 30,800 BTU/hr
η avg-8hr = 75.5 {(8 - 8.4) × [(80.1 - 75.5)/(6.4 - 8.4)]} = 76.4%
13.8 Carbon Monoxide Emissions
For each minute of the test period, the carbon monoxide emissions rate (g/min) shall be calculated as:

Total CO emissions for each of the four test periods (CO_ 1 , CO_ 2 , CO_ 3 , CO_ 4 ) shall be calculated as the sum of the emissions rates for each of the 1-minute intervals. Total CO emissions for the test run, CO T , shall be calculated as the sum of CO_ 1 , CO_ 2 , CO_ 3 and CO_ 4 .
14.0 Report
14.1.1 The report shall include the following.
14.1.2 Name and location of the laboratory conducting the test.
14.1.3 A description of the appliance tested and its condition, date of receipt and dates of tests.
14.1.4 A statement that the test results apply only to the specific appliance tested.
14.1.5 A statement that the test report shall not be reproduced except in full, without the written approval of the laboratory.
14.1.6 A description of the test procedures and test equipment including a schematic or other drawing showing the location of all required test equipment. Also, a description of test fuel sourcing, handling and storage practices shall be included.
14.1.7 Details of deviations from, additions to or exclusions from the test method, and their data quality implications on the test results (if any), as well as information on specific test conditions, such as environmental conditions.
14.1.8 A list of participants and observers present for the tests.
14.1.9 Data and drawings indicating the fire box size and location of the fuel charge.
14.1.10 Drawings and calculations used to determine firebox volume.
14.1.11 Information for each test run fuel charge including piece size, moisture content, and weight.
14.1.12 All required data for each test run shall be provided in spreadsheet format. Formulae used for all calculations shall be accessible for review.
14.1.13 Test run duration for each test.
14.1.14 Calculated results for delivered efficiency at each burn rate and the weighted average emissions reported as total emissions in grams, pounds per mm Btu of delivered heat, grams per MJ of delivered heat, grams per kilogram of dry fuel and grams per hour. Results shall be reported for each heat output category and the weighted average.
14.1.15 Tables 1A, 1B, 1C and Table 2 must be used for presentation of results in test reports.
14.1.16 A statement of the estimated uncertainty of measurement of the emissions and efficiency test results.
14.1.17 Raw data, calibration records, and other relevant documentation shall be retained by the laboratory for a minimum of 7 years.
15.0 Precision and Bias
15.1 Precision - It is not possible to specify the precision of the procedure in Method 28WHH because the appliance operation and fueling protocols and the appliances themselves produce variable amounts of emissions and cannot be used to determine reproducibility or repeatability of this measurement method.
15.2 Bias - No definitive information can be presented on the bias of the procedure in Method 28WHH for measuring solid fuel burning hydronic heater emissions because no material having an accepted reference value is available.
16.0 Keywords
16.1 Solid fuel, hydronic heating appliances, wood-burning hydronic heaters.
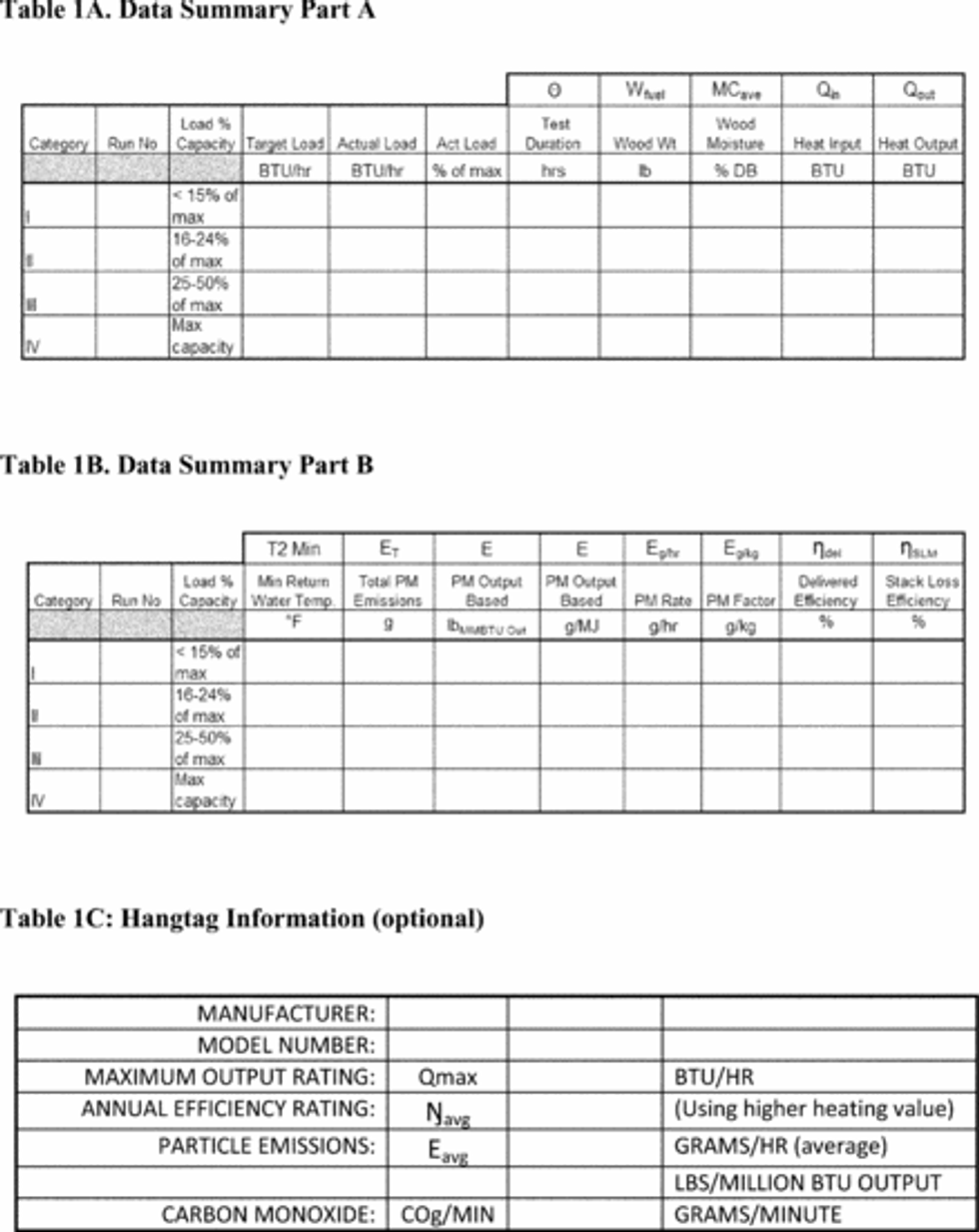
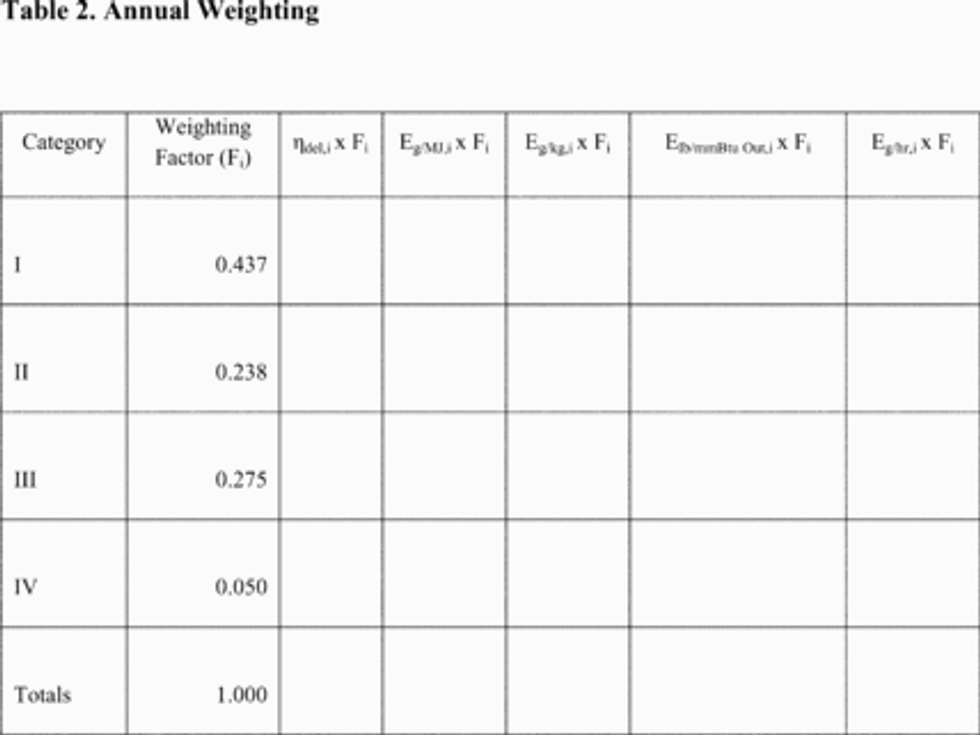
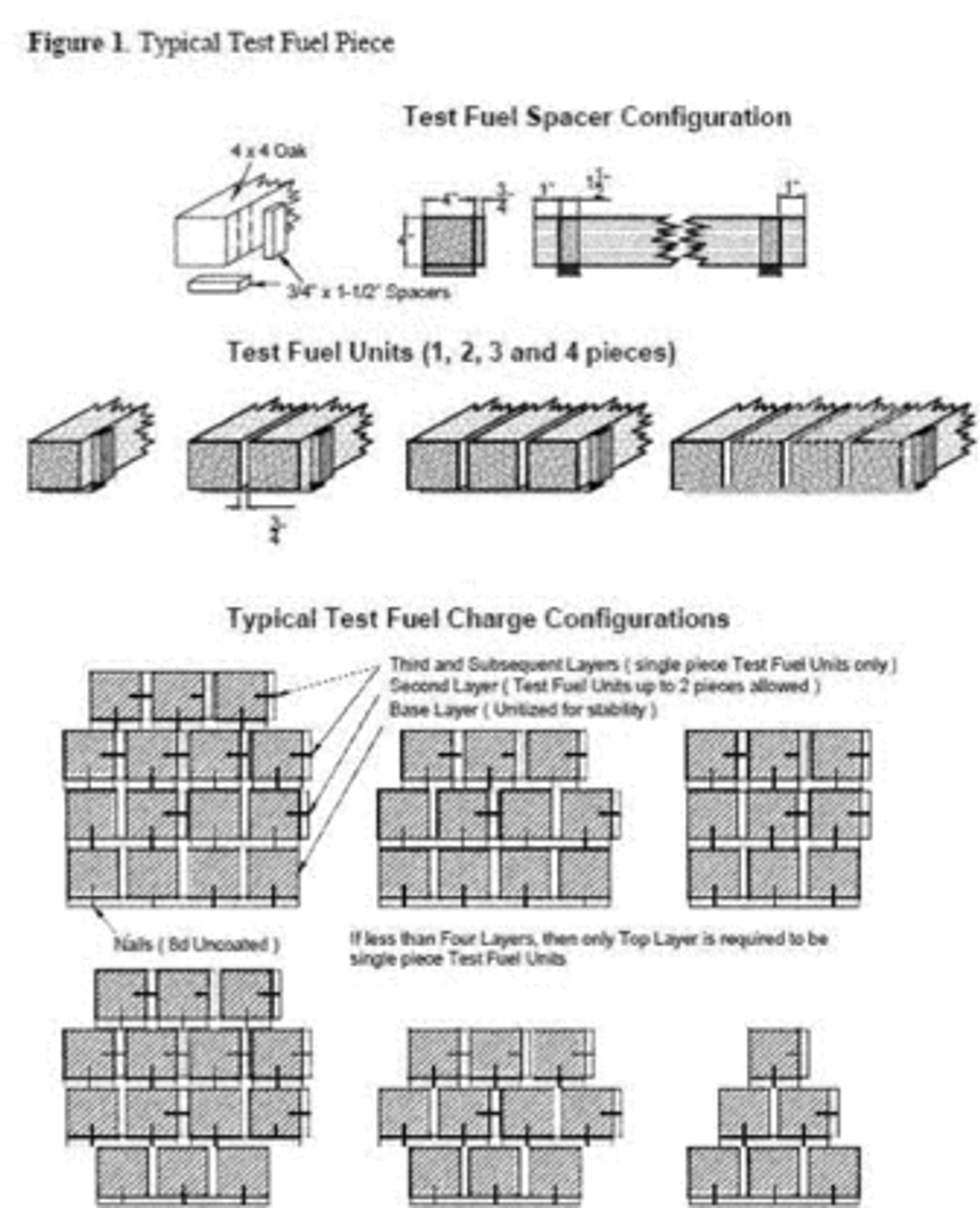
Test Method 28WHH for Certification of Cord Wood-Fired Hydronic Heating Appliances With Partial Thermal Storage: Measurement of Particulate Matter (PM) and Carbon Monoxide (CO) Emissions and Heating Efficiency of Wood-Fired Hydronic Heating Appliances With Partial Thermal Storage
1.0 Scope and Application
1.1 This test method applies to wood-fired hydronic heating appliances with heat storage external to the appliance. The units typically transfer heat through circulation of a liquid heat exchange media such as water or a water-antifreeze mixture. Throughout this document, the term “water” will be used to denote any of the heat transfer liquids approved for use by the manufacturer.
1.2 The test method measures PM and CO emissions and delivered heating efficiency at specified heat output rates referenced against the appliance's rated heating capacity as specified by the manufacturer and verified under this test method.
1.3 PM emissions are measured by the dilution tunnel method as specified in the EPA Method 28WHH and the standards referenced therein with the exceptions noted in section 12.5.9. Delivered efficiency is measured by determining the fuel energy input and appliance output. Heat output is determined through measurement of the flow rate and temperature change of water circulated through a heat exchanger external to the appliance and the increase in energy of the external storage. Heat input is determined from the mass of dry wood fuel and its higher heating value (HHV). Delivered efficiency does not attempt to account for pipeline loss.
1.4 Products covered by this test method include both pressurized and non-pressurized hydronic heating appliances intended to be fired with wood and for which the manufacturer specifies for indoor or outdoor installation. The system, which includes the heating appliance and external storage, is commonly connected to a heat exchanger by insulated pipes and normally includes a pump to circulate heated liquid. These systems are used to heat structures such as homes, barns and greenhouses. They also provide heat for domestic hot water, spas and swimming pools.
1.5 Distinguishing features of products covered by this standard include:
1.5.1 The manufacturer specifies the application for either indoor or outdoor installation.
1.5.2 A firebox with an access door for hand loading of fuel.
1.5.3 Typically an aquastat mounted as part of the appliance that controls combustion air supply to maintain the liquid in the appliance within a predetermined temperature range provided sufficient fuel is available in the firebox. The appliance may be equipped with other devices to control combustion.
1.5.4 A chimney or vent that exhausts combustion products from the appliance.
1.5.5 A liquid storage system, typically water, which is not large enough to accept all of the heat produced when a full load of wood is burned and the storage system starts a burn cycle at 125°F.
1.5.6 The heating appliances require external thermal storage and these units will only be installed as part of a system which includes thermal storage. The manufacturer specifies the minimum amount of thermal storage required. However, the storage system shall be large enough to ensure that the boiler (heater) does not cycle, slumber, or go into an off-mode when operated in a Category III load condition (See section 4.3).
1.6 The values stated are to be regarded as the standard whether in I-P or SI units. The values given in parentheses are for information only.
2.0 Summary of Method and References
2.1 PM and CO emissions are measured from a wood-fired hydronic heating appliance burning a prepared test fuel charge in a test facility maintained at a set of prescribed conditions. Procedures for determining heat output rates, PM and CO emissions, and efficiency and for reducing data are provided.
2.2 Referenced Documents
2.2.1 EPA Standards
2.2.1.1 Method 28 Certification and Auditing of Wood Heaters
2.2.1.2 Method 28WHH Measurement of Particulate Emissions and Heating Efficiency of Wood-Fired Hydronic Heating Appliances and the Standards Referenced therein.
2.2.2 Other Standards
2.2.2.1 CSA-B415.1-10 Performance Testing of Solid-Fuel-Burning Heating Appliances
3.0 Terminology
3.1 Definitions
3.1.1 Hydronic Heating - A heating system in which a heat source supplies energy to a liquid heat exchange media such as water that is circulated to a heating load and returned to the heat source through pipes.
3.1.2 Aquastat - A control device that opens or closes a circuit to control the rate of fuel consumption in response to the temperature of the heating media in the heating appliance.
3.1.3 Delivered Efficiency - The percentage of heat available in a test fuel charge that is delivered to a simulated heating load or the storage system as specified in this test method.
3.1.4 Emission Factor - The emission of a pollutant expressed in mass per unit of energy (typically) output from the boiler/heater.
3.1.5 Emission Index - The emission of a pollutant expressed in mass per unit mass of fuel used.
3.1.6 Emission Rate - The emission of a pollutant expressed in mass per unit time
3.1.7 Manufacturer's Rated Heat Output Capacity - The value in Btu/hr (MJ/hr) that the manufacturer specifies that a particular model of hydronic heating appliance is capable of supplying at its design capacity as verified by testing, in accordance with section 12.5.4.
3.1.8 Heat Output Rate - The average rate of energy output from the appliance during a specific test period in Btu/hr (MJ/hr).
3.1.9 Firebox - The chamber in the appliance in which the test fuel charge is placed and combusted.
3.1.10 NIST - National Institute of Standards and Technology.
3.1.11 Test Fuel Charge - The collection of test fuel placed in the appliance at the start of the emission test run.
3.1.12 Test Run - An individual emission test which encompasses the time required to consume the mass of the test fuel charge. The time of the test run also considers the time for the energy to be drawn from the thermal storage.
3.1.13 Test Run Under “Cold-to-Cold” Condition - Under this test condition the test fuel is added into an empty chamber along with kindling and ignition materials (paper). The boiler/heater at the start of this test is typically 125° to 130°F.
3.1.14 Test Run Under “Hot-to-Hot” Condition - Under this test condition the test fuel is added onto a still-burning bed of charcoals produced in a pre-burn period. The boiler/heater water is near its operating control limit at the start of the test.
3.1.15 Overall Efficiency, also known as Stack Loss Efficiency - The efficiency for each test run as determined using the CSA B415.1-10 (IBR, see §60.17 ) stack loss method (SLM).
3.1.16 Phases of a Burn Cycle - The “startup phase” is defined as the period from the start of the test until 15 percent of the test fuel charge is consumed. The “steady-state phase” is defined as the period from the end of the startup phase to a point at which 80 percent of the test fuel charge is consumed. The “end phase” is defined as the time from the end of the steady-state period to the end of the test.
3.1.17 Thermopile - A device consisting of a number of thermocouples connected in series, used for measuring differential temperature.
3.1.18 Slumber Mode - This is a mode in which the temperature of the water in the boiler/heater has exceeded the operating control limit and the control has changed the boiler/heater fan speed, dampers, and/or other operating parameters to minimize the heat output of the boiler/heater.
4.0 Summary of Test Method
4.1 Dilution Tunnel. Emissions are determined using the “dilution tunnel” method specified in EPA Method 28WHH and the standards referenced therein. The flow rate in the dilution tunnel is maintained at a constant level throughout the test cycle and accurately measured. Samples of the dilution tunnel flow stream are extracted at a constant flow rate and drawn through high efficiency filters. The filters are dried and weighed before and after the test to determine the emissions collected and this value is multiplied by the ratio of tunnel flow to filter flow to determine the total particulate emissions produced in the test cycle.
4.2 Efficiency. The efficiency test procedure takes advantage of the fact that this type of system delivers heat through circulation of the heated liquid (water) from the system to a remote heat exchanger ( e.g. baseboard radiators in a room) and back to the system. Measurements of the cooling water temperature difference as it enters and exits the test system heat exchanger along with the measured flow rate allow for an accurate determination of the useful heat output of the appliance. Also included in the heat output is the change in the energy content in the storage system during a test run. Energy input to the appliance during the test run is determined by weight of the test fuel charge, adjusted for moisture content, multiplied by the higher heating value. Additional measurements of the appliance weight and temperature at the beginning and end of a test cycle are used to correct for heat stored in the appliance. Overall efficiency (SLM) is determined using the CSA B415.1-10 (IBR, see §60.17 ) stack loss method for data quality assurance purposes.
4.3 Operation. Four test categories are defined for use in this method. These are:
4.3.1 Category I: A heat output of 15 percent or less of manufacturer's rated heat output capacity.
4.3.2 Category II: A heat output of 16 percent to 24 percent of manufacturer's rated heat output capacity.
4.3.3 Category III: A heat output of 25 percent to 50 percent of manufacturer's rated heat output capacity.
4.3.4 Category IV: Manufacturer's Rated Heat Output Capacity. These heat output categories refer to the output from the system by way of the load heat exchanger installed for the test. The output from just the boiler/heater part of the system may be higher for all or part of a test, as part of this boiler/heater output goes to storage.
For the Category III and IV runs, appliance operation is conducted on a hot-to-hot test cycle meaning that the appliance is brought to operating temperature and a coal bed is established prior to the addition of the test fuel charge and measurements are made for each test fuel charge cycle. The measurements are made under constant heat draw conditions within pre-determined ranges. No attempt is made to modulate the heat demand to simulate an indoor thermostat cycling on and off in response to changes in the indoor environment.
For the Category I and II runs, the unit is tested with a “cold start.” At the manufacturer's option, the Category II and III runs may be waived and it may be assumed that the particulate emission values and efficiency values determined in the startup, steady-state, and end phases of Category I are applicable in Categories II and III for the purpose of determining the annual averages in lb/mmBtu and g/MJ (See section 13). For the annual average in g/hr, the length of time for stored heat to be drawn from thermal storage shall be determined for the test load requirements of the respective category.
All test operations and measurements shall be conducted by personnel of the laboratory responsible for the submission of the test report.
5.0 Significance and Use
5.1 The measurement of particulate matter emission and CO rates is an important test method widely used in the practice of air pollution control.
5.1.1 These measurements, when approved by state or federal agencies, are often required for the purpose of determining compliance with regulations and statutes.
5.1.2 The measurements made before and after design modifications are necessary to demonstrate the effectiveness of design changes in reducing emissions and make this standard an important tool in manufacturers' research and development programs.
5.2 Measurement of heating efficiency provides a uniform basis for comparison of product performance that is useful to the consumer. It is also required to relate emissions produced to the useful heat production.
5.3 This is a laboratory method and is not intended to be fully representative of all actual field use. It is recognized that users of hand-fired, wood-burning equipment have a great deal of influence over the performance of any wood-burning appliance. Some compromises in realism have been made in the interest of providing a reliable and repeatable test method.
6.0 Test Equipment
6.1 Scale. A platform scale capable of weighing the boiler/heater under test and associated parts and accessories when completely filled with water to an accuracy of ±1.0 pound (±0.5 kg) and a readout resolution of ±0.2 pound (±0.1 kg).
6.2 Heat Exchanger. A water-to-water heat exchanger capable of dissipating the expected heat output from the system under test.
6.3 Water Temperature Difference Measurement. A Type - T 'special limits' thermopile with a minimum of 5 pairs of junctions shall be used to measure the temperature difference in water entering and leaving the heat exchanger. The temperature difference measurement uncertainty of this type of thermopile is equal to or less than ±0.50°F (±0.25°C). Other temperature measurement methods may be used if the temperature difference measurement uncertainty is equal to or less than ±0.50°F (±0.25°C). This measurement uncertainty shall include the temperature sensor, sensor well arrangement, piping arrangements, lead wire, and measurement/recording system. The response time of the temperature measurement system shall be less than half of the time interval at which temperature measurements are recorded.
6.4 Water Flow Meter. A water flow meter shall be installed in the inlet to the load side of the heat exchanger. The flow meter shall have an accuracy of ±1 percent of measured flow.
6.4.1 Optional - Appliance Side Water Flow Meter. A water flow meter with an accuracy of ±1 percent of the flow rate is recommended to monitor supply side water flow rate.
6.5 Optional Recirculation Pump. Circulating pump used during test to prevent stratification, in the boiler/heater, of liquid being heated.
6.6 Water Temperature Measurement. Thermocouples or other temperature sensors to measure the water temperature at the inlet and outlet of the load side of the heat exchanger must meet the calibration requirements specified in 10.1 of this method.
6.7 Lab Scale. For measuring the moisture content of wood slices as part of the overall wood moisture determination. Accuracy of ±0.01 pounds.
6.8 Flue Gas Temperature Measurement. Must meet the requirements of CSA B415.1-10 (IBR, see §60.17 ), clause 6.2.2.
6.9 Test Room Temperature Measurement. Must meet the requirements of CSA B415.1-10 (IBR, see §60.17 ), clause 6.2.1.
6.10 Flue Gas Composition Measurement. Must meet the requirements of CSA B415.1-10 (IBR, see §60.17 ), clauses 6.3.1 through 6.3.3.
6.11 Dilution Tunnel CO Measurement. In parallel with the flue gas composition measurements, the CO concentration in the dilution tunnel shall also be measured and reported at time intervals not to exceed one minute. This analyzer shall meet the zero and span drift requirements of CSA B415.1-10 (IBR, see §60.17 ). In addition the measurement repeatability shall be better than ±15 ppm over the range of CO levels observed in the dilution tunnel.
7.0 Safety
7.1 These tests involve combustion of wood fuel and substantial release of heat and products of combustion. The heating system also produces large quantities of very hot water and the potential for steam production and system pressurization. Appropriate precautions must be taken to protect personnel from burn hazards and respiration of products of combustion.
8.0 Sampling, Test Specimens and Test Appliances
8.1 Test specimens shall be supplied as complete appliances, as described in marketing materials, including all controls and accessories necessary for installation in the test facility. A full set of specifications, installation and operating instructions, and design and assembly drawings shall be provided when the product is to be placed under certification of a third-party agency. The manufacturer's written installation and operating instructions are to be used as a guide in the set-up and testing of the appliance and shall be part of the test record.
8.2 The size, connection arrangement, and control arrangement for the thermal storage shall be as specified in the manufacturer's documentation. It is not necessary to use the specific storage system that the boiler/heater will be marketed with. However, the capacity of the system used in the test cannot be greater than that specified as the minimum allowable for the boiler/heater.
8.3 All system control settings shall be the as-shipped, default settings. These default settings shall be the same as those communicated in a document to the installer or end user. These control settings and the documentation of the control settings as to be provided to the installer or end user shall be part of the test record.
8.4 Where the manufacturer defines several alternatives for the connection and loading arrangement, one shall be defined in the appliance documentation as the default or standard installation. It is expected that this will be the configuration for use with a simple baseboard heating system. This is the configuration to be followed for these tests. The manufacturer's documentation shall define the other arrangements as optional or alternative arrangements.
9.0 Preparation of Test Equipment
9.1 The appliance is to be placed on a scale capable of weighing the appliance fully loaded with a resolution of ±0.2 lb (0.1 kg).
9.2 The appliance shall be fitted with the type of chimney recommended or provided by the manufacturer and extending to 15 ±0.5 feet (4.6 ±0.15 m) from the upper surface of the scale. If no flue or chimney system is recommended or provided by the manufacturer, connect the appliance to a flue of a diameter equal to the flue outlet of the appliance. The flue section from the appliance flue collar to 8 ±0.5 feet above the scale shall be single wall stove pipe and the remainder of the flue shall be double wall insulated class A chimney.
9.3 Optional Equipment Use
9.3.1 A recirculation pump may be installed between connections at the top and bottom of the appliance to minimize thermal stratification if specified by the manufacturer. The pump shall not be installed in such a way as to change or affect the flow rate between the appliance and the heat exchanger.
9.3.2 If the manufacturer specifies that a thermal control valve or other device be installed and set to control the return water temperature to a specific set point, the valve or other device shall be installed and set per the manufacturer's written instructions.
9.4 Prior to filling the boiler/heater with water, weigh and record the appliance mass.
9.5 Heat Exchanger
9.5.1 Plumb the unit to a water-to-water heat exchanger with sufficient capacity to draw off heat at the maximum rate anticipated. Route hoses and electrical cables and instrument wires in a manner that does not influence the weighing accuracy of the scale as indicated by placing dead weights on the platform and verifying the scale's accuracy.
9.5.2 Locate thermocouples to measure the water temperature at the inlet and outlet of the load side of the heat exchanger.
9.5.3 Install a thermopile (or equivalent instrumentation) meeting the requirements of section 6.3 to measure the water temperature difference between the inlet and outlet of the load side of the heat exchanger
9.5.4 Install a calibrated water flow meter in the heat exchanger load side supply line. The water flow meter is to be installed on the cooling water inlet side of the heat exchanger so that it will operate at the temperature at which it is calibrated.
9.5.5 Place the heat exchanger in a box with 2 in. (50 mm) of expanded polystyrene (EPS) foam insulation surrounding it to minimize heat losses from the heat exchanger.
9.5.6 The reported efficiency and heat output rate shall be based on measurements made on the load side of the heat exchanger.
9.5.7 Temperature instrumentation per section 6.6 shall be installed in the appliance outlet and return lines. The average of the outlet and return water temperature on the supply side of the system shall be considered the average appliance temperature for calculation of heat storage in the appliance ( TF avg and TI avg ). Installation of a water flow meter in the supply side of the system is optional.
9.6 Storage Tank. The storage tank shall include a destratification pump as illustrated in Figure 1. The pump will draw from the bottom of the tank and return to the top as illustrated. Temperature sensors (TS1 and TS2 in Figure 1) shall be included to measure the temperature in the recirculation loop. The valve plan in Figure 1 allows the tank recirculation loop to operate and the boiler/heater-to-heat exchanger loop to operate at the same time but in isolation. This would typically be done before the start of a test or following completion of a test to determine the end of test average tank temperature. The nominal flow rate in the storage tank recirculation loop can be estimated based on pump manufacturers' performance curves and any significant restriction in the recirculation loop.
9.7 Fill the system with water. Determine the total weight of the water in the appliance when the water is circulating. Verify that the scale indicates a stable weight under operating conditions. Make sure air is purged properly.
10.0 Calibration and Standardization
10.1 Water Temperature Sensors. Temperature measuring equipment shall be calibrated before initial use and at least semi-annually thereafter. Calibrations shall be in compliance with National Institute of Standards and Technology (NIST) Monograph 175, Standard Limits of Error.
10.2 Heat Exchanger Load Side Water Flow Meter.
10.2.1 The heat exchanger load side water flow meter shall be calibrated within the flow range used for the test run using NIST-traceable methods. Verify the calibration of the water flow meter before and after each test run and at least once during each test run by comparing the water flow rate indicated by the flow meter to the mass of water collected from the outlet of the heat exchanger over a timed interval. Volume of the collected water shall be determined based on the water density calculated from section 13, Eq. 12, using the water temperature measured at the flow meter. The uncertainty in the verification procedure used shall be 1 percent or less. The water flow rate determined by the collection and weighing method shall be within 1 percent of the flow rate indicated by the water flow meter.
10.3 Scales. The scales used to weigh the appliance and test fuel charge shall be calibrated using NIST-traceable methods at least once every 6 months.
10.4 Flue Gas Analyzers - In accordance with CSA B415.1-10 (IBR, see §60.17 ), clause 6.8.
11.0 Conditioning
11.1 Prior to testing, an appliance is to be operated for a minimum of 50 hours using a medium heat draw rate. The conditioning may be at the manufacturer's facility prior to the certification test. If the conditioning is at the certification test laboratory, the pre-burn for the first test can be included as part of the conditioning requirement. If conditioning is included in pre-burn, then the appliance shall be aged with fuel meeting the specifications outlined in section 12.2 with a moisture content between 19 and 25 percent on a dry basis. Operate the appliance at a medium heat output rate (Category II or III) for at least 10 hours for non-catalytic appliances and 50 hours for catalytic appliances. Record and report hourly flue gas exit temperature data and the hours of operation. The aging procedure shall be conducted and documented by a testing laboratory.
12.0 Procedure
12.1 Appliance Installation. Assemble the appliance and parts in conformance with the manufacturer's written installation instructions. Clean the flue with an appropriately sized, wire chimney brush before each certification test series.
12.2 Fuel. Test fuel charge fuel shall be red ( Quercus ruba L. ) or white ( Quercus Alba ) oak 19 to 25 percent moisture content on a dry basis. Piece length shall be 80 percent of the firebox depth rounded down to the nearest 1 inch (25mm) increment. For example, if the firebox depth is 46 inches (1168mm) the piece length would be 36 inches (46 inches × 0.8 = 36.8 inches, rounded down to 36 inches). Pieces are to be placed in the firebox parallel to the longest firebox dimension. For fireboxes with sloped surfaces that create a non-uniform firebox length, the piece length shall be adjusted for each layer based on 80 percent of the length at the level where the layer is placed. The test fuel shall be cord wood with cross section dimensions and weight limits as defined in CSA B415.1-10 (IBR, see §60.17 ), section 8.3, Table 4. The use of dimensional lumber is not allowed.
12.2.1 Select three pieces of cord wood from the same batch of wood as the test fuel and the same weight as the average weight of the pieces in the test load ±1.0 lb. From each of these three pieces, cut three slices. Each slice shall be 1/2 inch to 3/4 inch thick. One slice shall be cut across the center of the length of the piece. The other two slices shall be cut half way between the center and the end. Immediately measure the mass of each piece in pounds. Dry each slice in an oven at 220°F for 24 hours or until no further weight change occurs. The slices shall be arranged in the oven so as to provide separation between faces. Remove from the oven and measure the mass of each piece again as soon as practical, in pounds.
The moisture content of each slice, on a dry basis, shall be calculated as:

Where:
W SliceWet = weight of the slice before drying in pounds
W SliceDry = weight of the slice after drying in pounds
MC Slice = moisture content of the slice in % dry basis
The average moisture content of the entire test load (MC) shall be determined using Eq. 6. Each individual slice shall have a moisture content in the range of 18 percent to 28 percent on a dry basis. The average moisture content for the test fuel load shall be in the range of 19 percent to 25 percent. Moisture shall not be added to previously dried fuel pieces except by storage under high humidity conditions and temperature up to 100°F. Fuel moisture measurement shall begin within 4 hours of using the fuel batch for a test. Use of a pin-type meter to estimate the moisture content prior to a test is recommended.
12.2.2 Firebox Volume. Determine the firebox volume in cubic feet. Firebox volume shall include all areas accessible through the fuel loading door where firewood could reasonably be placed up to the horizontal plane defined by the top of the loading door. A drawing of the firebox showing front, side and plan views or an isometric view with interior dimensions shall be provided by the manufacturer and verified by the laboratory. Calculations for firebox volume from computer aided design (CAD) software programs are acceptable and shall be included in the test report if used. If the firebox volume is calculated by the laboratory the firebox drawings and calculations shall be included in the test report.
12.2.3 Test Fuel charge. Test fuel charges shall be determined by multiplying the firebox volume by 10 pounds (4.54 kg) per ft 3 (28L), or a higher load density as recommended by the manufacturer's printed operating instructions, of wood (as used wet weight). Select the number of pieces of cord wood that most nearly match this target weight. However, the test fuel charge cannot be less than the target of 10 pounds (4.54 kg) per ft 3 (28L).
12.3 Sampling Equipment. Prepare the particulate emission sampling equipment as defined by EPA Method 28WHH and the standards referenced therein.
12.4 Appliance Startup. The appliance shall be fired with wood fuel of any species, size and moisture content, at the laboratory's discretion, to bring it up to operating temperature. Operate the appliance until the water is heated to the upper operating control limit and has cycled at least two times. Then remove all unburned fuel, zero the scale and verify the scales accuracy using dead weights.
12.4.1 Startup Procedure for Category III and IV Test Runs, “Hot-to-Hot.”
12.4.1.1 Pretest t Burn Cycle. Following appliance startup (section 12.4), reload appliance with oak cord wood and allow it to burn down to the specified coal bed weight. The pre-test burn cycle fuel charge weight shall be within ±10 percent of the test fuel charge weight. Piece size and length shall be selected such that charcoalization is achieved by the time the fuel charge has burned down to the required coal bed weight. Pieces with a maximum thickness of approximately 2 inches have been found to be suitable. Charcoalization is a general condition of the test fuel bed evidenced by an absence of large pieces of burning wood in the coal bed and the remaining fuel pieces being brittle enough to be broken into smaller charcoal pieces with a metal poker. Manipulations to the fuel bed prior to the start of the test run are to be done to achieve charcoalization while maintaining the desired heat output rate. During the pre-test burn cycle and at least one hour prior to starting the test run, adjust water flow to the heat exchanger to establish the target heat draw for the test. For the first test run the heat draw rate shall be equal to the manufacturer's rated heat output capacity.
12.4.1.2 Allowable Adjustments. Fuel addition or subtractions, and coal bed raking shall be kept to a minimum but are allowed up to 15 minutes prior to the start of the test run. For the purposes of this method, coal bed raking is the use of a metal tool (poker) to stir coals, break burning fuel into smaller pieces, dislodge fuel pieces from positions of poor combustion, and check for the condition of charcoalization. Record all adjustments to and additions or subtractions of fuel, and any other changes to the appliance operations that occur during pretest ignition period. During the 15-minute period prior to the start of the test run, the wood heater loading door shall not be open more than a total of 1 minute. Coal bed raking is the only adjustment allowed during this period.
12.4.1.3 Coal Bed Weight. The appliance is to be loaded with the test fuel charge when the coal bed weight is between 10 percent and 20 percent of the test fuel charge weight. Coals may be raked as necessary to level the coal bed but may only be raked and stirred once between 15 to 20 minutes prior to the addition of the test fuel charge.
12.4.1.4 Storage. The Category III and IV test runs may be done either with or without the thermal storage. If thermal storage is used, the initial temperature of the storage must be 125°F or greater at the start of the test. The storage may be heated during the pre-test burn cycle or it may be heated by external means. If thermal storage is used, prior to the start of the test run, the storage tank destratification pump, shown in Figure 1, shall be operated until the total volume pumped exceeds 1.5 times the tank volume and the difference between the temperature at the top and bottom of the storage tank (TS 1 and TS 2 ) is less than 1°F. These two temperatures shall then be recorded to determine the starting average tank temperature. The total volume pumped may be based on the nominal flow rate of the destratification pump (See section 9.6). If the Category III and IV runs are done with storage, it is recognized that during the last hour of the pre-burn cycle the storage tank must be mixed to achieve a uniform starting temperature and cannot receive heat from the boiler/heater during this time. During this time period, the boiler/heater might cycle or go into a steady reduced output mode. (Note - this would happen, for example, in a Category IV run if the actual maximum output of the boiler/heater exceed the manufacturer's rated output.) A second storage tank may be used temporarily to enable the boiler/heater to operate during this last hour of the pre-burn period as it will during the test period. The temperature of this second storage tank is not used in the calculations but the return water to the boiler/heater (after mixing device if used) must be 125°F or greater.
12.4.2 Startup Procedure for Category I and II Test Runs, “Cold-to-Cold.”
12.4.2.1 Initial Temperatures. This test shall be started with both the boiler/heater and the storage at a minimum temperature of 125°F. The boiler/heater maximum temperature at the start of this test shall be 135°F. The boiler/heater and storage may be heated through a pre-burn or it may be heated by external means.
12.4.2.2 Firebox Condition at Test Start. Prior to the start of this test remove all ash and charcoal from the combustion chamber(s). The loading of the test fuel and kindling should follow the manufacturer's recommendations, subject to the following constraints: Up to 10 percent kindling and paper may be used which is in addition to the fuel load. Further, up to 10 percent of the fuel load ( i.e., included in the 10 lb/ft 3 ) may be smaller than the main fuel. This startup fuel shall still be larger than 2 inches.
12.4.2.3 Storage. The Category I and II test runs shall be done with thermal storage. The initial temperature of the storage must be 125°F or greater at the start of the test. The storage may be heated during the pre-test burn cycle or it may be heated by external means. Prior to the start of the test run, the storage tank destratification pump, shown in Figure 1, shall be operated until the total volume pumped exceeds 1.5 times the tank volume and the difference between the temperature at the top and bottom of the storage tank (TS 1 and TS 2 ) is less than 1°F. These two temperatures shall then be recorded to determine the starting average tank temperature. The total volume pumped may be based on the nominal flow rate of the destratification pump (See section 9.6).
12.5 Test Runs. For all test runs, the return water temperature to the hydronic heater must be equal to or greater than 120°F (this is lower than the initial tank temperature to allow for any pipeline losses). Where the storage system is used, flow of water from the boiler/heater shall be divided between the storage tank and the heat exchanger such that the temperature change of the circulating water across the heat exchanger shall be 30 ±5°F, averaged over the entire test run. This is typically adjusted using the system valves.
Complete a test run in each heat output rate category, as follows:
12.5.1 Test Run Start. For Category III and IV runs: Once the appliance is operating normally and the pretest coal bed weight has reached the target value per section 12.4.1, tare the scale and load the full test charge into the appliance. Time for loading shall not exceed 5 minutes. The actual weight of the test fuel charge shall be measured and recorded within 30 minutes prior to loading. Start all sampling systems.
For Category I and II runs: Once the appliance has reached the starting temperature, tare the scale and load the full test charge, including kindling into the appliance. The actual weight of the test fuel charge shall be measured and recorded within 30 minutes prior to loading. Light the fire following the manufacturer's written normal startup procedure. Start all sampling systems.
12.5.1.1 Record all water temperatures, differential water temperatures and water flow rates at time intervals of one minute or less.
12.5.1.2 Record particulate emissions data per the requirements of EPA Method 28WHH and the standards referenced therein.
12.5.1.3 Record data needed to determine overall efficiency (SLM) per the requirements of CSA B415.1-10 (IBR, see §60.17 ) clauses 6.2.1, 6.2.2, 6.3, 8.5.7, 10.4.3(a), 10.4.3(f), and 13.7.9.3
12.5.1.3.1 Measure and record the test room air temperature in accordance with the requirements of CSA B415.1-10 (IBR, see §60.17 ), clauses 6.2.1, 8.5.7 and 10.4.3(g).
12.5.1.3.2 Measure and record the flue gas temperature in accordance with the requirements of CSA B415.1-10 (IBR, see §60.17 ), clauses 6.2.2, 8.5.7 and 10.4.3(f).
12.5.1.3.3 Determine and record the carbon monoxide (CO) and carbon dioxide (CO 2 ) concentrations in the flue gas in accordance with CSA B415.1-10 (IBR, see §60.17 ), clauses 6.3, 8.5.7 and 10.4.3(i) and (j).
12.5.1.3.4 Measure and record the test fuel weight per the requirements of CSA B415.1-10 (IBR, see §60.17 ), clauses 8.5.7 and 10.4.3(h).
12.5.1.3.5 Record the test run time per the requirements of CSA B415.1-10 (IBR, see §60.17 ), clause 10.4.3(a).
12.5.1.3.6 Record and document all settings and adjustments, if any, made to the boiler/heater as recommended/required by manufacturer's instruction manual for different combustion conditions or heat loads. These may include temperature setpoints, under and over-fire air adjustment, or other adjustments that could be made by an operator to optimize or alter combustion. All such settings shall be included in the report for each test run.
12.5.1.4 Monitor the average heat output rate on the load side of the heat exchanger based on water temperatures and flow. If the heat output rate over a 10 minute averaging period gets close to the upper or lower limit of the target range (±5 percent), adjust the water flow through the heat exchanger to compensate. Make changes as infrequently as possible while maintaining the target heat output rate. The first test run shall be conducted at the Category IV heat output rate to validate that the appliance is capable of producing the manufacturer's rated heat output capacity.
12.5.2 Test Fuel Charge Adjustment. It is acceptable to adjust the test fuel charge ( i.e., reposition) once during a test run if more than 60 percent of the initial test fuel charge weight has been consumed and more than 10 minutes have elapsed without a measurable (1 lb or 0.5 kg) weight change while the operating control is in the demand mode. The time used to make this adjustment shall be less than 60 seconds.
12.5.3 Test Run Completion. For the Category III and IV, “hot-to-hot” test runs, the test run is completed when the remaining weight of the test fuel charge is 0.0 lb (0.0 kg). ( W FuelBurned = W fuel ) End the test run when the scale has indicated a test fuel charge weight of 0.0 lb (0.0 kg) or less for 30 seconds.
For the Category I and II “cold-to-cold” test runs, the test run is completed; and the end of a test is defined at the first occurrence of any one of the following:
(a) The remaining weight of the test fuel charge is less than 1 percent of the total test fuel weight ( W FuelBurned > 0.99 · W fuel );
(b) The automatic control system on the boiler/heater switches to an off mode. In this case, the boiler/heater fan (if used) is typically stopped and all air flow dampers are closed by the control system. Note that this off mode cannot be an “overheat” or emergency shutdown which typically requires a manual reset; or
(c) If the boiler/heater does not have an automatic off mode: After 90 percent of the fuel load has been consumed and the scale has indicated a rate of change of the test fuel charge of less than 1.0 lb/hr for a period of 10 minutes or longer. Note - this is not considered “stopped fuel combustion,” See section 12.5.6.1.
12.5.3.1 At the end of the test run, stop the particulate sampling train and overall efficiency (SLM) measurements, and record the run time, and all final measurement values.
12.5.3.2 At the end of the test run, continue to operate the storage tank destratification pump until the total volume pumped exceeds 1.5 times the tank volume. The maximum average of the top and bottom temperatures measured after this time may be taken as the average tank temperature at the end of the tests (TFSavg, See section 13.1). The total volume pumped may be based on the nominal flow rate of the destratification pump (See section 9.6).
12.5.3.3 For the Category I and II test runs, there is a need to determine the energy content of the unburned fuel remaining in the chamber if the remaining mass in the chamber is greater than 1 percent of the test fuel weight. Following the completion of the test, as soon as safely practical, this remaining fuel is removed from the chamber, separated from the remaining ash and weighed. This separation could be implemented with a slotted “scoop” or similar tool. A 1/4 inch opening size in the separation tool shall be used to separate the ash and charcoal. This separated char is assigned a heating value of 12,500 Btu/lb.
12.5.4 Heat Output Capacity Validation. The first test run must produce a heat output rate that is within 10 percent of the manufacturer's rated heat output capacity (Category IV) throughout the test run and an average heat output rate within 5 percent of the manufacturer's rated heat output capacity. If the appliance is not capable of producing a heat output within these limits, the manufacturer's rated heat output capacity is considered not validated and testing is to be terminated. In such cases, the tests may be restarted using a lower heat output capacity if requested by the manufacturer. Alternatively, during the Category IV run, if the rated output cannot be maintained for a 15 minute interval, the manufacturer may elect to reduce the rated output to match the test and complete the Category IV run on this basis. The target outputs for Categories I, II, and III shall then be recalculated based on this change in rated output capacity.
12.5.5 Additional Test Runs. Using the manufacturer's rated heat output capacity as a basis, conduct a test for additional heat output categories as specified in section 4.3. It is not required to run these tests in any particular order.
12.5.6 Alternative Heat Output Rate for Category I. If an appliance cannot be operated in the Category I heat output range due to stopped combustion, two test runs shall be conducted at heat output rates within Category II. When this is the case, the weightings for the weighted averages indicated in section 14.1.15 shall be the average of the Category I and II weighting's and shall be applied to both Category II results. Appliances that are not capable of operation within Category II (<25 percent of maximum) cannot be evaluated by this test method.
12.5.6.1 Stopped Fuel Combustion. Evidence that an appliance cannot be operated at a Category I heat output rate due to stopped fuel combustion shall include documentation of two or more attempts to operate the appliance in heat output rate Category I and fuel combustion has stopped prior to complete consumption of the test fuel charge. Stopped fuel combustion is evidenced when an elapsed time of 60 minutes or more has occurred without a measurable (1 lb or 0.5 kg) weight change in the test fuel charge while the appliance operating control is in the demand mode. Report the evidence and the reasoning used to determine that a test in heat output rate Category I cannot be achieved. For example, two unsuccessful attempts to operate at an output rate of 10 percent of the rated output capacity are not sufficient evidence that heat output rate Category I cannot be achieved.
12.5.7 Appliance Overheating. Appliances with their associated thermal storage shall be capable of operating in all heat output categories without overheating to be rated by this test method. Appliance overheating occurs when the rate of heat withdrawal from the appliance is lower than the rate of heat production when the unit control is in the idle mode. This condition results in the water in the appliance continuing to increase in temperature well above the upper limit setting of the operating control. Evidence of overheating includes: 1 hour or more of appliance water temperature increase above the upper temperature set-point of the operating control, exceeding the temperature limit of a safety control device (independent from the operating control - typically requires manual reset), boiling water in a non-pressurized system or activation of a pressure or temperature relief valve in a pressurized system.
12.5.8 Option to Eliminate Tests in Category II and III. Following successful completion of a test run in Category I, the manufacturer may eliminate the Category II and III tests. For the purpose of calculating the annual averages for particulates and efficiency, the values obtained in the Category I run shall be assumed to apply also to Category II and Category III. It is envisioned that this option would be applicable to systems which have sufficient thermal storage such that the fuel load in the Category I test can be completely consumed without the system reaching its upper operating temperature limit. In this case, the boiler/heater would likely be operating at maximum thermal output during the entire test and this output rate may be higher than the manufacturer's rated heat output capacity. The Category II and III runs would then be the same as the Category I run. It may be assumed that the particulate emission values and efficiency values determined in the startup, steady-state, and end phases of Category I are applicable in Categories II and III, for the purpose of determining the annual averages in lb/mmBtu and g/MJ (See section 13). For the annual average in g/hr, the length of time for stored heat to be drawn from thermal storage shall be determined for the test load requirements of the respective category.
12.5.9 Modification to Measurement Procedure in EPA Method 28WHH to Determine Emissions Separately During the Startup, Steady-State and End Phases. With one of the two particulate sampling trains used, filter changes shall be made at the end of the startup phase and the steady-state phase (See section 3.0). This shall be done to determine the particulate emission rate and particulate emission index for the startup, steady-state, and end phases individually. For this one train, the particulates measured during each of these three phases shall be added together to also determine the particulate emissions for the whole run.
12.5.10 Modification to Measurement Procedure in EPA Method 28WHH and the Standards Referenced therein on Averaging Period for Determination of Efficiency by the Stack Loss Method. The methods currently defined in Method 28WHH allow averaging over 10-minute time periods for flue gas temperature, flue gas CO 2 , and flue gas CO for the determination of the efficiency with the stack loss method. However, under some cycling conditions the “on” period may be short relative to this 10-minute period. For this reason, during cycling operation the averaging period for these parameters may not be longer than the burner on period divided by 10. The averaging period need not be shorter than one minute. During the off period, under cycling operation, averaging periods as specified in EPA Method 28WHH and the standards referenced therein, may be used. Where short averaging times are used, however, the averaging period for fuel consumption may still be at 10 minutes. This average wood consumption rate shall be applied to all of the smaller time intervals included.
12.6 Additional Test Runs. The testing laboratory may conduct more than one test run in each of the heat output categories specified in section 4.3. If more than one test run is conducted at a specified heat output rate, the results from at least two-thirds of the test runs in that heat output rate category shall be used in calculating the weighted average emission rate. The measurement data and results of all test runs shall be reported regardless of which values are used in calculating the weighted average emission rate.
13.0 Calculation of Results
13.1 Nomenclature
CO s - Carbon monoxide measured in the dilution tunnel at arbitrary time in ppm dry basis.
CO g/min - Carbon monoxide emission rate in g/min.
CO T - Total carbon monoxide emission for the full test run in grams.
CO _1 - Startup period carbon monoxide emissions in grams.
CO _2 - Steady-state period carbon monoxide emission in grams.
CO _3 - End period carbon monoxide emission in grams.
E T - Total particulate emissions for the full test run as determined per EPA Method 28WHH and the standards referenced therein in grams.
E 1 - Startup period particulate emissions in grams.
E 2 - Steady-state period particulate emissions in grams.
E 3 - End period particulate emissions in grams.
E 1_g/kg - Startup period particulate emission index in grams per kg fuel.
E 2_g/kg - Steady-state period particulate emission index in grams per kg fuel.
E 3_g/kg - End period particulate emission index in grams per kg fuel.
E 1_g/hr - Startup period particulate emission rate in grams per hour.
E 2_g/hr - Steady-state period particulate emission rate in grams per hour.
E 3_g/hr - End period particulate emission rate in grams per hour.
E g/MJ - Emission rate in grams per MJ of heat output.
E lb/mmBtu output - Emissions rate in pounds per million Btu of heat output.
E g/kg - Emissions factor in grams per kilogram of dry fuel burned.
E g/hr - Emission factor in grams per hour.
HHV - Higher heating value of fuel = 8600 Btu/lb (19.990 MJ/kg).
LHV - Lower heating value of fuel = 7988 Btu/lb (18.567 MJ/kg).
ΔT - Temperature difference between cooling water entering and exiting the heat exchanger.
Q out - Total heat output in Btu (MJ).
Q in - Total heat input available in test fuel charge in Btu's (MJ).
Q std - Volumetric flow rate in dilution tunnel in dscfm.
M - Mass flow rate of water in lb/min (kg/min).
V i - Volume of water indicated by a totalizing flow meter at the i th reading in gallons (liters).
V f - Volumetric flow rate of water in heat exchange system in gallons per minute (liters/min).
Θ - Total length of burn period in hours (Θ 1 Θ 2 Θ 3 ).
Θ 1 - Length of time of the startup period in hours.
Θ 2 - Length of time of the steady-state period in hours.
Θ 3 - Length of time of the end period in hours.
Θ 4 - Length of time for stored heat to be used following a burn period in hours.
t i - Data sampling interval in minutes.
η del - Delivered heating efficiency in percent.
F i - Weighting factor for heat output category i. (See Table 2.)
T1 - Temperature of water at the inlet on the supply side of the heat exchanger,°F.
T2 - Temperature of the water at the outlet on the supply side of the heat exchanger,°F.
T3 - Temperature of cooling water at the inlet to the load side of the heat exchanger,°F.
T4 - Temperature of cooling water at the outlet of the load side of the heat exchanger,°F.
T5 - Temperature of the hot water supply as it leaves the boiler/heater,°F.
T6 - Temperature of return water as it enters the boiler/heater,°F.
T7 - Temperature in the boiler/heater optional destratification loop at the top of the boiler/heater,°F.
T8 - Temperature in the boiler/heater optional destratification loop at the bottom of the boiler/heater,°F.
TI avg - Average temperature of the appliance and water at start of the test.
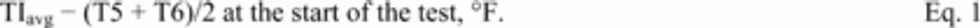
TF avg - Average temperature of the appliance and water at the end of the test.
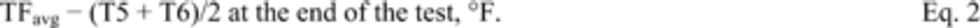
TIS 1 - Temperature at the inlet to the storage system at the start of the test.
TIS 2 - Temperature at the outlet from the storage system at the start of the test.
TFS 1 - Temperature at the inlet to the storage system at the end of the test.
TFS 2 - Temperature at the outlet from the storage system at the end of the test.
TIS avg - Average temperature of the storage system at the start of the test.
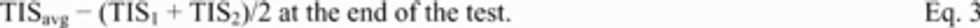
TFS avg - Average temperature of the storage system at the end of the test.

MC - Fuel moisture content in percent dry basis.
σ - Density of water in pounds per gallon.
σ Initial - Density of water in the boiler/heater system at the start of the test in pounds per gallons.
σ boiler/heater - Density of water in the boiler/heater system at an arbitrary time during the test in pounds per gallon.
C p - Specific heat of water in Btu/lb,°F.
C steel - Specific heat of steel (0.1 Btu/lb,°F).
V boiler/heater - total volume of water in the boiler/heater system on the weight scale in gallons.
W fuel - Fuel charge weight, as-fired or “wet”, in pounds (kg).
W fuel_1 - Fuel consumed during the startup period in pounds (kg).
W fuel_2 - Fuel consumed during the steady state period in pounds (kg).
W fuel_3 - Fuel consumed during the end period in pounds (kg).
W FuelBurned - Weight of fuel that has been burned from the start of the test to an arbitrary time, including the needed correction for the change in density and weight of the water in the boiler/heater system on the scale in pounds (kg).
W RemainingFuel - Weight of unburned fuel separated from the ash at the end of a test. Useful only for Category I and Category II tests.
W app - Weight of empty appliance in pounds (kg).
W wat - Weight of water in supply side of the system in pounds (kg).
W ScaleInitial - Weight reading on the scale at the start of the test, just after the test load has been added in pounds (kg).
W Scale - Reading of the weight scale at an arbitrary time during the test run in pounds (kg).
W StorageTank - Weight of the storage tank empty in pounds (kg).
W WaterStorage - Weight of the water in the storage tank at TIS avg in pounds (kg).
13.2 After the test is completed, determine the particulate emissions E T in accordance with EPA Method 28WHH and the standards referenced therein.
13.3 Determination of the weight of fuel that has been burned at an arbitrary time.
For the purpose of tracking the consumption of the test fuel load during a test run the following may be used to calculate the weight of fuel that burned since the start of the test:

Water density, σ, is calculated using Equation 12.
13.4 Determine Average Fuel Load Moisture Content.

13.5 Determine Heat Input.
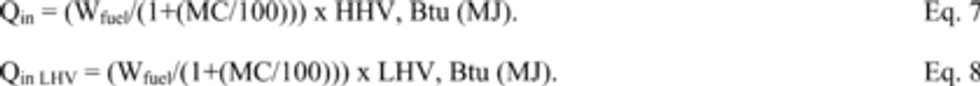
13.5.1 Correction to Q in for the Category I and II tests, where there is greater than 1 percent of the test fuel charge in the chamber at the end of the test period.

13.6 Determine Heat Output, Efficiency, and Emissions.
13.6.1 Determine heat output as:
Q out = Σ [Heat output determined for each sampling time interval] Change in heat stored in the appliance Change in heat in storage tank.
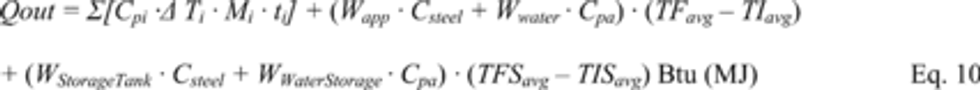
Note:
The subscript (i) indicates the parameter value for sampling time interval t i .M i = Mass flow rate = gal/min × density of water (lb/gal) = lb/min.
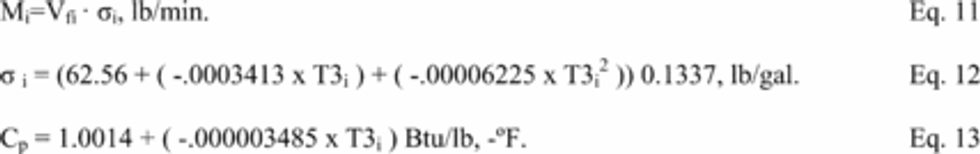
C steel = 0.1 Btu/lb, -° F.
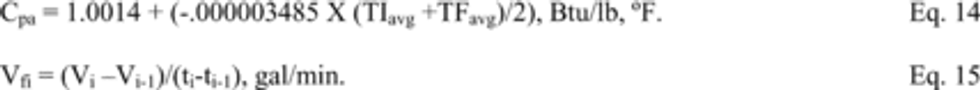
Note:
V i is the total water volume at the end of interval i and V i-1 is the total water volume at the beginning of the time interval. This calculation is necessary when a totalizing type water meter is used.
13.6.2 Determine Heat Output Rate Over Burn Period (Θ 1 Θ 2 Θ 3 ) as:

13.6.3 Determine Emission Rates and Emission Factors as:

If thermal storage is not used in a Category III or IV run, then Θ 4 = 0.
E 1_g/kg = E 1 /(W fuel_1 /(1 MC/100)), g/dry kg.
E 2_g/kg = E 2 /(W fuel_2 /(1 MC/100)), g/dry kg.
E 3_g/kg = E 3 /(W fuel_3 /(1 MC/100)), g/dry kg.
E 1_g/hr = E 1 /Θ 1 , g/hr.
E 2_g/hr = E 2 /Θ 2 , g/hr.
E 3_g/hr = E 3 /Θ 3 , g/hr.
13.6.4 Determine delivered efficiency as:

13.6.5 Determine η SLM - Overall Efficiency, also known as Stack Loss Efficiency, using stack loss method (SLM).
For determination of the average overall thermal efficiency (η SLM ) for the test run, use the data collected over the full test run and the calculations in accordance with CSA B415.1-10 (IBR, see §60.17 ), clause 13.7 except for 13.7.2(e), (f), (g), and (h), use the following average fuel properties for oak: %C = 50.0, %H = 6.6, %O = 43.2, %Ash = 0.2.
13.6.5.1 Whenever the CSA B415.1-10 (IBR, see §60.17 ) overall efficiency is found to be lower than the overall efficiency based on load side measurements, as determined by Eq. 22 of this method, section 14.1.7 of the test report must include a discussion of the reasons for this result. For a test where the CSA B415.1-10 overall efficiency SLM is less than 2 percentage points lower than the overall efficiency based on load side measurements, the efficiency based on load side measurements shall be considered invalid. [Note on the rationale for the 2 percentage points limit. The SLM method does not include boiler/heater jacket losses and, for this reason, should provide an efficiency which is actually higher than the efficiency based on the energy input and output measurements or “delivered efficiency.” A delivered efficiency that is higher than the efficiency based on the SLM could be considered suspect. A delivered efficiency greater than 2 percentage points higher than the efficiency based on the SLM, then, clearly indicates a measurement error.]
13.6.6 Carbon Monoxide Emissions
For each minute of the test period, the carbon monoxide emission rate shall be calculated as:

Total CO emissions for each of the three test periods (CO _1 , CO _2 , CO _3 ) shall be calculated as the sum of the emission rates for each of the 1-minute intervals. Total CO emission for the test run, CO T , shall be calculated as the sum of CO _1 , CO _2 , and CO _3 .
13.7 Weighted Average Emissions and Efficiency.
13.7.1 Determine the weighted average emission rate and delivered efficiency from the individual tests in the specified heat output categories. The weighting factors (F i ) are derived from an analysis of ASHRAE bin data which provides details of normal building heating requirements in terms of percent of design capacity and time in a particular capacity range - or “bin” - over the course of a heating season. The values used in this method represent an average of data from several cities located in the northern United States.
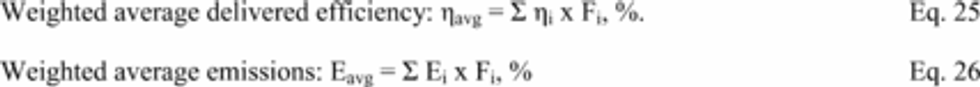
If, as discussed in section 12.5.8, the option to eliminate tests in Category II and III is elected, the values of efficiency and particulate emission rate as measured in Category I, shall be assigned also to Category II and III for the purpose of determining the annual averages.
14.0 Report
14.1.1 The report shall include the following:
14.1.2 Name and location of the laboratory conducting the test.
14.1.3 A description of the appliance tested and its condition, date of receipt and dates of tests.
14.1.4 A description of the minimum amount of external thermal storage that is required for use with this system. This shall be specified both in terms of volume in gallons and stored energy content in Btu with a storage temperature ranging from 125°F to the manufacturer's specified setpoint temperature.
14.1.5 A statement that the test results apply only to the specific appliance tested.
14.1.6 A statement that the test report shall not be reproduced except in full, without the written approval of the laboratory.
14.1.7 A description of the test procedures and test equipment including a schematic or other drawing showing the location of all required test equipment. Also, a description of test fuel sourcing, handling and storage practices shall be included.
14.1.8 Details of deviations from, additions to or exclusions from the test method, and their data quality implications on the test results (if any), as well as information on specific test conditions, such as environmental conditions.
14.1.9 A list of participants and their roles and observers present for the tests.
14.1.10 Data and drawings indicating the fire box size and location of the fuel charge.
14.1.11 Drawings and calculations used to determine firebox volume.
14.1.12 Information for each test run fuel charge including piece size, moisture content and weight.
14.1.13 All required data and applicable blanks for each test run shall be provided in spreadsheet format both in the printed report and in a computer file such that the data can be easily analyzed and calculations easily verified. Formulas used for all calculations shall be accessible for review.
14.1.14 For each test run, Θ 1 ,Θ 2 , Θ 3 , the total CO and particulate emission for each of these three periods, and Θ 4 .
14.1.15 Calculated results for delivered efficiency at each heat output rate and the weighted average emissions reported as total emissions in grams, pounds per mm Btu of delivered heat, grams per MJ of delivered heat, grams per kilogram of dry fuel and grams per hour. Results shall be reported for each heat output category and the weighted average.
14.1.16 Tables 1A, 1B, 1C, 1D, 1E and Table 2 must be used for presentation of results in test reports.
14.1.17 A statement of the estimated uncertainty of measurement of the emissions and efficiency test results.
14.1.18 A plot of CO emission rate in grams/minute vs. time, based on 1 minute averages, for the entire test period, for each run.
14.1.19 A plot of estimated boiler/heater energy release rate in Btu/hr based on 10 minute averages, for the entire test period, for each run. This will be calculated from the fuel used, the wood heating value and moisture content, and the SLM efficiency during each 10 minute period.
14.1.20 Raw data, calibration records, and other relevant documentation shall be retained by the laboratory for a minimum of 7 years.
15.0 Precision and Bias
15.1 Precision - It is not possible to specify the precision of the procedure in this test method because the appliance operation and fueling protocols and the appliances themselves produce variable amounts of emissions and cannot be used to determine reproducibility or repeatability of this test method.
15.2 Bias - No definitive information can be presented on the bias of the procedure in this test method for measuring solid fuel burning hydronic heater emissions because no material having an accepted reference value is available.
16.0 Keywords
16.1 Solid fuel, hydronic heating appliances, wood-burning hydronic heaters, partial thermal storage.
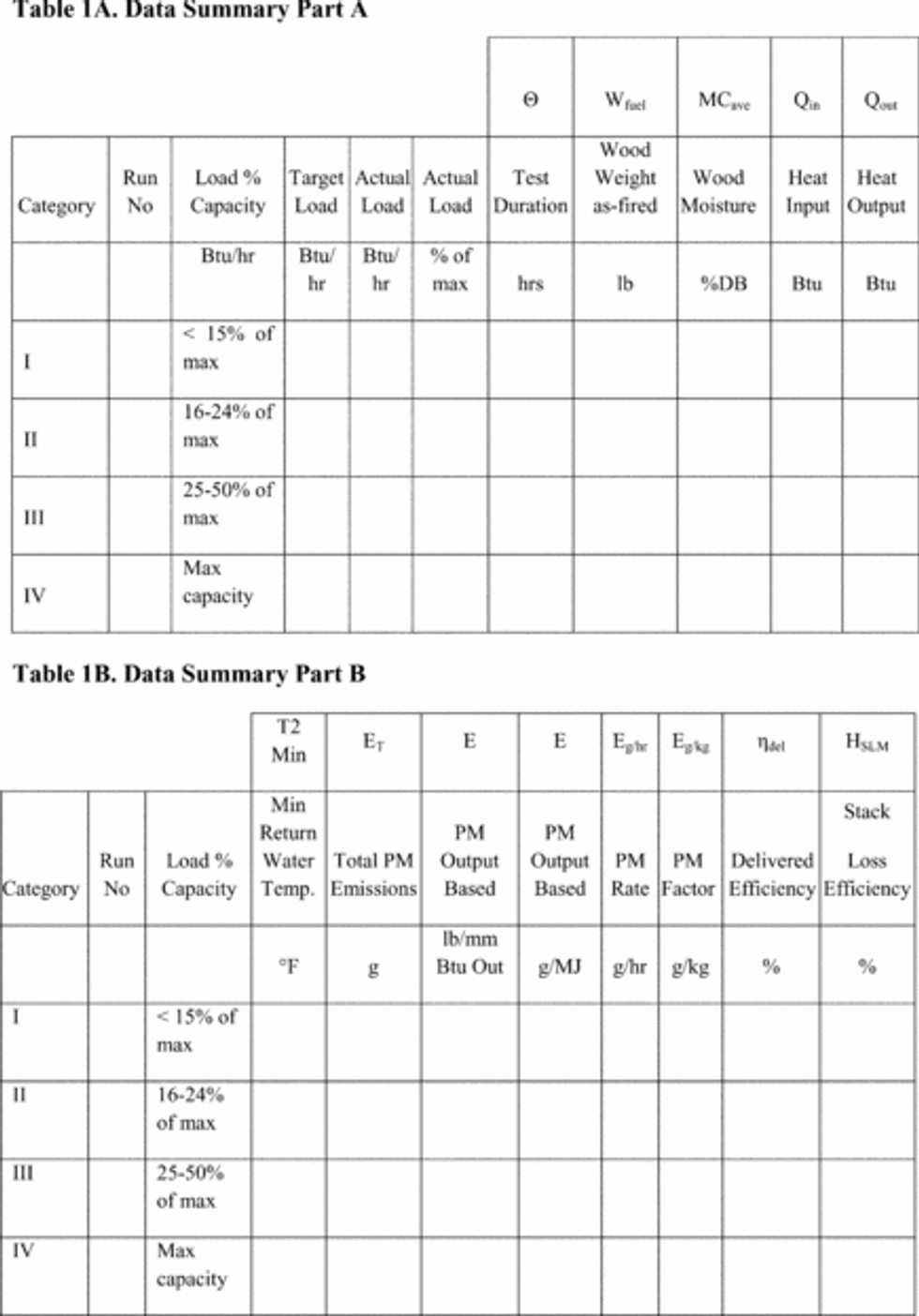
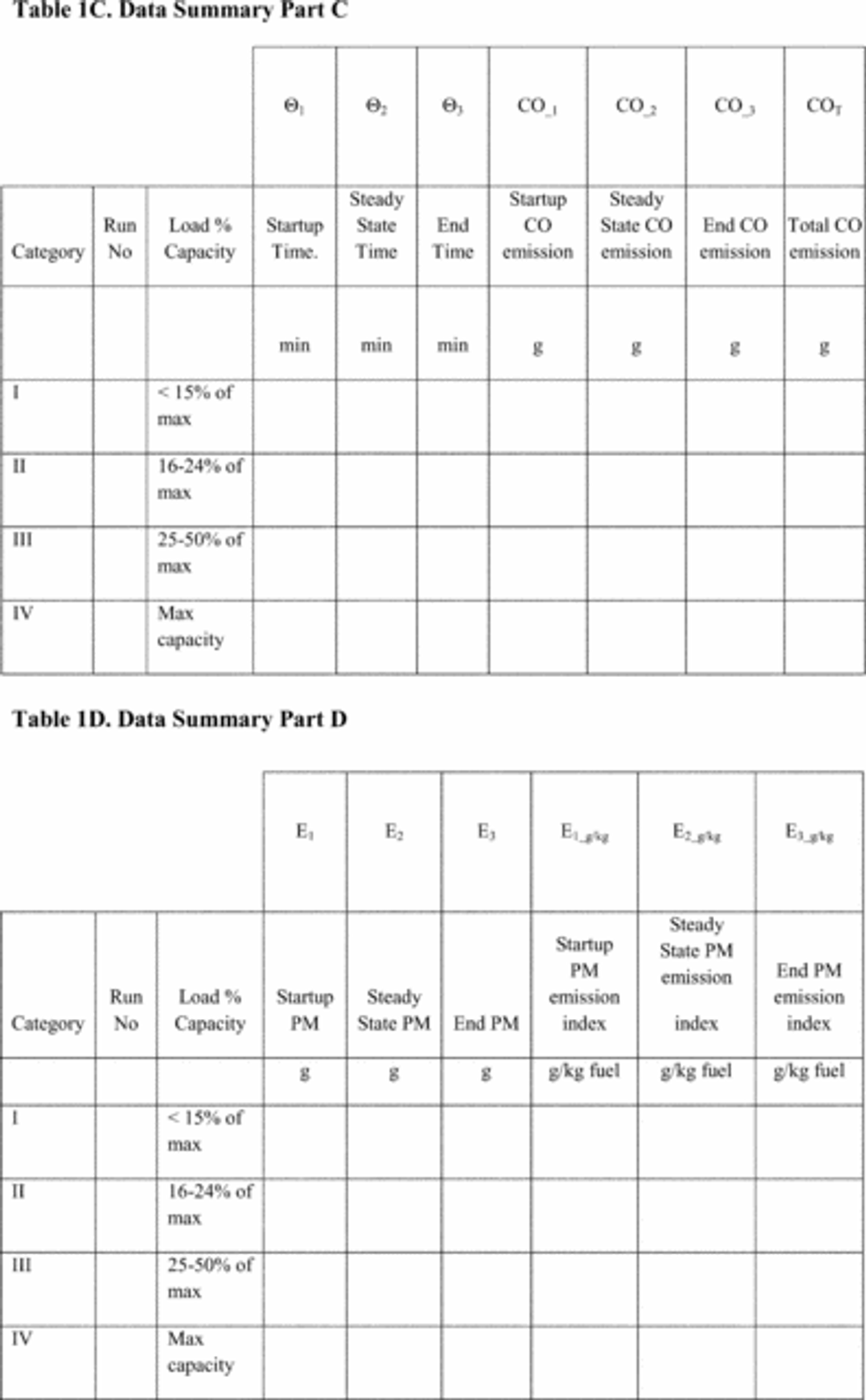
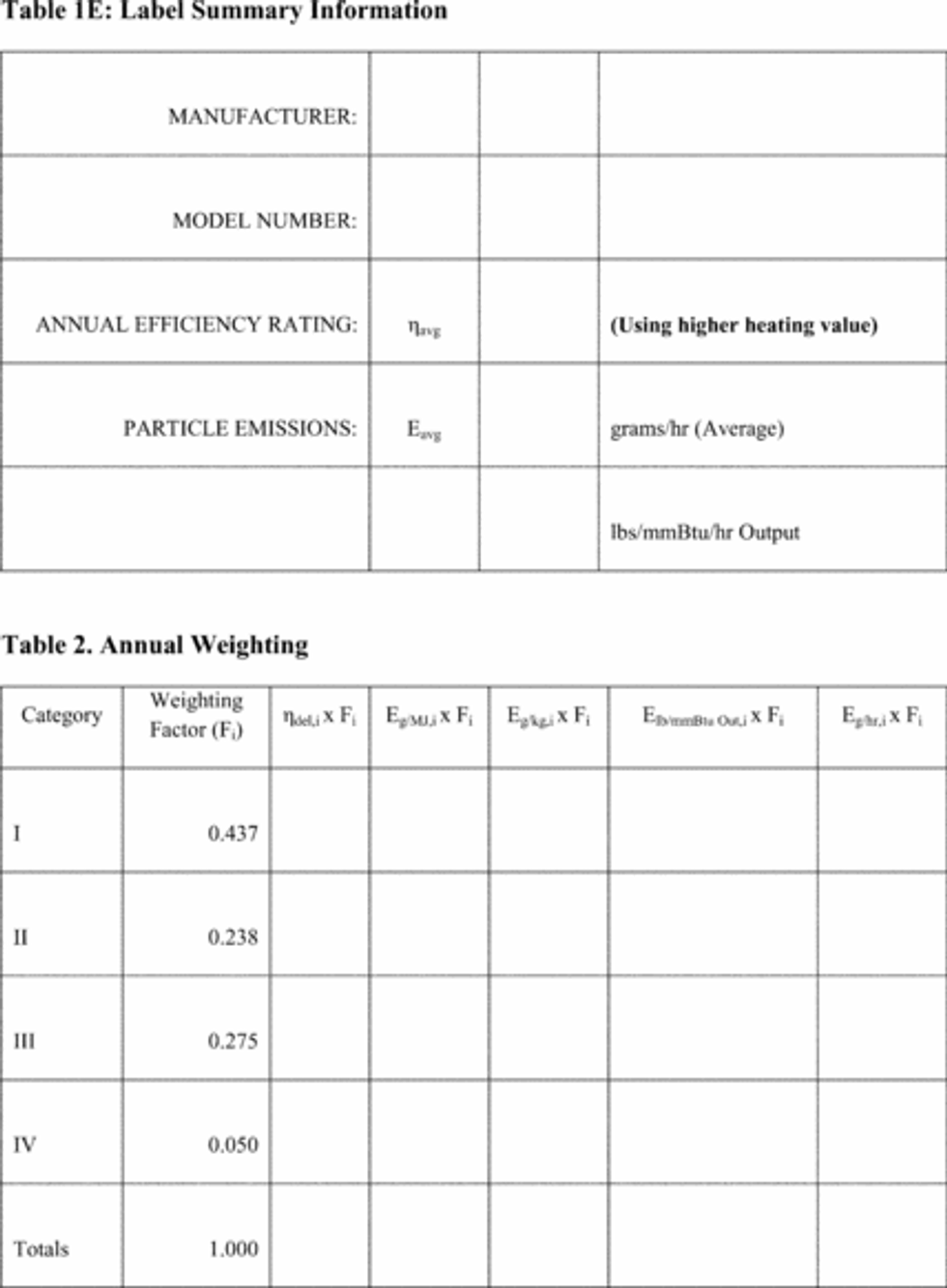
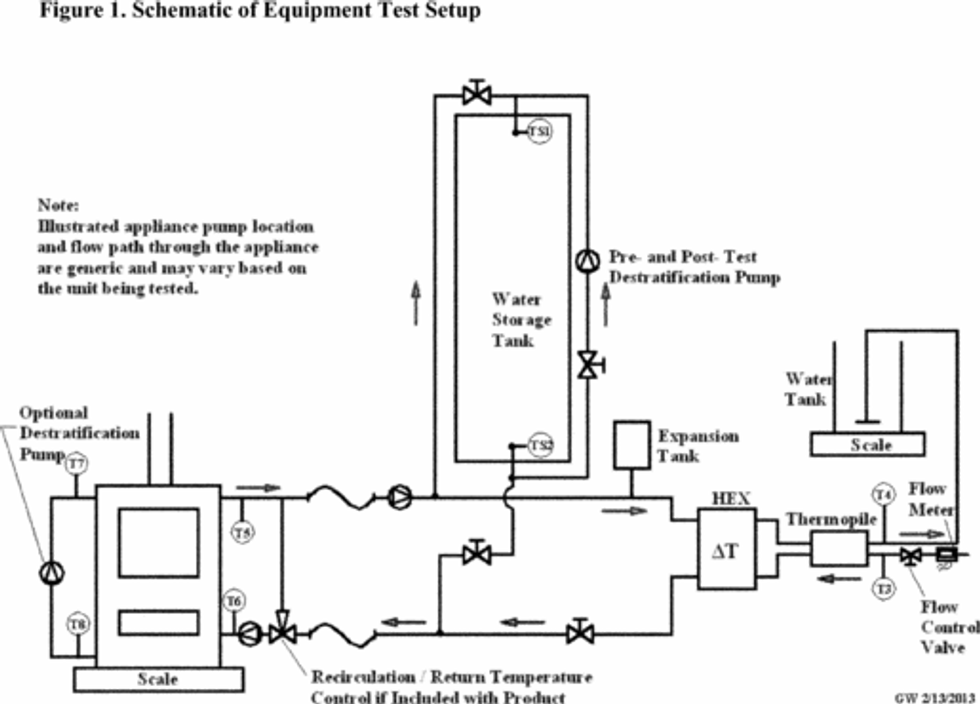
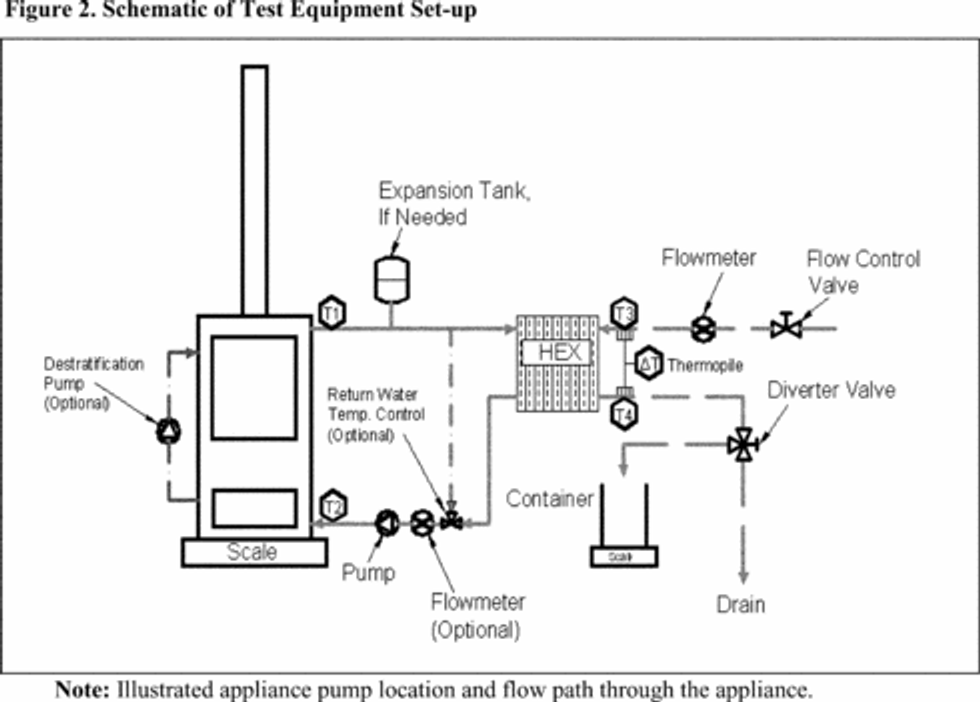
Method 29 - Determination of Metals Emissions From Stationary Sources
Note:
This method does not include all of the specifications ( e.g., equipment and supplies) and procedures ( e.g., sampling and analytical) essential to its performance. Some material is incorporated by reference from other methods in this part. Therefore, to obtain reliable results, persons using this method should have a thorough knowledge of at least the following additional test methods: Method 5 and Method 12.
1.0 Scope and Application
1.1 Analytes.
| Analyte | CAS No. |
|---|---|
| Antimony (Sb) | 7440-36-0 |
| Arsenic (As) | 7440-38-2 |
| Barium (Ba) | 7440-39-3 |
| Beryllium (Be) | 7440-41-7 |
| Cadmium (Cd) | 7440-43-9 |
| Chromium (Cr) | 7440-47-3 |
| Cobalt (Co) | 7440-48-4 |
| Copper (Cu) | 7440-50-8 |
| Lead (Pb) | 7439-92-1 |
| Manganese (Mn) | 7439-96-5 |
| Mercury (Hg) | 7439-97-6 |
| Nickel (Ni) | 7440-02-0 |
| Phosphorus (P) | 7723-14-0 |
| Selenium (Se) | 7782-49-2 |
| Silver (Ag) | 7440-22-4 |
| Thallium (Tl) | 7440-28-0 |
| Zinc (Zn) | 7440-66-6 |
1.2 Applicability. This method is applicable to the determination of metals emissions from stationary sources. This method may be used to determine particulate emissions in addition to the metals emissions if the prescribed procedures and precautions are followed.
1.2.1 Hg emissions can be measured, alternatively, using EPA Method 101A of Appendix B, 40 CFR Part 61. Method 101-A measures only Hg but it can be of special interest to sources which need to measure both Hg and Mn emissions.
2.0 Summary of Method
2.1 Principle. A stack sample is withdrawn isokinetically from the source, particulate emissions are collected in the probe and on a heated filter, and gaseous emissions are then collected in an aqueous acidic solution of hydrogen peroxide (analyzed for all metals including Hg) and an aqueous acidic solution of potassium permanganate (analyzed only for Hg). The recovered samples are digested, and appropriate fractions are analyzed for Hg by cold vapor atomic absorption spectroscopy (CVAAS) and for Sb, As, Ba, Be, Cd, Cr, Co, Cu, Pb, Mn, Ni, P, Se, Ag, Tl, and Zn by inductively coupled argon plasma emission spectroscopy (ICAP) or atomic absorption spectroscopy (AAS). Graphite furnace atomic absorption spectroscopy (GFAAS) is used for analysis of Sb, As, Cd, Co, Pb, Se, and Tl if these elements require greater analytical sensitivity than can be obtained by ICAP. If one so chooses, AAS may be used for analysis of all listed metals if the resulting in-stack method detection limits meet the goal of the testing program. Similarly, inductively coupled plasma-mass spectroscopy (ICP-MS) may be used for analysis of Sb, As, Ba, Be, Cd, Cr, Co, Cu, Pb, Mn, Ni, Ag, Tl and Zn.
3.0 Definitions [Reserved]
4.0 Interferences
4.1 Iron (Fe) can be a spectral interference during the analysis of As, Cr, and Cd by ICAP. Aluminum (Al) can be a spectral interference during the analysis of As and Pb by ICAP. Generally, these interferences can be reduced by diluting the analytical sample, but such dilution raises the in-stack detection limits. Background and overlap corrections may be used to adjust for spectral interferences. Refer to Method 6010 of Reference 2 in section 16.0 or the other analytical methods used for details on potential interferences to this method. For all GFAAS analyses, use matrix modifiers to limit interferences, and matrix match all standards.
5.0 Safety
5.1 Disclaimer. This method may involve hazardous materials, operations, and equipment. This test method may not address all of the safety problems associated with its use. It is the responsibility of the user of this test method to establish appropriate safety and health practices and to determine the applicability of regulatory limitations prior to performing this test method.
5.2 Corrosive Reagents. The following reagents are hazardous. Personal protective equipment and safe procedures are useful in preventing chemical splashes. If contact occurs, immediately flush with copious amounts of water at least 15 minutes. Remove clothing under shower and decontaminate. Treat residual chemical burn as thermal burn.
5.2.1 Nitric Acid (HNO 3 ). Highly corrosive to eyes, skin, nose, and lungs. Vapors cause bronchitis, pneumonia, or edema of lungs. Reaction to inhalation may be delayed as long as 30 hours and still be fatal. Provide ventilation to limit exposure. Strong oxidizer. Hazardous reaction may occur with organic materials such as solvents.
5.2.2 Sulfuric Acid (H 2 SO 4 ). Rapidly destructive to body tissue. Will cause third degree burns. Eye damage may result in blindness. Inhalation may be fatal from spasm of the larynx, usually within 30 minutes. May cause lung tissue damage with edema. 1 mg/m 3 for 8 hours will cause lung damage or, in higher concentrations, death. Provide ventilation to limit inhalation. Reacts violently with metals and organics.
5.2.3 Hydrochloric Acid (HC1). Highly corrosive liquid with toxic vapors. Vapors are highly irritating to eyes, skin, nose, and lungs, causing severe damage. May cause bronchitis, pneumonia, or edema of lungs. Exposure to concentrations of 0.13 to 0.2 percent can be lethal to humans in a few minutes. Provide ventilation to limit exposure. Reacts with metals, producing hydrogen gas.
5.2.4 Hydrofluoric Acid (HF). Highly corrosive to eyes, skin, nose, throat, and lungs. Reaction to exposure may be delayed by 24 hours or more. Provide ventilation to limit exposure.
5.2.5 Hydrogen Peroxide (H 2 O 2 ). Irritating to eyes, skin, nose, and lungs. 30% H 2 O 2 is a strong oxidizing agent. Avoid contact with skin, eyes, and combustible material. Wear gloves when handling.
5.2.6 Potassium Permanganate (KMnO 4 ). Caustic, strong oxidizer. Avoid bodily contact with.
5.2.7 Potassium Persulfate. Strong oxidizer. Avoid bodily contact with. Keep containers well closed and in a cool place.
5.3 Reaction Pressure. Due to the potential reaction of the potassium permanganate with the acid, there could be pressure buildup in the acidic KMnO 4 absorbing solution storage bottle. Therefore these bottles shall not be fully filled and shall be vented to relieve excess pressure and prevent explosion potentials. Venting is required, but not in a manner that will allow contamination of the solution. A No. 70-72 hole drilled in the container cap and Teflon liner has been used.
6.0 Equipment and Supplies
6.1 Sampling. A schematic of the sampling train is shown in Figure 29-1. It has general similarities to the Method 5 train.
6.1.1 Probe Nozzle (Probe Tip) and Borosilicate or Quartz Glass Probe Liner. Same as Method 5, sections 6.1.1.1 and 6.1.1.2, except that glass nozzles are required unless alternate tips are constructed of materials that are free from contamination and will not interfere with the sample. If a probe tip other than glass is used, no correction to the sample test results to compensate for the nozzle's effect on the sample is allowed. Probe fittings of plastic such as Teflon, polypropylene, etc. are recommended instead of metal fittings to prevent contamination. If one chooses to do so, a single glass piece consisting of a combined probe tip and probe liner may be used.
6.1.2 Pitot Tube and Differential Pressure Gauge. Same as Method 2, sections 6.1 and 6.2, respectively.
6.1.3 Filter Holder. Glass, same as Method 5, section 6.1.1.5, except use a Teflon filter support or other non-metallic, non-contaminating support in place of the glass frit.
6.1.4 Filter Heating System. Same as Method 5, section 6.1.1.6.
6.1.5 Condenser. Use the following system for condensing and collecting gaseous metals and determining the moisture content of the stack gas. The condensing system shall consist of four to seven impingers connected in series with leak-free ground glass fittings or other leak-free, non-contaminating fittings. Use the first impinger as a moisture trap. The second impinger (which is the first HNO 3 /H 2 O 2 impinger) shall be identical to the first impinger in Method 5. The third impinger (which is the second HNO 3 /H 2 O 2 impinger) shall be a Greenburg Smith impinger with the standard tip as described for the second impinger in Method 5, section 6.1.1.8. The fourth (empty) impinger and the fifth and sixth (both acidified KMnO 4 ) impingers are the same as the first impinger in Method 5. Place a temperature sensor capable of measuring to within 1°C (2°F) at the outlet of the last impinger. If no Hg analysis is planned, then the fourth, fifth, and sixth impingers are not used.
6.1.6 Metering System, Barometer, and Gas Density Determination Equipment. Same as Method 5, sections 6.1.1.9, 6.1.2, and 6.1.3, respectively.
6.1.7 Teflon Tape. For capping openings and sealing connections, if necessary, on the sampling train.
6.2 Sample Recovery. Same as Method 5, sections 6.2.1 through 6.2.8 (Probe-Liner and Probe-Nozzle Brushes or Swabs, Wash Bottles, Sample Storage Containers, Petri Dishes, Glass Graduated Cylinder, Plastic Storage Containers, Funnel and Rubber Policeman, and Glass Funnel), respectively, with the following exceptions and additions:
6.2.1 Non-metallic Probe-Liner and Probe-Nozzle Brushes or Swabs. Use non-metallic probe-liner and probe-nozzle brushes or swabs for quantitative recovery of materials collected in the front-half of the sampling train.
6.2.2 Sample Storage Containers. Use glass bottles (see section 8.1 of this Method) with Teflon-lined caps that are non-reactive to the oxidizing solutions, with capacities of 1000- and 500-ml, for storage of acidified KMnO 4 - containing samples and blanks. Glass or polyethylene bottles may be used for other sample types.
6.2.3 Graduated Cylinder. Glass or equivalent.
6.2.4 Funnel. Glass or equivalent.
6.2.5 Labels. For identifying samples.
6.2.6 Polypropylene Tweezers and/or Plastic Gloves. For recovery of the filter from the sampling train filter holder.
6.3 Sample Preparation and Analysis.
6.3.1 Volumetric Flasks, 100-ml, 250-ml, and 1000-ml. For preparation of standards and sample dilutions.
6.3.2 Graduated Cylinders. For preparation of reagents.
6.3.3 Parr Bombs or Microwave Pressure Relief Vessels with Capping Station (CEM Corporation model or equivalent). For sample digestion.
6.3.4 Beakers and Watch Glasses. 250-ml beakers, with watch glass covers, for sample digestion.
6.3.5 Ring Stands and Clamps. For securing equipment such as filtration apparatus.
6.3.6 Filter Funnels. For holding filter paper.
6.3.7 Disposable Pasteur Pipets and Bulbs.
6.3.8 Volumetric Pipets.
6.3.9 Analytical Balance. Accurate to within 0.1 mg.
6.3.10 Microwave or Conventional Oven. For heating samples at fixed power levels or temperatures, respectively.
6.3.11 Hot Plates.
6.3.12 Atomic Absorption Spectrometer (AAS). Equipped with a background corrector.
6.3.12.1 Graphite Furnace Attachment. With Sb, As, Cd, Co, Pb, Se, and Tl hollow cathode lamps (HCLs) or electrodeless discharge lamps (EDLs). Same as Reference 2 in section 16.0. Methods 7041 (Sb), 7060 (As), 7131 (Cd), 7201 (Co), 7421 (Pb), 7740 (Se), and 7841 (Tl).
6.3.12.2 Cold Vapor Mercury Attachment. With a mercury HCL or EDL, an air recirculation pump, a quartz cell, an aerator apparatus, and a heat lamp or desiccator tube. The heat lamp shall be capable of raising the temperature at the quartz cell by 10°C above ambient, so that no condensation forms on the wall of the quartz cell. Same as Method 7470 in Reference 2 in section 16.0. See note 2: section 11.1.3 for other acceptable approaches for analysis of Hg in which analytical detection limits of 0.002 ng/ml were obtained.
6.3.13 Inductively Coupled Argon Plasma Spectrometer. With either a direct or sequential reader and an alumina torch. Same as EPA Method 6010 in Reference 2 in section 16.0.
6.3.14 Inductively Coupled Plasma-Mass Spectrometer.
Same as EPA Method 6020 in Reference 2 in section 16.0.
7.0 Reagents and Standards
7.1 Unless otherwise indicated, it is intended that all reagents conform to the specifications established by the Committee on Analytical Reagents of the American Chemical Society, where such specifications are available. Otherwise, use the best available grade.
7.2 Sampling Reagents.
7.2.1 Sample Filters. Without organic binders. The filters shall contain less than 1.3 µg/in. 2 of each of the metals to be measured. Analytical results provided by filter manufacturers stating metals content of the filters are acceptable. However, if no such results are available, analyze filter blanks for each target metal prior to emission testing. Quartz fiber filters meeting these requirements are recommended. However, if glass fiber filters become available which meet these requirements, they may be used. Filter efficiencies and unreactiveness to sulfur dioxide (SO 2 ) or sulfur trioxide (SO 3 ) shall be as described in section 7.1.1 of Method 5.
7.2.2 Water. To conform to ASTM Specification D1193-77 or 91, Type II (incorporated by reference - see §60.17 ). If necessary, analyze the water for all target metals prior to field use. All target metals should be less than 1 ng/ml.
7.2.3 HNO 3 , Concentrated. Baker Instra-analyzed or equivalent.
7.2.4 HCl, Concentrated. Baker Instra-analyzed or equivalent.
7.2.5 H 2 O 2 , 30 Percent (V/V).
7.2.6 KMnO 4 .
7.2.7 H 2 SO 4 , Concentrated.
7.2.8 Silica Gel and Crushed Ice. Same as Method 5, sections 7.1.2 and 7.1.4, respectively.
7.3 Pretest Preparation of Sampling Reagents.
7.3.1 HNO 3 /H 2 O 2 Absorbing Solution, 5 Percent HNO 3 /10 Percent H 2 O 2 . Add carefully with stirring 50 ml of concentrated HNO 3 to a 1000-ml volumetric flask containing approximately 500 ml of water, and then add carefully with stirring 333 ml of 30 percent H 2 O 2 . Dilute to volume with water. Mix well. This reagent shall contain less than 2 ng/ml of each target metal.
7.3.2 Acidic KMnO 4 Absorbing Solution, 4 Percent KMnO 4 (W/V), 10 Percent H 2 SO 4 (V/V). Prepare fresh daily. Mix carefully, with stirring, 100 ml of concentrated H 2 SO 4 into approximately 800 ml of water, and add water with stirring to make a volume of 1 liter: this solution is 10 percent H 2 SO 4 (V/V). Dissolve, with stirring, 40 g of KMnO 4 into 10 percent H 2 SO 4 (V/V) and add 10 percent H 2 SO 4 (V/V) with stirring to make a volume of 1 liter. Prepare and store in glass bottles to prevent degradation. This reagent shall contain less than 2 ng/ml of Hg.
Precaution: To prevent autocatalytic decomposition of the permanganate solution, filter the solution through Whatman 541 filter paper.
7.3.3 HNO 3 , 0.1 N. Add with stirring 6.3 ml of concentrated HNO 3 (70 percent) to a flask containing approximately 900 ml of water. Dilute to 1000 ml with water. Mix well. This reagent shall contain less than 2 ng/ml of each target metal.
7.3.4 HCl, 8 N. Carefully add with stirring 690 ml of concentrated HCl to a flask containing 250 ml of water. Dilute to 1000 ml with water. Mix well. This reagent shall contain less than 2 ng/ml of Hg.
7.4 Glassware Cleaning Reagents.
7.4.1 HNO 3 , Concentrated. Fisher ACS grade or equivalent.
7.4.2 Water. To conform to ASTM Specifications D1193, Type II.
7.4.3 HNO 3 , 10 Percent (V/V). Add with stirring 500 ml of concentrated HNO 3 to a flask containing approximately 4000 ml of water. Dilute to 5000 ml with water. Mix well. This reagent shall contain less than 2 ng/ml of each target metal.
7.5 Sample Digestion and Analysis Reagents. The metals standards, except Hg, may also be made from solid chemicals as described in Reference 3 in section 16.0. Refer to References 1, 2, or 5 in section 16.0 for additional information on Hg standards. The 1000 µg/ml Hg stock solution standard may be made according to section 7.2.7 of Method 101A.
7.5.1 HCl, Concentrated.
7.5.2 HF, Concentrated.
7.5.3 HNO 3 , Concentrated. Baker Instra-analyzed or equivalent.
7.5.4 HNO 3 , 50 Percent (V/V). Add with stirring 125 ml of concentrated HNO 3 to 100 ml of water. Dilute to 250 ml with water. Mix well. This reagent shall contain less than 2 ng/ml of each target metal.
7.5.5 HNO 3 , 5 Percent (V/V). Add with stirring 50 ml of concentrated HNO 3 to 800 ml of water. Dilute to 1000 ml with water. Mix well. This reagent shall contain less than 2 ng/ml of each target metal.
7.5.6 Water. To conform to ASTM Specifications D1193, Type II.
7.5.7 Hydroxylamine Hydrochloride and Sodium Chloride Solution. See Reference 2 In section 16.0 for preparation.
7.5.8 Stannous Chloride. See Reference 2 in section 16.0 for preparation.
7.5.9 KMnO 4 , 5 Percent (W/V). See Reference 2 in section 16.0 for preparation.
7.5.10 H 2 SO 4 , Concentrated.
7.5.11 Potassium Persulfate, 5 Percent (W/V). See Reference 2 in section 16.0 for preparation.
7.5.12 Nickel Nitrate, Ni(N0 3 ) 2 6H 2 0.
7.5.13 Lanthanum Oxide, La 2 0 3 .
7.5.14 Hg Standard (AAS Grade), 1000 µg/ml.
7.5.15 Pb Standard (AAS Grade), 1000 µg/ml.
7.5.16 As Standard (AAS Grade), 1000 µg/ml.
7.5.17 Cd Standard (AAS Grade), 1000 µg/ml.
7.5.18 Cr Standard (AAS Grade), 1000 µg/ml.
7.5.19 Sb Standard (AAS Grade), 1000 µg/ml.
7.5.20 Ba Standard (AAS Grade), 1000 µg/ml.
7.5.21 Be Standard (AAS Grade), 1000 µg/ml.
7.5.22 Co Standard (AAS Grade), 1000 µg/ml.
7.5.23 Cu Standard (AAS Grade), 1000 µg/ml.
7.5.24 Mn Standard (AAS Grade), 1000 µg/ml.
7.5.25 Ni Standard (AAS Grade), 1000 µg/ml.
7.5.26 P Standard (AAS Grade), 1000 µg/ml.
7.5.27 Se Standard (AAS Grade), 1000 µg/ml.
7.5.28 Ag Standard (AAS Grade), 1000 µg/ml.
7.5.29 Tl Standard (AAS Grade), 1000 µg/ml.
7.5.30 Zn Standard (AAS Grade), 1000 µg/ml.
7.5.31 Al Standard (AAS Grade), 1000 µg/ml.
7.5.32 Fe Standard (AAS Grade), 1000 µg/ml.
7.5.33 Hg Standards and Quality Control Samples. Prepare fresh weekly a 10 µg/ml intermediate Hg standard by adding 5 ml of 1000 µg/ml Hg stock solution prepared according to Method 101A to a 500-ml volumetric flask; dilute with stirring to 500 ml by first carefully adding 20 ml of 15 percent HNO 3 and then adding water to the 500-ml volume. Mix well. Prepare a 200 ng/ml working Hg standard solution fresh daily: add 5 ml of the 10 µg/ml intermediate standard to a 250-ml volumetric flask, and dilute to 250 ml with 5 ml of 4 percent KMnO 4 , 5 ml of 15 percent HNO 3 , and then water. Mix well. Use at least five separate aliquots of the working Hg standard solution and a blank to prepare the standard curve. These aliquots and blank shall contain 0.0, 1.0, 2.0, 3.0, 4.0, and 5.0 ml of the working standard solution containing 0, 200, 400, 600, 800, and 1000 ng Hg, respectively. Prepare quality control samples by making a separate 10 µg/ml standard and diluting until in the calibration range.
7.5.34 ICAP Standards and Quality Control Samples. Calibration standards for ICAP analysis can be combined into four different mixed standard solutions as follows:
| Solution | Elements |
|---|---|
| I | As, Be, Cd, Mn, Pb, Se, Zn. |
| II | Ba, Co, Cu, Fe. |
| III | Al, Cr, Ni. |
| IV | Ag, P, Sb, Tl. |
Prepare these standards by combining and diluting the appropriate volumes of the 1000 µg/ml solutions with 5 percent HNO 3 . A minimum of one standard and a blank can be used to form each calibration curve. However, prepare a separate quality control sample spiked with known amounts of the target metals in quantities in the mid-range of the calibration curve. Suggested standard levels are 25 µg/ml for Al, Cr and Pb, 15 µg/ml for Fe, and 10 µg/ml for the remaining elements. Prepare any standards containing less than 1 µg/ml of metal on a daily basis. Standards containing greater than 1 µg/ml of metal should be stable for a minimum of 1 to 2 weeks. For ICP-MS, follow Method 6020 in EPA Publication SW-846 Third Edition (November 1986) including updates I, II, IIA, IIB and III, as incorporated by reference in §60.17(i) .
7.5.35 GFAAS Standards. Sb, As, Cd, Co, Pb, Se, and Tl. Prepare a 10 µg/ml standard by adding 1 ml of 1000 µg/ml standard to a 100-ml volumetric flask. Dilute with stirring to 100 ml with 10 percent HNO 3 . For GFAAS, matrix match the standards. Prepare a 100 ng/ml standard by adding 1 ml of the 10 µg/ml standard to a 100-ml volumetric flask, and dilute to 100 ml with the appropriate matrix solution. Prepare other standards by diluting the 100 ng/ml standards. Use at least five standards to make up the standard curve. Suggested levels are 0, 10, 50, 75, and 100 ng/ml. Prepare quality control samples by making a separate 10 µg/ml standard and diluting until it is in the range of the samples. Prepare any standards containing less than 1 µg/ml of metal on a daily basis. Standards containing greater than 1 µg/ml of metal should be stable for a minimum of 1 to 2 weeks.
7.5.36 Matrix Modifiers.
7.5.36.1 Nickel Nitrate, 1 Percent (V/V). Dissolve 4.956 g of Ni(N0 3 ) 2 ·6H 2 0 or other nickel compound suitable for preparation of this matrix modifier in approximately 50 ml of water in a 100-ml volumetric flask. Dilute to 100 ml with water.
7.5.36.2 Nickel Nitrate, 0.1 Percent (V/V). Dilute 10 ml of 1 percent nickel nitrate solution to 100 ml with water. Inject an equal amount of sample and this modifier into the graphite furnace during GFAAS analysis for As.
7.5.36.3 Lanthanum. Carefully dissolve 0.5864 g of La 2 0 3 in 10 ml of concentrated HN0 3 , and dilute the solution by adding it with stirring to approximately 50 ml of water. Dilute to 100 ml with water, and mix well. Inject an equal amount of sample and this modifier into the graphite furnace during GFAAS analysis for Pb.
7.5.37 Whatman 40 and 541 Filter Papers (or equivalent). For filtration of digested samples.
8.0 Sample Collection, Preservation, Transport, and Storage
8.1 Sampling. The complexity of this method is such that, to obtain reliable results, both testers and analysts must be trained and experienced with the test procedures, including source sampling; reagent preparation and handling; sample handling; safety equipment and procedures; analytical calculations; reporting; and the specific procedural descriptions throughout this method.
8.1.1 Pretest Preparation. Follow the same general procedure given in Method 5, section 8.1, except that, unless particulate emissions are to be determined, the filter need not be desiccated or weighed. First, rinse all sampling train glassware with hot tap water and then wash in hot soapy water. Next, rinse glassware three times with tap water, followed by three additional rinses with water. Then soak all glassware in a 10 percent (V/V) nitric acid solution for a minimum of 4 hours, rinse three times with water, rinse a final time with acetone, and allow to air dry. Cover all glassware openings where contamination can occur until the sampling train is assembled for sampling.
8.1.2 Preliminary Determinations. Same as Method 5, section 8.1.2.
8.1.3 Preparation of Sampling Train.
8.1.3.1 Set up the sampling train as shown in Figure 29-1. Follow the same general procedures given in Method 5, section 8.3, except place 100 ml of the HNO 3 /H 2 O 2 solution (Section 7.3.1 of this method) in each of the second and third impingers as shown in Figure 29-1. Place 100 ml of the acidic KMnO 4 absorbing solution (Section 7.3.2 of this method) in each of the fifth and sixth impingers as shown in Figure 29-1, and transfer approximately 200 to 300 g of pre-weighed silica gel from its container to the last impinger. Alternatively, the silica gel may be weighed directly in the impinger just prior to final train assembly.
8.1.3.2 Based on the specific source sampling conditions, the use of an empty first impinger can be eliminated if the moisture to be collected in the impingers will be less than approximately 100 ml.
8.1.3.3 If Hg analysis will not be performed, the fourth, fifth, and sixth impingers as shown in Figure 29-1 are not required.
8.1.3.4 To insure leak-free sampling train connections and to prevent possible sample contamination problems, use Teflon tape or other non-contaminating material instead of silicone grease.
Precaution: Exercise extreme care to prevent contamination within the train. Prevent the acidic KMnO 4 from contacting any glassware that contains sample material to be analyzed for Mn. Prevent acidic H 2 O 2 from mixing with the acidic KMnO 4 .
8.1.4 Leak-Check Procedures. Follow the leak-check procedures given in Method 5, section 8.4.2 (Pretest Leak-Check), section 8.4.3 (Leak-Checks During the Sample Run), and section 8.4.4 (Post-Test Leak-Checks).
8.1.5 Sampling Train Operation. Follow the procedures given in Method 5, section 8.5. When sampling for Hg, use a procedure analogous to that described in section 8.1 of Method 101A, 40 CFR Part 61, Appendix B, if necessary to maintain the desired color in the last acidified permanganate impinger. For each run, record the data required on a data sheet such as the one shown in Figure 5-3 of Method 5.
8.1.6 Calculation of Percent Isokinetic. Same as Method 5, section 12.11.
8.2 Sample Recovery.
8.2.1 Begin cleanup procedures as soon as the probe is removed from the stack at the end of a sampling period. The probe should be allowed to cool prior to sample recovery. When it can be safely handled, wipe off all external particulate matter near the tip of the probe nozzle and place a rinsed, non-contaminating cap over the probe nozzle to prevent losing or gaining particulate matter. Do not cap the probe tip tightly while the sampling train is cooling; a vacuum can form in the filter holder with the undesired result of drawing liquid from the impingers onto the filter.
8.2.2 Before moving the sampling train to the cleanup site, remove the probe from the sampling train and cap the open outlet. Be careful not to lose any condensate that might be present. Cap the filter inlet where the probe was fastened. Remove the umbilical cord from the last impinger and cap the impinger. Cap the filter holder outlet and impinger inlet. Use non-contaminating caps, whether ground-glass stoppers, plastic caps, serum caps, or Teflon ® tape to close these openings.
8.2.3 Alternatively, the following procedure may be used to disassemble the train before the probe and filter holder/oven are completely cooled: Initially disconnect the filter holder outlet/impinger inlet and loosely cap the open ends. Then disconnect the probe from the filter holder or cyclone inlet and loosely cap the open ends. Cap the probe tip and remove the umbilical cord as previously described.
8.2.4 Transfer the probe and filter-impinger assembly to a cleanup area that is clean and protected from the wind and other potential causes of contamination or loss of sample. Inspect the train before and during disassembly and note any abnormal conditions. Take special precautions to assure that all the items necessary for recovery do not contaminate the samples. The sample is recovered and treated as follows (see schematic in Figures 29-2a and 29-2b):
8.2.5 Container No. 1 (Sample Filter). Carefully remove the filter from the filter holder and place it in its labeled petri dish container. To handle the filter, use either acid-washed polypropylene or Teflon coated tweezers or clean, disposable surgical gloves rinsed with water and dried. If it is necessary to fold the filter, make certain the particulate cake is inside the fold. Carefully transfer the filter and any particulate matter or filter fibers that adhere to the filter holder gasket to the petri dish by using a dry (acid-cleaned) nylon bristle brush. Do not use any metal-containing materials when recovering this train. Seal the labeled petri dish.
8.2.6 Container No. 2 (Acetone Rinse). Perform this procedure only if a determination of particulate emissions is to be made. Quantitatively recover particulate matter and any condensate from the probe nozzle, probe fitting, probe liner, and front half of the filter holder by washing these components with a total of 100 ml of acetone, while simultaneously taking great care to see that no dust on the outside of the probe or other surfaces gets in the sample. The use of exactly 100 ml is necessary for the subsequent blank correction procedures. Distilled water may be used instead of acetone when approved by the Administrator and shall be used when specified by the Administrator; in these cases, save a water blank and follow the Administrator's directions on analysis.
8.2.6.1 Carefully remove the probe nozzle, and clean the inside surface by rinsing with acetone from a wash bottle while brushing with a non-metallic brush. Brush until the acetone rinse shows no visible particles, then make a final rinse of the inside surface with acetone.
8.2.6.2 Brush and rinse the sample exposed inside parts of the probe fitting with acetone in a similar way until no visible particles remain. Rinse the probe liner with acetone by tilting and rotating the probe while squirting acetone into its upper end so that all inside surfaces will be wetted with acetone. Allow the acetone to drain from the lower end into the sample container. A funnel may be used to aid in transferring liquid washings to the container. Follow the acetone rinse with a non-metallic probe brush. Hold the probe in an inclined position, squirt acetone into the upper end as the probe brush is being pushed with a twisting action three times through the probe. Hold a sample container underneath the lower end of the probe, and catch any acetone and particulate matter which is brushed through the probe until no visible particulate matter is carried out with the acetone or until none remains in the probe liner on visual inspection. Rinse the brush with acetone, and quantitatively collect these washings in the sample container. After the brushing, make a final acetone rinse of the probe as described above.
8.2.6.3 It is recommended that two people clean the probe to minimize sample losses. Between sampling runs, keep brushes clean and protected from contamination. Clean the inside of the front-half of the filter holder by rubbing the surfaces with a non-metallic brush and rinsing with acetone. Rinse each surface three times or more if needed to remove visible particulate. Make a final rinse of the brush and filter holder. After all acetone washings and particulate matter have been collected in the sample container, tighten the lid so that acetone will not leak out when shipped to the laboratory. Mark the height of the fluid level to determine whether or not leakage occurred during transport. Clearly label the container to identify its contents.
8.2.7 Container No. 3 (Probe Rinse). Keep the probe assembly clean and free from contamination during the probe rinse. Rinse the probe nozzle and fitting, probe liner, and front-half of the filter holder thoroughly with a total of 100 ml of 0.1 N HNO 3 , and place the wash into a sample storage container. Perform the rinses as applicable and generally as described in Method 12, section 8.7.1. Record the volume of the rinses. Mark the height of the fluid level on the outside of the storage container and use this mark to determine if leakage occurs during transport. Seal the container, and clearly label the contents. Finally, rinse the nozzle, probe liner, and front-half of the filter holder with water followed by acetone, and discard these rinses.
Note:
The use of a total of exactly 100 ml is necessary for the subsequent blank correction procedures.
8.2.8 Container No. 4 (Impingers 1 through 3, Moisture Knockout Impinger, when used, HNO 3 /H 2 O 2 Impingers Contents and Rinses). Due to the potentially large quantity of liquid involved, the tester may place the impinger solutions from impingers 1 through 3 in more than one container, if necessary. Measure the liquid in the first three impingers to within 0.5 ml using a graduated cylinder. Record the volume. This information is required to calculate the moisture content of the sampled flue gas. Clean each of the first three impingers, the filter support, the back half of the filter housing, and connecting glassware by thoroughly rinsing with 100 ml of 0.1 N HNO 3 using the procedure as applicable in Method 12, section 8.7.3.
Note:
The use of exactly 100 ml of 0.1 N HNO 3 rinse is necessary for the subsequent blank correction procedures. Combine the rinses and impinger solutions, measure and record the final total volume. Mark the height of the fluid level, seal the container, and clearly label the contents.
8.2.9 Container Nos. 5A (0.1 N HNO 3 ), 5B (KMnO 4 /H 2 SO 4 absorbing solution), and 5C (8 N HCl rinse and dilution).
8.2.9.1 When sampling for Hg, pour all the liquid from the impinger (normally impinger No. 4) that immediately preceded the two permanganate impingers into a graduated cylinder and measure the volume to within 0.5 ml. This information is required to calculate the moisture content of the sampled flue gas. Place the liquid in Container No. 5A. Rinse the impinger with exactly 100 ml of 0.1 N HNO 3 and place this rinse in Container No. 5A.
8.2.9.2 Pour all the liquid from the two permanganate impingers into a graduated cylinder and measure the volume to within 0.5 ml. This information is required to calculate the moisture content of the sampled flue gas. Place this acidic KMnO 4 solution into Container No. 5B. Using a total of exactly 100 ml of fresh acidified KMnO 4 solution for all rinses (approximately 33 ml per rinse), rinse the two permanganate impingers and connecting glassware a minimum of three times. Pour the rinses into Container No. 5B, carefully assuring transfer of all loose precipitated materials from the two impingers. Similarly, using 100 ml total of water, rinse the permanganate impingers and connecting glass a minimum of three times, and pour the rinses into Container 5B, carefully assuring transfer of any loose precipitated material. Mark the height of the fluid level, and clearly label the contents. Read the Precaution: in section 7.3.2.
Note:
Due to the potential reaction of KMnO 4 with acid, pressure buildup can occur in the sample storage bottles. Do not fill these bottles completely and take precautions to relieve excess pressure. A No. 70-72 hole drilled in the container cap and Teflon liner has been used successfully.
8.2.9.3 Wash the two permanganate impingers with 25 ml of 8 N HCl, and place the wash in a separate sample container labeled No. 5C containing 200 ml of water. First, place 200 ml of water in the container. Then wash the impinger walls and stem with the 8 N HCl by turning the impinger on its side and rotating it so that the HCl contacts all inside surfaces. Use a total of only 25 ml of 8 N HCl for rinsing both permanganate impingers combined. Rinse the first impinger, then pour the actual rinse used for the first impinger into the second impinger for its rinse. Finally, pour the 25 ml of 8 N HCl rinse carefully into the container with the 200 ml of water. Mark the height of the fluid level on the outside of the container in order to determine if leakage occurs during transport.
8.2.10 Container No. 6 (Silica Gel). Note the color of the indicating silica gel to determine whether it has been completely spent and make a notation of its condition. Transfer the silica gel from its impinger to its original container and seal it. The tester may use a funnel to pour the silica gel and a rubber policeman to remove the silica gel from the impinger. The small amount of particles that might adhere to the impinger wall need not be removed. Do not use water or other liquids to transfer the silica gel since weight gained in the silica gel impinger is used for moisture calculations. Alternatively, if a balance is available in the field, record the weight of the spent silica gel (or silica gel plus impinger) to the nearest 0.5 g.
8.2.11 Container No. 7 (Acetone Blank). If particulate emissions are to be determined, at least once during each field test, place a 100-ml portion of the acetone used in the sample recovery process into a container labeled No. 7. Seal the container.
8.2.12 Container No. 8A (0.1 N HNO 3 Blank). At least once during each field test, place 300 ml of the 0.1 N HNO 3 solution used in the sample recovery process into a container labeled No. 8A. Seal the container.
8.2.13 Container No. 8B (Water Blank). At least once during each field test, place 100 ml of the water used in the sample recovery process into a container labeled No. 8B. Seal the container.
8.2.14 Container No. 9 (5 Percent HNO 3 /10 Percent H 2 O 2 Blank). At least once during each field test, place 200 ml of the 5 Percent HNO 3 /10 Percent H 2 O 2 solution used as the nitric acid impinger reagent into a container labeled No. 9. Seal the container.
8.2.15 Container No. 10 (Acidified KMnO 4 Blank). At least once during each field test, place 100 ml of the acidified KMnO 4 solution used as the impinger solution and in the sample recovery process into a container labeled No. 10. Prepare the container as described in section 8.2.9.2. Read the Precaution : in section 7.3.2 and read the note in section 8.2.9.2.
8.2.16 Container No. 11 (8 N HCl Blank). At least once during each field test, place 200 ml of water into a sample container labeled No. 11. Then carefully add with stirring 25 ml of 8 N HCl. Mix well and seal the container.
8.2.17 Container No. 12 (Sample Filter Blank). Once during each field test, place into a petri dish labeled No. 12 three unused blank filters from the same lot as the sampling filters. Seal the petri dish.
8.3 Sample Preparation. Note the level of the liquid in each of the containers and determine if any sample was lost during shipment. If a noticeable amount of leakage has occurred, either void the sample or use methods, subject to the approval of the Administrator, to correct the final results. A diagram illustrating sample preparation and analysis procedures for each of the sample train components is shown in Figure 29-3.
8.3.1 Container No. 1 (Sample Filter).
8.3.1.1 If particulate emissions are being determined, first desiccate the filter and filter catch without added heat (do not heat the filters to speed the drying) and weigh to a constant weight as described in section 11.2.1 of Method 5.
8.3.1.2 Following this procedure, or initially, if particulate emissions are not being determined in addition to metals analysis, divide the filter with its filter catch into portions containing approximately 0.5 g each. Place the pieces in the analyst's choice of either individual microwave pressure relief vessels or Parr Bombs. Add 6 ml of concentrated HNO 3 and 4 ml of concentrated HF to each vessel. For microwave heating, microwave the samples for approximately 12 to 15 minutes total heating time as follows: heat for 2 to 3 minutes, then turn off the microwave for 2 to 3 minutes, then heat for 2 to 3 minutes, etc., continue this alternation until the 12 to 15 minutes total heating time are completed (this procedure should comprise approximately 24 to 30 minutes at 600 watts). Microwave heating times are approximate and are dependent upon the number of samples being digested simultaneously. Sufficient heating is evidenced by sorbent reflux within the vessel. For conventional heating, heat the Parr Bombs at 140°C (285°F) for 6 hours. Then cool the samples to room temperature, and combine with the acid digested probe rinse as required in section 8.3.3.
8.3.1.3 If the sampling train includes an optional glass cyclone in front of the filter, prepare and digest the cyclone catch by the procedures described in section 8.3.1.2 and then combine the digestate with the digested filter sample.
8.3.2 Container No. 2 (Acetone Rinse). Note the level of liquid in the container and confirm on the analysis sheet whether or not leakage occurred during transport. If a noticeable amount of leakage has occurred, either void the sample or use methods, subject to the approval of the Administrator, to correct the final results. Measure the liquid in this container either volumetrically within 1 ml or gravimetrically within 0.5 g. Transfer the contents to an acid-cleaned, tared 250-ml beaker and evaporate to dryness at ambient temperature and pressure. If particulate emissions are being determined, desiccate for 24 hours without added heat, weigh to a constant weight according to the procedures described in section 11.2.1 of Method 5, and report the results to the nearest 0.1 mg. Redissolve the residue with 10 ml of concentrated HNO 3 . Quantitatively combine the resultant sample, including all liquid and any particulate matter, with Container No. 3 before beginning section 8.3.3.
8.3.3 Container No. 3 (Probe Rinse). Verify that the pH of this sample is 2 or lower. If it is not, acidify the sample by careful addition with stirring of concentrated HNO 3 to pH 2. Use water to rinse the sample into a beaker, and cover the beaker with a ribbed watch glass. Reduce the sample volume to approximately 20 ml by heating on a hot plate at a temperature just below boiling. Digest the sample in microwave vessels or Parr Bombs by quantitatively transferring the sample to the vessel or bomb, carefully adding the 6 ml of concentrated HNO 3 , 4 ml of concentrated HF, and then continuing to follow the procedures described in section 8.3.1.2. Then combine the resultant sample directly with the acid digested portions of the filter prepared previously in section 8.3.1.2. The resultant combined sample is referred to as “Sample Fraction 1”. Filter the combined sample using Whatman 541 filter paper. Dilute to 300 ml (or the appropriate volume for the expected metals concentration) with water. This diluted sample is “Analytical Fraction 1”. Measure and record the volume of Analytical Fraction 1 to within 0.1 ml. Quantitatively remove a 50-ml aliquot and label as “Analytical Fraction 1B”. Label the remaining 250-ml portion as “Analytical Fraction 1A”. Analytical Fraction 1A is used for ICAP or AAS analysis for all desired metals except Hg. Analytical Fraction 1B is used for the determination of front-half Hg.
8.3.4 Container No. 4 (Impingers 1-3). Measure and record the total volume of this sample to within 0.5 ml and label it “Sample Fraction 2”. Remove a 75- to 100-ml aliquot for Hg analysis and label the aliquot “Analytical Fraction 2B”. Label the remaining portion of Container No. 4 as “Sample Fraction 2A”. Sample Fraction 2A defines the volume of Analytical Fraction 2A prior to digestion. All of Sample Fraction 2A is digested to produce “Analytical Fraction 2A”. Analytical Fraction 2A defines the volume of Sample Fraction 2A after its digestion and the volume of Analytical Fraction 2A is normally 150 ml. Analytical Fraction 2A is analyzed for all metals except Hg. Verify that the pH of Sample Fraction 2A is 2 or lower. If necessary, use concentrated HNO 3 by careful addition and stirring to lower Sample Fraction 2A to pH 2. Use water to rinse Sample Fraction 2A into a beaker and then cover the beaker with a ribbed watchglass. Reduce Sample Fraction 2A to approximately 20 ml by heating on a hot plate at a temperature just below boiling. Then follow either of the digestion procedures described in sections 8.3.4.1 or 8.3.4.2.
8.3.4.1 Conventional Digestion Procedure. Add 30 ml of 50 percent HNO 3 , and heat for 30 minutes on a hot plate to just below boiling. Add 10 ml of 3 percent H 2 O 2 and heat for 10 more minutes. Add 50 ml of hot water, and heat the sample for an additional 20 minutes. Cool, filter the sample, and dilute to 150 ml (or the appropriate volume for the expected metals concentrations) with water. This dilution produces Analytical Fraction 2A. Measure and record the volume to within 0.1 ml.
8.3.4.2 Microwave Digestion Procedure. Add 10 ml of 50 percent HNO 3 and heat for 6 minutes total heating time in alternations of 1 to 2 minutes at 600 Watts followed by 1 to 2 minutes with no power, etc., similar to the procedure described in section 8.3.1. Allow the sample to cool. Add 10 ml of 3 percent H 2 O 2 and heat for 2 more minutes. Add 50 ml of hot water, and heat for an additional 5 minutes. Cool, filter the sample, and dilute to 150 ml (or the appropriate volume for the expected metals concentrations) with water. This dilution produces Analytical Fraction 2A. Measure and record the volume to within 0.1 ml.
Note:
All microwave heating times given are approximate and are dependent upon the number of samples being digested at a time. Heating times as given above have been found acceptable for simultaneous digestion of up to 12 individual samples. Sufficient heating is evidenced by solvent reflux within the vessel.
8.3.5 Container No. 5A (Impinger 4), Container Nos. 5B and 5C (Impingers 5 and 6). Keep the samples in Containers Nos. 5A, 5B, and 5C separate from each other. Measure and record the volume of 5A to within 0.5 ml. Label the contents of Container No. 5A to be Analytical Fraction 3A. To remove any brown MnO 2 precipitate from the contents of Container No. 5B, filter its contents through Whatman 40 filter paper into a 500 ml volumetric flask and dilute to volume with water. Save the filter for digestion of the brown MnO 2 precipitate. Label the 500 ml filtrate from Container No. 5B to be Analytical Fraction 3B. Analyze Analytical Fraction 3B for Hg within 48 hours of the filtration step. Place the saved filter, which was used to remove the brown MnO 2 precipitate, into an appropriately sized vented container, which will allow release of any gases including chlorine formed when the filter is digested. In a laboratory hood which will remove any gas produced by the digestion of the MnO 2 , add 25 ml of 8 N HCl to the filter and allow to digest for a minimum of 24 hours at room temperature. Filter the contents of Container No. 5C through a Whatman 40 filter into a 500-ml volumetric flask. Then filter the result of the digestion of the brown MnO 2 from Container No. 5B through a Whatman 40 filter into the same 500-ml volumetric flask, and dilute and mix well to volume with water. Discard the Whatman 40 filter. Mark this combined 500-ml dilute HCl solution as Analytical Fraction 3C.
8.3.6 Container No. 6 (Silica Gel). Weigh the spent silica gel (or silica gel plus impinger) to the nearest 0.5 g using a balance.
9.0 Quality Control
9.1 Field Reagent Blanks, if analyzed. Perform the digestion and analysis of the blanks in Container Nos. 7 through 12 that were produced in sections 8.2.11 through 8.2.17, respectively. For Hg field reagent blanks, use a 10 ml aliquot for digestion and analysis.
9.1.1 Digest and analyze one of the filters from Container No. 12 per section 8.3.1, 100 ml from Container No. 7 per section 8.3.2, and 100 ml from Container No. 8A per section 8.3.3. This step produces blanks for Analytical Fractions 1A and 1B.
9.1.2 Combine 100 ml of Container No. 8A with 200 ml from Container No. 9, and digest and analyze the resultant volume per section 8.3.4. This step produces blanks for Analytical Fractions 2A and 2B.
9.1.3 Digest and analyze a 100-ml portion of Container No. 8A to produce a blank for Analytical Fraction 3A.
9.1.4 Combine 100 ml from Container No. 10 with 33 ml from Container No. 8B to produce a blank for Analytical Fraction 3B. Filter the resultant 133 ml as described for Container No. 5B in section 8.3.5, except do not dilute the 133 ml. Analyze this blank for Hg within 48 hr of the filtration step, and use 400 ml as the blank volume when calculating the blank mass value. Use the actual volumes of the other analytical blanks when calculating their mass values.
9.1.5 Digest the filter that was used to remove any brown MnO 2 precipitate from the blank for Analytical Fraction 3B by the same procedure as described in section 8.3.5 for the similar sample filter. Filter the digestate and the contents of Container No. 11 through Whatman 40 paper into a 500-ml volumetric flask, and dilute to volume with water. These steps produce a blank for Analytical Fraction 3C.
9.1.6 Analyze the blanks for Analytical Fraction Blanks 1A and 2A per section 11.1.1 and/or section 11.1.2. Analyze the blanks for Analytical Fractions 1B, 2B, 3A, 3B, and 3C per section 11.1.3. Analysis of the blank for Analytical Fraction 1A produces the front-half reagent blank correction values for the desired metals except for Hg; Analysis of the blank for Analytical Fraction 1B produces the front-half reagent blank correction value for Hg. Analysis of the blank for Analytical Fraction 2A produces the back-half reagent blank correction values for all of the desired metals except for Hg, while separate analyses of the blanks for Analytical Fractions 2B, 3A, 3B, and 3C produce the back-half reagent blank correction value for Hg.
9.2 Quality Control Samples. Analyze the following quality control samples.
9.2.1 ICAP and ICP-MS Analysis. Follow the respective quality control descriptions in section 8 of Methods 6010 and 6020 in EPA Publication SW-846 Third Edition (November 1986) including updates I, II, IIA, IIB and III, as incorporated by reference in §60.17(i) . For the purposes of a source test that consists of three sample runs, modify those requirements to include the following: two instrument check standard runs, two calibration blank runs, one interference check sample at the beginning of the analysis (analyze by Method of Standard Additions unless within 25 percent), one quality control sample to check the accuracy of the calibration standards (required to be within 25 percent of calibration), and one duplicate analysis (required to be within 20 percent of average or repeat all analyses).
9.2.2 Direct Aspiration AAS and/or GFAAS Analysis for Sb, As, Ba, Be, Cd, Cu, Cr, Co, Pb, Ni, Mn, Hg, P, Se, Ag, Tl, and Zn. Analyze all samples in duplicate. Perform a matrix spike on at least one front-half sample and one back-half sample, or one combined sample. If recoveries of less than 75 percent or greater than 125 percent are obtained for the matrix spike, analyze each sample by the Method of Standard Additions. Analyze a quality control sample to check the accuracy of the calibration standards. If the results are not within 20 percent, repeat the calibration.
9.2.3 CVAAS Analysis for Hg. Analyze all samples in duplicate. Analyze a quality control sample to check the accuracy of the calibration standards (if not within 15 percent, repeat calibration). Perform a matrix spike on one sample (if not within 25 percent, analyze all samples by the Method of Standard Additions). Additional information on quality control can be obtained from Method 7470 in EPA Publication SW-846 Third Edition (November 1986) including updates I, II, IIA, IIB and III, as incorporated by reference in §60.17(i) , or in Standard Methods for Water and Wastewater Method 303F.
10.0 Calibration and Standardization
Note:
Maintain a laboratory log of all calibrations.
10.1 Sampling Train Calibration. Calibrate the sampling train components according to the indicated sections of Method 5: Probe Nozzle (Section 10.1); Pitot Tube (Section 10.2); Metering System (Section 10.3); Probe Heater (Section 10.4); Temperature Sensors (Section 10.5); Leak-Check of the Metering System (Section 8.4.1); and Barometer (Section 10.6).
10.2 Inductively Coupled Argon Plasma Spectrometer Calibration. Prepare standards as outlined in section 7.5. Profile and calibrate the instrument according to the manufacturer's recommended procedures using those standards. Check the calibration once per hour. If the instrument does not reproduce the standard concentrations within 10 percent, perform the complete calibration procedures. Perform ICP-MS analysis by following Method 6020 in EPA Publication SW-846 Third Edition (November 1986) including updates I, II, IIA, IIB and III, as incorporated by reference in §60.17(i) .
10.3 Atomic Absorption Spectrometer - Direct Aspiration AAS, GFAAS, and CVAAS analyses. Prepare the standards as outlined in section 7.5 and use them to calibrate the spectrometer. Calibration procedures are also outlined in the EPA methods referred to in Table 29-2 and in Method 7470 in EPA Publication SW-846 Third Edition (November 1986) including updates I, II, IIA, IIB and III, as incorporated by reference in §60.17(i) , or in Standard Methods for Water and Wastewater Method 303F (for Hg). Run each standard curve in duplicate and use the mean values to calculate the calibration line. Recalibrate the instrument approximately once every 10 to 12 samples.
10.4 Field Balance Calibration Check. Check the calibration of the balance used to weigh impingers with a weight that is at least 500g or within 50g of a loaded impinger. The weight must be ASTM E617-13 “Standard Specification for Laboratory Weights and Precision Mass Standards” (incorporated by reference-see 40 CFR 60.17) Class 6 (or better). Daily before use, the field balance must measure the weight within ±0.5g of the certified mass. If the daily balance calibration check fails, perform corrective measures and repeat the check before using balance.
10.5 Analytical Balance Calibration. Perform a multipoint calibration (at least five points spanning the operational range) of the analytical balance before the first use, and semiannually thereafter. The calibration of the analytical balance must be conducted using ASTM E617-13 “Standard Specification for Laboratory Weights and Precision Mass Standards” (incorporated by reference - see 40 CFR 60.17) Class 2 (or better) tolerance weights. Audit the balance each day it is used for gravimetric measurements by weighing at least one ASTM E617-13 Class 2 tolerance (or better) calibration weight that corresponds to 50 to 150 percent of the weight of one filter or between 1g and 5g. If the scale cannot reproduce the value of the calibration weight to within 0.5 mg of the certified mass, perform corrective measures, and conduct the multipoint calibration before use.
11.0 Analytical Procedure
11.1 Sample Analysis. For each sampling train sample run, seven individual analytical samples are generated; two for all desired metals except Hg, and five for Hg. A schematic identifying each sample container and the prescribed analytical preparation and analysis scheme is shown in Figure 29-3. The first two analytical samples, labeled Analytical Fractions 1A and 1B, consist of the digested samples from the front-half of the train. Analytical Fraction 1A is for ICAP, ICP-MS or AAS analysis as described in sections 11.1.1 and 11.1.2, respectively. Analytical Fraction 1B is for front-half Hg analysis as described in section 11.1.3. The contents of the back-half of the train are used to prepare the third through seventh analytical samples. The third and fourth analytical samples, labeled Analytical Fractions 2A and 2B, contain the samples from the moisture removal impinger No. 1, if used, and HNO 3 /H 2 O 2 impingers Nos. 2 and 3. Analytical Fraction 2A is for ICAP, ICP-MS or AAS analysis for target metals, except Hg. Analytical Fraction 2B is for analysis for Hg. The fifth through seventh analytical samples, labeled Analytical Fractions 3A, 3B, and 3C, consist of the impinger contents and rinses from the empty impinger No. 4 and the H 2 SO 4 /KMnO 4 Impingers Nos. 5 and 6. These analytical samples are for analysis for Hg as described in section 11.1.3. The total back-half Hg catch is determined from the sum of Analytical Fractions 2B, 3A, 3B, and 3C. Analytical Fractions 1A and 2A can be combined proportionally prior to analysis.
11.1.1 ICAP and ICP-MS Analysis. Analyze Analytical Fractions 1A and 2A by ICAP using Method 6010 or Method 200.7 (40 CFR 136, Appendix C). Calibrate the ICAP, and set up an analysis program as described in Method 6010 or Method 200.7. Follow the quality control procedures described in section 9.2.1. Recommended wavelengths for analysis are as shown in Table 29-2. These wavelengths represent the best combination of specificity and potential detection limit. Other wavelengths may be substituted if they can provide the needed specificity and detection limit, and are treated with the same corrective techniques for spectral interference. Initially, analyze all samples for the target metals (except Hg) plus Fe and Al. If Fe and Al are present, the sample might have to be diluted so that each of these elements is at a concentration of less than 50 ppm so as to reduce their spectral interferences on As, Cd, Cr, and Pb. Perform ICP-MS analysis by following Method 6020 in EPA Publication SW-846 Third Edition (November 1986) including updates I, II, IIA, IIB and III, as incorporated by reference in §60.17(i) .
Note:
When analyzing samples in a HF matrix, an alumina torch should be used; since all front-half samples will contain HF, use an alumina torch.
11.1.2 AAS by Direct Aspiration and/or GFAAS. If analysis of metals in Analytical Fractions 1A and 2A by using GFAAS or direct aspiration AAS is needed, use Table 29-3 to determine which techniques and procedures to apply for each target metal. Use Table 29-3, if necessary, to determine techniques for minimization of interferences. Calibrate the instrument according to section 10.3 and follow the quality control procedures specified in section 9.2.2.
11.1.3 CVAAS Hg analysis. Analyze Analytical Fractions 1B, 2B, 3A, 3B, and 3C separately for Hg using CVAAS following the method outlined in Method 7470 in EPA Publication SW-846 Third Edition (November 1986) including updates I, II, IIA, IIB and III, as incorporated by reference in §60.17(i) , or in Standard Methods for Water and Wastewater Analysis, 15th Edition, Method 303F, or, optionally using note no. 2 at the end of this section. Set up the calibration curve (zero to 1000 ng) as described in Method 7470 or similar to Method 303F using 300-ml BOD bottles instead of Erlenmeyers. Perform the following for each Hg analysis. From each original sample, select and record an aliquot in the size range from 1 ml to 10 ml. If no prior knowledge of the expected amount of Hg in the sample exists, a 5 ml aliquot is suggested for the first dilution to 100 ml (see note no. 1 at end of this section). The total amount of Hg in the aliquot shall be less than 1 µg and within the range (zero to 1000 ng) of the calibration curve. Place the sample aliquot into a separate 300-ml BOD bottle, and add enough water to make a total volume of 100 ml. Next add to it sequentially the sample digestion solutions and perform the sample preparation described in the procedures of Method 7470 or Method 303F. (See note no. 2 at the end of this section). If the maximum readings are off-scale (because Hg in the aliquot exceeded the calibration range; including the situation where only a 1-ml aliquot of the original sample was digested), then dilute the original sample (or a portion of it) with 0.15 percent HNO 3 (1.5 ml concentrated HNO 3 per liter aqueous solution) so that when a 1- to 10-ml aliquot of the “0.15 HNO 3 percent dilution of the original sample” is digested and analyzed by the procedures described above, it will yield an analysis within the range of the calibration curve.
Note No. 1:
When Hg levels in the sample fractions are below the in-stack detection limit given in Table 29-1, select a 10 ml aliquot for digestion and analysis as described.
Note No. 2:
Optionally, Hg can be analyzed by using the CVAAS analytical procedures given by some instrument manufacturer's directions. These include calibration and quality control procedures for the Leeman Model PS200, the Perkin Elmer FIAS systems, and similar models, if available, of other instrument manufacturers. For digestion and analyses by these instruments, perform the following two steps: (1), Digest the sample aliquot through the addition of the aqueous hydroxylamine hydrochloride/sodium chloride solution the same as described in this section: ( The Leeman, Perkin Elmer, and similar instruments described in this note add automatically the necessary stannous chloride solution during the automated analysis of Hg.) ; (2), Upon completion of the digestion described in (1), analyze the sample according to the instrument manufacturer's directions. This approach allows multiple (including duplicate) automated analyses of a digested sample aliquot.
12.0 Data Analysis and Calculations
12.1 Nomenclature.
A = Analytical detection limit, µg/ml.
B = Liquid volume of digested sample prior to aliquotting for analysis, ml.
C = Stack sample gas volume, dsm 3 .
C a1 = Concentration of metal in Analytical Fraction 1A as read from the standard curve, µg/ml.
C a2 = Concentration of metal in Analytical Fraction 2A as read from the standard curve, (µg/ml).
C s = Concentration of a metal in the stack gas, mg/dscm.
D = In-stack detection limit, µg/m 3 .
F a = Aliquot factor, volume of Sample Fraction 2 divided by volume of Sample Fraction 2A (see section 8.3.4.)
F d = Dilution factor (F d = the inverse of the fractional portion of the concentrated sample in the solution actually used in the instrument to produce the reading C a1 . For example, if a 2 ml aliquot of Analytical Fraction 1A is diluted to 10 ml to place it in the calibration range, F d = 5).
Hg bh = Total mass of Hg collected in the back-half of the sampling train, µg.
Hg bh2 = Total mass of Hg collected in Sample Fraction 2, µg.
Hg bh3(A,B,C) = Total mass of Hg collected separately in Fraction 3A, 3B, or 3C, µg.
Hg bhb = Blank correction value for mass of Hg detected in back-half field reagent blanks, µg.
Hg fh = Total mass of Hg collected in the front-half of the sampling train (Sample Fraction 1), µg.
Hg fhb = Blank correction value for mass of Hg detected in front-half field reagent blank, µg.
Hg t = Total mass of Hg collected in the sampling train, µg.
M bh = Total mass of each metal (except Hg) collected in the back-half of the sampling train (Sample Fraction 2), µg.
M bhb = Blank correction value for mass of metal detected in back-half field reagent blank, µg.
M fh = Total mass of each metal (except Hg) collected in the front half of the sampling train (Sample Fraction 1), µg.
M fhb = Blank correction value for mass of metal detected in front-half field reagent blank, µg.
M t = Total mass of each metal (separately stated for each metal) collected in the sampling train, µg.
M t = Total mass of that metal collected in the sampling train, µg; (substitute Hg t for M t for the H g calculation).
Q bh2 = Quantity of Hg, µg, TOTAL in the ALIQUOT of Analytical Fraction 2B selected for digestion and analysis .
Note:
For example, if a 10 ml aliquot of Analytical Fraction 2B is taken and digested and analyzed (according to section 11.1.3 and its notes nos. 1 and 2), then calculate and use the total amount of Hg in the 10 ml aliquot for Q bh2 .
Q bh3(A,B,C) = Quantity of Hg, µg, TOTAL, separately, in the ALIQUOT of Analytical Fraction 3A, 3B, or 3C selected for digestion and analysis (see notes in sections 12.7.1 and 12.7.2 describing the quantity “Q” and calculate similarly).
Q fh = Quantity of Hg, µg, TOTAL in the ALIQUOT of Analytical Fraction 1B selected for digestion and analysis.
Note:
For example, if a 10 ml aliquot of Analytical Fraction 1B is taken and digested and analyzed (according to section 11.1.3 and its notes nos. 1 and 2), then calculate and use the total amount of Hg in the 10 ml aliquot for Q fh .
V a = Total volume of digested sample solution (Analytical Fraction 2A), ml (see section 8.3.4.1 or 8.3.4.2, as applicable).
V f1B = Volume of aliquot of Analytical Fraction 1B analyzed, ml.
Note:
For example, if a 1 ml aliquot of Analytical Fraction 1B was diluted to 50 ml with 0.15 percent HNO 3 as described in section 11.1.3 to bring it into the proper analytical range, and then 1 ml of that 50-ml was digested according to section 11.1.3 and analyzed, V f1B would be 0.02 ml.
V f2B = Volume of Analytical Fraction 2B analyzed, ml.
Note:
For example, if 1 ml of Analytical Fraction 2B was diluted to 10 ml with 0.15 percent HNO 3 as described in section 11.1.3 to bring it into the proper analytical range, and then 5 ml of that 10 ml was analyzed, V f2B would be 0.5 ml.
V f3(A,B,C) = Volume, separately, of Analytical Fraction 3A, 3B, or 3C analyzed, ml (see previous notes in sections 12.7.1 and 12.7.2, describing the quantity “V” and calculate similarly).
V m(std) = Volume of gas sample as measured by the dry gas meter, corrected to dry standard conditions, dscm.
V soln,1 = Total volume of digested sample solution (Analytical Fraction 1), ml.
V soln,1 = Total volume of Analytical Fraction 1, ml.
V soln,2 = Total volume of Sample Fraction 2, ml.
V soln,3(A,B,C) = Total volume, separately, of Analytical Fraction 3A, 3B, or 3C, ml.
K 4 = 10 −3 mg/µg.
12.2 Dry Gas Volume. Using the data from this test, calculate V m(std) , the dry gas sample volume at standard conditions as outlined in section 12.3 of Method 5.
12.3 Volume of Water Vapor and Moisture Content. Using the total volume of condensate collected during the source sampling, calculate the volume of water vapor V w(std) and the moisture content B ws of the stack gas. Use Equations 5-2 and 5-3 of Method 5.
12.4 Stack Gas Velocity. Using the data from this test and Equation 2-9 of Method 2, calculate the average stack gas velocity.
12.5 In-Stack Detection Limits. Calculate the in-stack method detection limits shown in Table 29-4 using the conditions described in section 13.3.1 as follows:
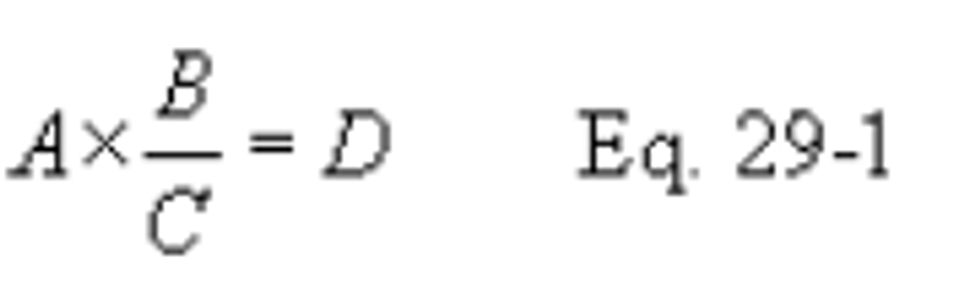
12.6 Metals (Except Hg) in Source Sample.
12.6.1 Analytical Fraction 1A, Front-Half, Metals (except Hg). Calculate separately the amount of each metal collected in Sample Fraction 1 of the sampling train using the following equation:
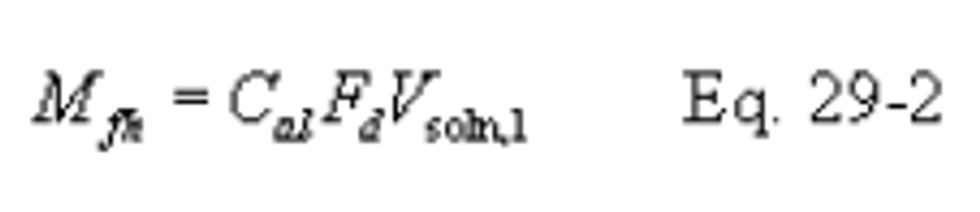
Note:
If Analytical Fractions 1A and 2A are combined, use proportional aliquots. Then make appropriate changes in Equations 29-2 through 29-4 to reflect this approach.
12.6.2 Analytical Fraction 2A, Back-Half, Metals (except Hg). Calculate separately the amount of each metal collected in Fraction 2 of the sampling train using the following equation:
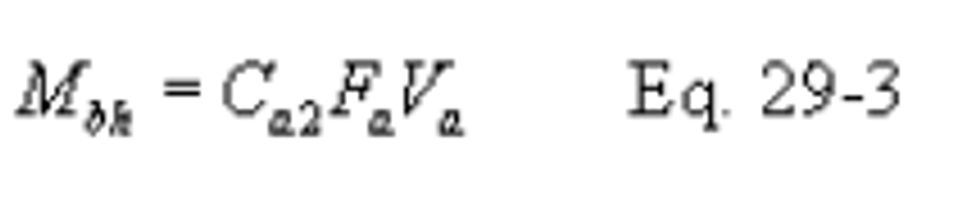
12.6.3 Total Train, Metals (except Hg). Calculate the total amount of each of the quantified metals collected in the sampling train as follows:
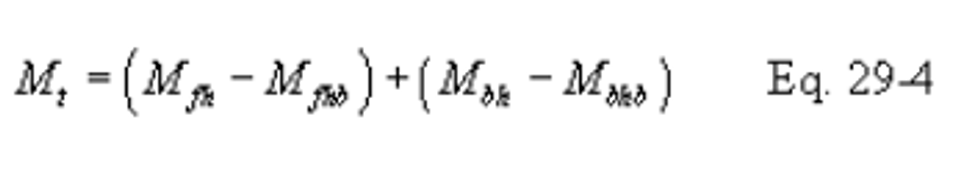
Note:
If the measured blank value for the front half (M fhb ) is in the range 0.0 to “A” µg (where “A” µg equals the value determined by multiplying 1.4 µg/in. 2 times the actual area in in. 2 of the sample filter), use M fhb to correct the emission sample value (M fh) ; if M fhb exceeds “A” µg, use the greater of I or II:
I. “A” µg.
II. The lesser of (a) M fhb , or (b) 5 percent of M fh . If the measured blank value for the back-half (M bhb ) is in the range 0.0 to 1 µg, use M bhb to correct the emission sample value (M bh ); if M bhb exceeds 1 µg, use the greater of I or II:
I. 1 µg.
II. The lesser of (a) M bhb , or (b) 5 percent of M bh .
12.7 Hg in Source Sample.
12.7.1 Analytical Fraction 1B; Front-Half Hg. Calculate the amount ofHg collected in the front-half, Sample Fraction 1, of the sampling train by using Equation 29-5:
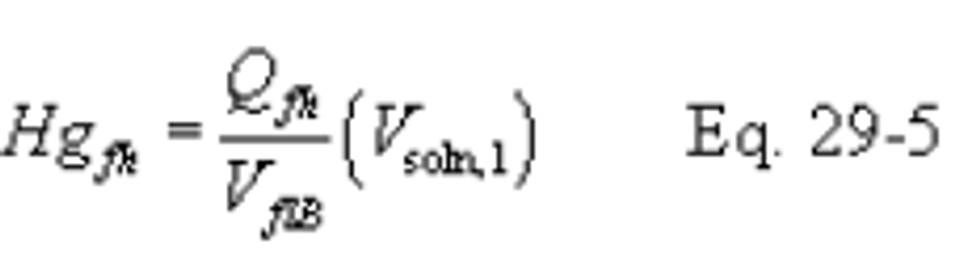
12.7.2 Analytical Fractions 2B, 3A, 3B, and 3C; Back Half Hg.
12.7.2.1 Calculate the amount of Hg collected in Sample Fraction 2 by using Equation 29-6:
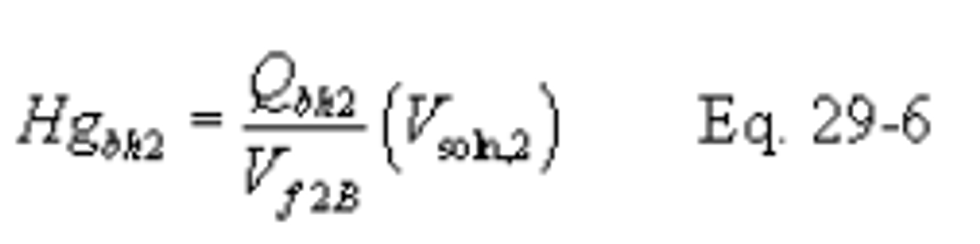
12.7.2.2 Calculate each of the back-half Hg values for Analytical Fractions 3A, 3B, and 3C by using Equation 29-7:
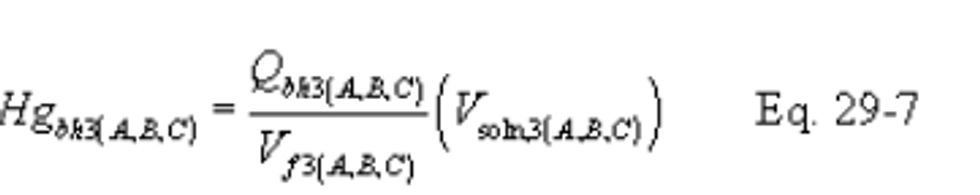
12.7.2.3 Calculate the total amount of Hg collected in the back-half of the sampling train by using Equation 29-8:
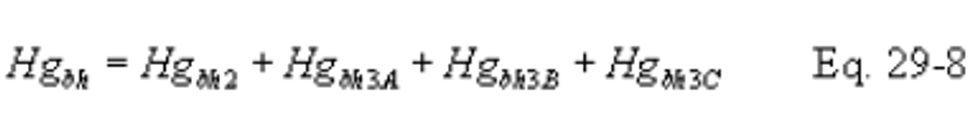
12.7.3 Total Train Hg Catch. Calculate the total amount of Hg collected in the sampling train by using Equation 29-9:

Note:
If the total of the measured blank values (Hg fhb Hg bhb ) is in the range of 0.0 to 0.6 µg, then use the total to correct the sample value (Hg fh Hg bh ); if it exceeds 0.6 µg, use the greater of I. or II:
I. 0.6 µg.
II. The lesser of (a) (Hg fhb Hg bhb ), or (b) 5 percent of the sample value (Hg fh Hg bh ).
12.8 Individual Metal Concentrations in Stack Gas. Calculate the concentration of each metal in the stack gas (dry basis, adjusted to standard conditions) by using Equation 29-10:
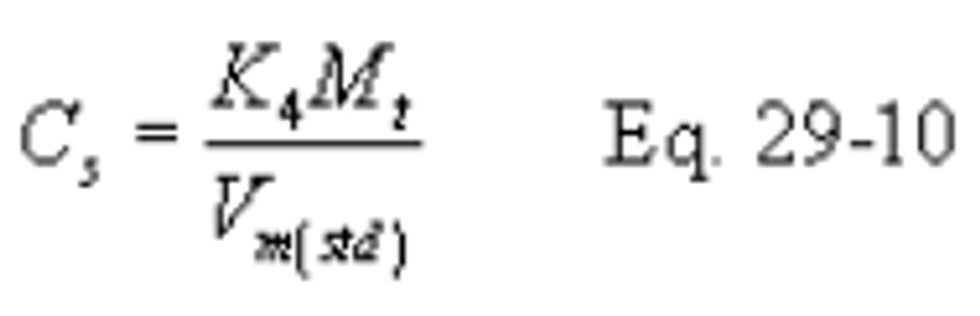
12.9 Isokinetic Variation and Acceptable Results. Same as Method 5, sections 12.11 and 12.12, respectively.
13.0 Method Performance
13.1 Range. For the analysis described and for similar analyses, the ICAP response is linear over several orders of magnitude. Samples containing metal concentrations in the nanograms per ml (ng/ml) to micrograms per ml (µg/ml) range in the final analytical solution can be analyzed using this method. Samples containing greater than approximately 50 µg/ml As, Cr, or Pb should be diluted to that level or lower for final analysis. Samples containing greater than approximately 20 µg/ml of Cd should be diluted to that level before analysis.
13.2 Analytical Detection Limits.
Note:
See section 13.3 for the description of in-stack detection limits.
13.2.1 ICAP analytical detection limits for the sample solutions (based on SW-846, Method 6010) are approximately as follows: Sb (32 ng/ml), As (53 ng/ml), Ba (2 ng/ml), Be (0.3 ng/ml), Cd (4 ng/ml), Cr (7 ng/ml), Co (7 ng/ml), Cu (6 ng/ml), Pb (42 ng/ml), Mn (2 ng/ml), Ni (15 ng/ml), P (75 ng/ml), Se (75 ng/ml), Ag (7 ng/ml), Tl (40 ng/ml), and Zn (2 ng/ml). ICP-MS analytical detection limits (based on SW-846, Method 6020) are lower generally by a factor of ten or more. Be is lower by a factor of three. The actual sample analytical detection limits are sample dependent and may vary due to the sample matrix.
13.2.2 The analytical detection limits for analysis by direct aspiration AAS (based on SW-846 , Method 7000 series) are approximately as follows: Sb (200 ng/ml), As (2 ng/ml), Ba (100 ng/ml), Be (5 ng/ml), Cd (5 ng/ml), Cr (50 ng/ml), Co (50 ng/ml), Cu (20 ng/ml), Pb (100 ng/ml), Mn (10 ng/ml), Ni (40 ng/ml), Se (2 ng/ml), Ag (10 ng/ml), Tl (100 ng/ml), and Zn (5 ng/ml).
13.2.3 The detection limit for Hg by CVAAS (on the resultant volume of the digestion of the aliquots taken for Hg analyses) can be approximately 0.02 to 0.2 ng/ml, depending upon the type of CVAAS analytical instrument used. 13.2.4 The use of GFAAS can enhance the detection limits compared to direct aspiration AAS as follows: Sb (3 ng/ml), As (1 ng/ml), Be (0.2 ng/ml), Cd (0.1 ng/ml), Cr (1 ng/ml), Co (1 ng/ml), Pb (1 ng/ml), Se (2 ng/ml), and Tl (1 ng/ml).
13.3 In-stack Detection Limits.
13.3.1 For test planning purposes in-stack detection limits can be developed by using the following information: (1) The procedures described in this method, (2) the analytical detection limits described in section 13.2 and in SW-846, (3) the normal volumes of 300 ml (Analytical Fraction 1) for the front-half and 150 ml (Analytical Fraction 2A) for the back-half samples, and (4) a stack gas sample volume of 1.25 m 3 . The resultant in-stack method detection limits for the above set of conditions are presented in Table 29-1 and were calculated by using Eq. 29-1 shown in section 12.5.
13.3.2 To ensure optimum precision/resolution in the analyses, the target concentrations of metals in the analytical solutions should be at least ten times their respective analytical detection limits. Under certain conditions, and with greater care in the analytical procedure, these concentrations can be as low as approximately three times the respective analytical detection limits without seriously impairing the precision of the analyses. On at least one sample run in the source test, and for each metal analyzed, perform either repetitive analyses, Method of Standard Additions, serial dilution, or matrix spike addition, etc., to document the quality of the data.
13.3.3 Actual in-stack method detection limits are based on actual source sampling parameters and analytical results as described above. If required, the method in-stack detection limits can be improved over those shown in Table 29-1 for a specific test by either increasing the sampled stack gas volume, reducing the total volume of the digested samples, improving the analytical detection limits, or any combination of the three. For extremely low levels of Hg only, the aliquot size selected for digestion and analysis can be increased to as much as 10 ml, thus improving the in-stack detection limit by a factor of ten compared to a 1 ml aliquot size.
13.3.3.1 A nominal one hour sampling run will collect a stack gas sampling volume of about 1.25 m 3 . If the sampling time is increased to four hours and 5 m 3 are collected, the in-stack method detection limits would be improved by a factor of four compared to the values shown in Table 29-1.
13.3.3.2 The in-stack detection limits assume that all of the sample is digested and the final liquid volumes for analysis are the normal values of 300 ml for Analytical Fraction 1, and 150 ml for Analytical Fraction 2A. If the volume of Analytical Fraction 1 is reduced from 300 to 30 ml, the in-stack detection limits for that fraction of the sample would be improved by a factor of ten. If the volume of Analytical Fraction 2A is reduced from 150 to 25 ml, the in-stack detection limits for that fraction of the sample would be improved by a factor of six. Matrix effect checks are necessary on sample analyses and typically are of much greater significance for samples that have been concentrated to less than the normal original sample volume. Reduction of Analytical Fractions 1 and 2A to volumes of less than 30 and 25 ml, respectively, could interfere with the redissolving of the residue and could increase interference by other compounds to an intolerable level.
13.3.3.3 When both of the modifications described in sections 13.3.3.1 and 13.3.3.2 are used simultaneously on one sample, the resultant improvements are multiplicative. For example, an increase in stack gas volume by a factor of four and a reduction in the total liquid sample digested volume of both Analytical Fractions 1 and 2A by a factor of six would result in an improvement by a factor of twenty-four of the in-stack method detection limit.
13.4 Precision. The precision (relative standard deviation) for each metal detected in a method development test performed at a sewage sludge incinerator were found to be as follows:
Sb (12.7 percent), As (13.5 percent), Ba (20.6 percent), Cd (11.5 percent), Cr (11.2 percent), Cu (11.5 percent), Pb (11.6 percent), P (14.6 percent), Se (15.3 percent), Tl (12.3 percent), and Zn (11.8 percent). The precision for Ni was 7.7 percent for another test conducted at a source simulator. Be, Mn, and Ag were not detected in the tests. However, based on the analytical detection limits of the ICAP for these metals, their precisions could be similar to those for the other metals when detected at similar levels.
14.0 Pollution Prevention [Reserved]
15.0 Waste Management [Reserved]
16.0 Alternative Procedures
16.1 Alternative Analyzer. Samples may also be analyzed by cold vapor atomic fluorescence spectrometry.
16.2 [Reserved]
17.0 References
1. Method 303F in Standard Methods for the Examination of Water Wastewater, 15th Edition, 1980. Available from the American Public Health Association, 1015 18th Street N.W., Washington, D.C. 20036.
2. EPA Methods 6010, 6020, 7000, 7041, 7060, 7131, 7421, 7470, 7740, and 7841, Test Methods for Evaluating Solid Waste: Physical/Chemical Methods. SW-846, Third Edition, November 1986, with updates I, II, IIA, IIB and III. Office of Solid Waste and Emergency Response, U. S. Environmental Protection Agency, Washington, DC 20460.
3. EPA Method 200.7, Code of Federal Regulations, Title 40, Part 136, Appendix C. July 1, 1987.
4. EPA Methods 1 through 5, Code of Federal Regulations, Title 40, Part 60, Appendix A, July 1, 1991.
5. EPA Method 101A, Code of Federal Regulations, Title 40, Part 61, Appendix B, July 1, 1991.
18.0 Tables, Diagrams, Flowcharts, and Validation Data
| Metal | Front-half: probe and filter | Back-half: impinters 1-3 | Back-half: impringers 4-6 a | Total train |
|---|---|---|---|---|
| Antimony | 1 7.7 (0.7) | 1 3.8 (0.4) | 1 11.5 (1.1) | |
| Arsenic | 1 12.7 (0.3) | 1 6.4 (0.1) | 1 19.1 (0.4) | |
| Barium | 0.5 | 0.3 | 0.8 | |
| Beryllium | 1 0.07 (0.05) | 1 0.04 (0.03) | 1 0.11 (0.08) | |
| Cadmium | 1 1.0 (0.02) | 1 0.5 (0.01) | 1 1.5 (0.03) | |
| Chromium | 1 1.7 (0.2) | 1 0.8 (0.1) | 1 2.5 (0.3) | |
| Cobalt | 1 1.7 (0.2) | 1 0.8 (0.1) | 1 2.5 (0.3) | |
| Copper | 1.4 | 0.7 | 2.1 | |
| Lead | 1 10.1 (0.2) | 1 5.0 (0.1) | 1 15.1 (0.3) | |
| Manganese | 1 0.5 (0.2) | 1 0.2 (0.1) | 1 0.7 (0.3) | |
| Mercury | 2 0.06 | 2 0.3 | 2 0.2 | 2 0.56 |
| Nickel | 3.6 | 1.8 | 5.4 | |
| Phosphorus | 18 | 9 | 27 | |
| Selenium | 1 18 (0.5) | 1 9 (0.3) | 1 27 (0.8) | |
| Silver | 1.7 | 0.9 (0.7) | 2.6 | |
| Thallium | 1 9.6 (0.2) | 1 4.8 (0.1) | 1 14.4 (0.3) | |
| Zinc | 0.5 | 0.3 | 0.8 | |
| a Mercury analysis only. 1 Detection limit when analyzed by ICAP or GFAAS as shown in parentheses (see section 11.1.2). 2 Detection limit when anaylzed by CVAAS, estimated for Back-half and Total Train. See sections 13.2 and 11.1.3. Note: Actual method in-stack detection limits may vary from these values, as described in section 13.3.3. | ||||
| Analyte | Wavelength (nm) |
|---|---|
| Aluminum (Al) | 308.215 |
| Antimony (Sb) | 206.833 |
| Arsenic (As) | 193.696 |
| Barium (Ba) | 455.403 |
| Beryllium (Be) | 313.042 |
| Cadmium (Cd) | 226.502 |
| Chromium (Cr) | 267.716 |
| Cobalt (Co) | 228.616 |
| Copper (Cu) | 328.754 |
| Iron (Fe) | 259.940 |
| Lead (Pb) | 220.353 |
| Manganese (Mn) | 257.610 |
| Nickel (Ni) | 231.604 |
| Phosphorus (P) | 214.914 |
| Selenium (Se) | 196.026 |
| Silver (Ag) | 328.068 |
| Thallium (T 1 ) | 190,864 |
| Zinc (Zn) | 213,856 |
| Metal | Technique | SW-846 1 Methods No. | Wavelength (nm) | Interferences | |
|---|---|---|---|---|---|
| Cause | Minimization | ||||
| Fe | Aspiration | 7380 | 248.3 | Contamination | Great care taken to avoid contamination. |
| Pb | Aspiration | 7420 | 283.3 | 217.0 nm alternate | Background correction required. |
| Pb | Furnace | 7421 | 283.3 | Poor recoveries | Matrix modifier, add 10 µl of phosphorus acid to 1 ml of prepared sample in sampler cup. |
| Mn | Aspiration | 7460 | 279.5 | 403.1 nm alternate | Background correction required. |
| Ni | Aspiration | 7520 | 232.0 | 352.4 nm alternate Fe, Co, and Cr Nonlinear response | Background correction required. Matrix matching or nitrous-oxide/acetylene flame Sample dilution or use 352.3 nm line |
| Se | Furnace | 7740 | 196.0 | Volatility | Spike samples and reference materials and add nickel nitrate to minimize volatilization. |
| Adsorption & scatter | Background correction is required and Zeeman background correction can be useful. | ||||
| Ag | Aspiration | 7760 | 328.1 | Adsorption & scatter AgCl insoluble | Background correction is required. Avoid hydrochloric acid unless silver is in solution as a chloride complex. Sample and standards monitored for aspiration rate. |
| Tl | Aspiration | 7840 | 276.8 | Background correction is required. Hydrochloric acid should not be used. | |
| Tl | Furnace | 7841 | 276.8 | Hydrochloric acid or chloride | Background correction is required. Verify that losses are not occurring for volatilization by spiked samples or standard addition; Palladium is a suitable matrix modifier. |
| Zn | Aspiration | 7950 | 213.9 | High Si, Cu, & P Contamination | Strontium removes Cu and phosphate. Great care taken to avoid contamination. |
| Sb | Aspiration | 7040 | 217.6 | 1000 mg/ml Pb, Ni, Cu, or acid | Use secondary wavelength of 231.1 nm; match sample & standards acid concentration or use nitrous oxide/acetylene flame. |
| Sb | Furnace | 7041 | 217.6 | High Pb | Secondary wavelength or Zeeman correction. |
| As | Furnace | 7060 | 193.7 | Arsenic Volatilization Aluminum | Spike samples and add nickel nitrate solution to digestates prior to analysis. Use Zeeman background correction. |
| Ba | Aspiration | 7080 | 553.6 | ||
| Calcium | |||||
| Barium Ionization | High hollow cathode current and narrow band set. 2 ml of KCl per 100 m1 of sample. | ||||
| Be | Aspiration | 7090 | 234.9 | 500 ppm Al. High Mg and Si | Add 0.1% fluoride. |
| Be | Furnace | 7091 | 234.9 | Be in optical path | Optimize parameters to minimize effects. |
| Cd | Aspiration | 7130 | 228.8 | Absorption and light scattering | Background correction is required. |
| Cd | Furnace | 7131 | 228.8 | As above Excess Chloride Pipet Tips | As above. Ammonium phosphate used as a matrix modifier. Use cadmium-free tips. |
| Cr | Aspiration | 7190 | 357.9 | Alkali metal | KCl ionization suppressant in samples and standards - Consult mfgs' literature. |
| Co | Furnace | 7201 | 240.7 | Excess chloride | Use Method of Standard Additions. |
| Cr | Furnace | 7191 | 357.9 | 200 mg/L Ca and P | All calcium nitrate for a know constant effect and to eliminate effect of phosphate. |
| Cu | Aspiration | 7210 | 324.7 | Absorption and Scatter | Consult manufacturer's manual. |
| 1 Refer to EPA publication SW-846 (Reference 2 in section 16.0). | |||||
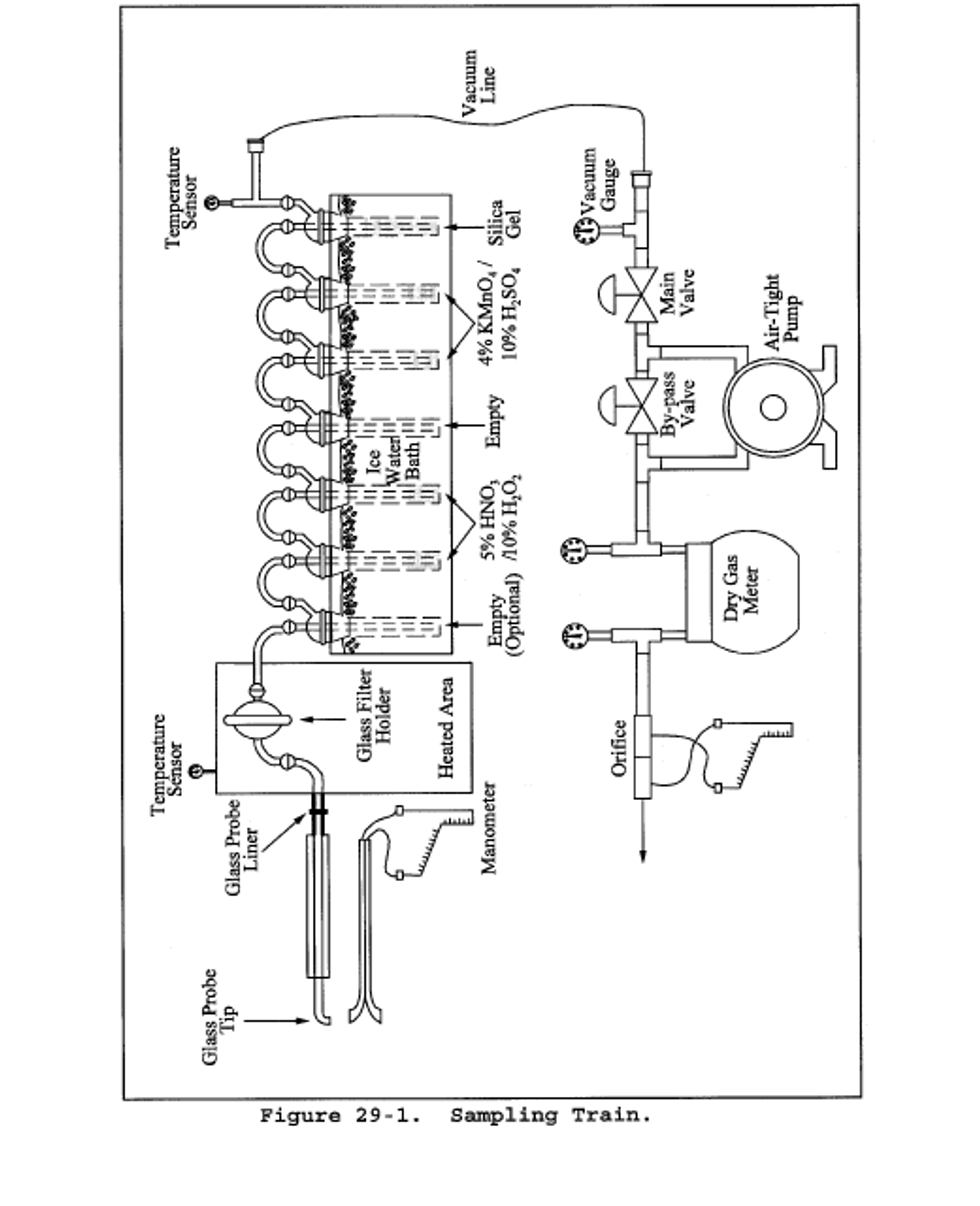
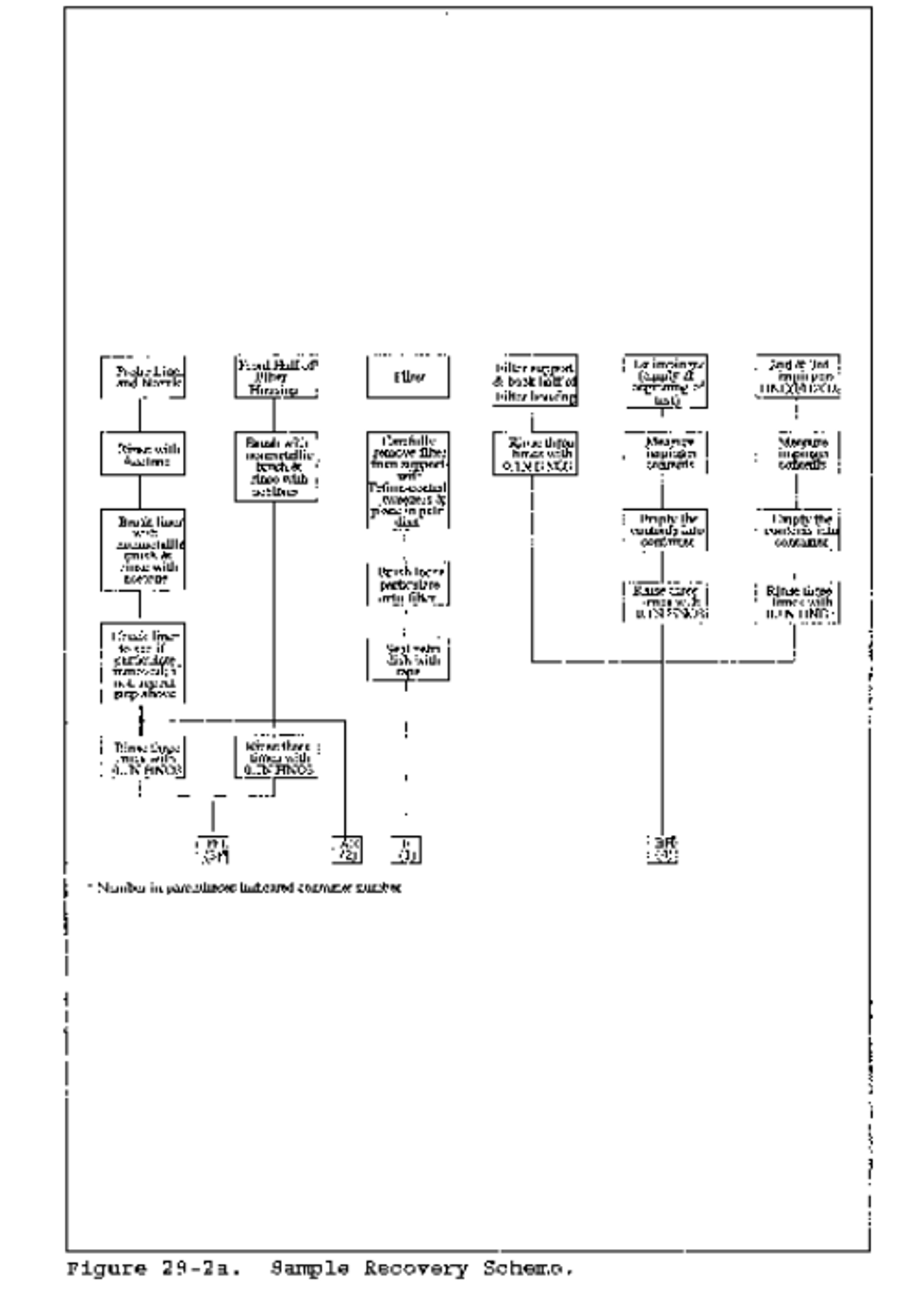

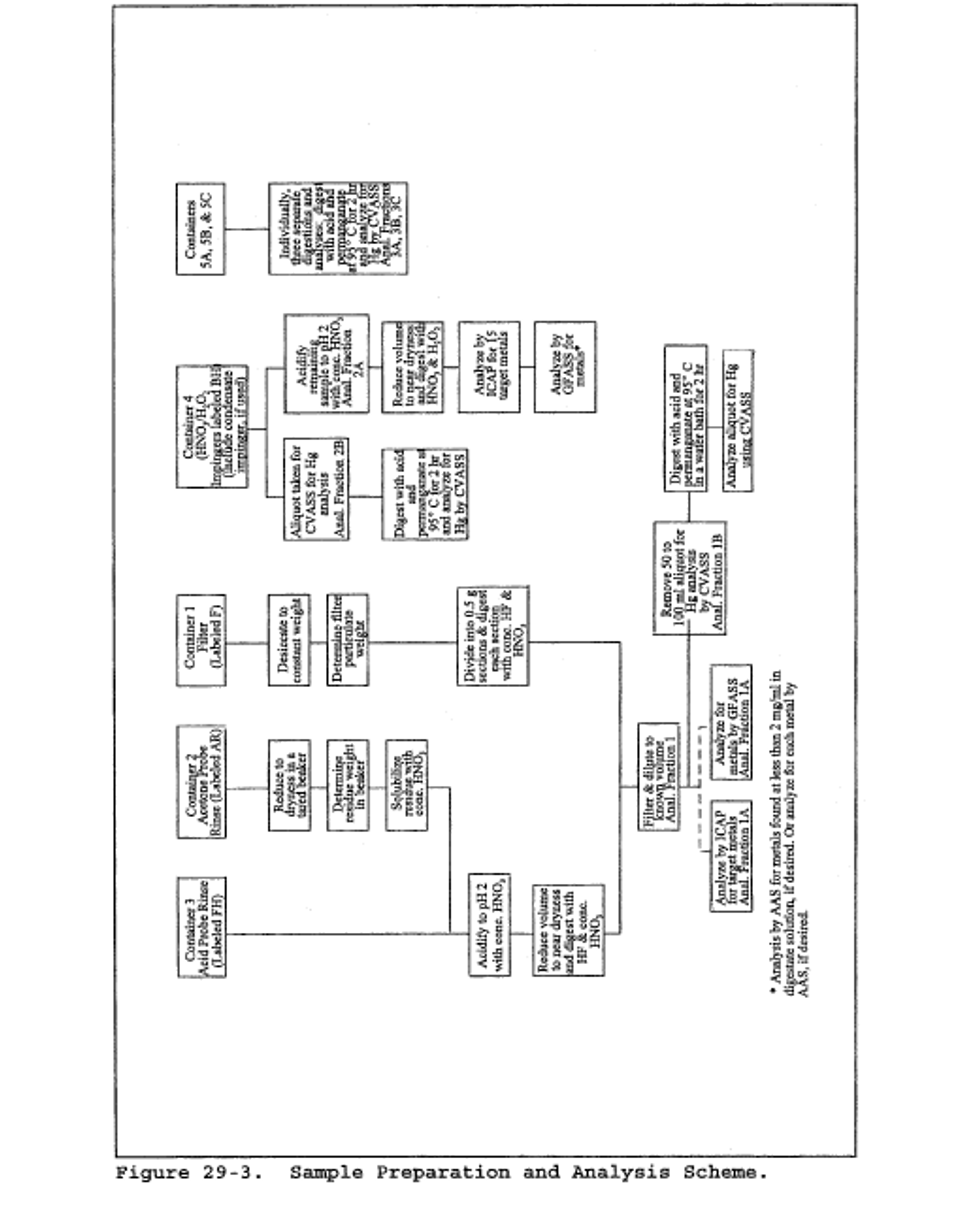
Method 30A - Determination of Total Vapor Phase Mercury Emissions From Stationary Sources (Instrumental Analyzer Procedure)
1.0 Scope and Application
What Is Method 30A?
Method 30A is a procedure for measuring total vapor phase mercury (Hg) emissions from stationary sources using an instrumental analyzer. This method is particularly appropriate for performing emissions testing and for conducting relative accuracy test audits (RATAs) of mercury continuous emissions monitoring systems (Hg CEMS) and sorbent trap monitoring systems at coal-fired combustion sources. Quality assurance and quality control requirements are included to assure that you, the tester, collect data of known and acceptable quality for each testing site. This method does not completely describe all equipment, supplies, and sampling procedures and analytical procedures you will need but refers to other test methods for some of the details. Therefore, to obtain reliable results, you should also have a thorough knowledge of these additional methods which are also found in appendices A-1 and A-3 to this part:
(a) Method 1 - Sample and Velocity Traverses for Stationary Sources.
(b) Method 4 - Determination of Moisture Content in Stack Gases.
1.1 Analytes. What does this method determine? This method is designed to measure the mass concentration of total vapor phase Hg in flue gas, which represents the sum of elemental Hg (Hg 0 ) and oxidized forms of Hg (Hg 2 ), in mass concentration units of micrograms per cubic meter (µg/m 3 ).
| Analyte | CAS No. | Sensitivity |
|---|---|---|
| Elemental Hg (Hg 0 ) | 7439-97-6 | Typically <2% of Calibration Span. |
| Oxidized Hg (Hg 2 ) | (Same). |
1.2 Applicability. When is this method required? Method 30A is offered as a reference method for emission testing and for RATAs of Hg CEMS and sorbent trap monitoring systems at coal-fired boilers. Method 30A may also be specified for other source categories in the future, either by New Source Performance Standards (NSPS), National Emission Standards for Hazardous Air Pollutants (NESHAP), emissions trading programs, State Implementation Plans (SIP), or operating permits that require measurement of Hg concentrations in stationary source emissions to determine compliance with an applicable emission standard or limit, or to conduct RATAs of Hg CEMS and sorbent trap monitoring systems.
1.3 Data Quality Objectives (DQO). How good must my collected data be? Method 30A has been designed to provide data of high and known quality for Hg emission testing and for relative accuracy testing of Hg monitoring systems including Hg CEMS and sorbent trap monitoring systems. In these and other applications, the principle objective is to ensure the accuracy of the data at the actual emission levels encountered. To meet this objective, calibration standards prepared according to an EPA traceability protocol must be used and measurement system performance tests are required.
2.0 Summary of Method
In this method, a sample of the effluent gas is continuously extracted and conveyed to an analyzer capable of measuring the total vapor phase Hg concentration. Elemental and oxidized mercury (i.e., Hg 0 and Hg 2 ) may be measured separately or simultaneously but, for purposes of this method, total vapor phase Hg is the sum of Hg 0 and Hg 2 . You must meet the performance requirements of this method (i.e., system calibration, interference testing, dynamic spiking, and system integrity/drift checks) to validate your data. The dynamic spiking requirement is deferred until January 1, 2009.
3.0 Definitions
3.1 Calibration Curve means the relationship between an analyzer's response to the injection of a series of calibration gases and the actual concentrations of those gases.
3.2 Calibration Gas means a gas standard containing Hg 0 or HgCl 2 at a known concentration that is produced and certified in accordance with an EPA traceability protocol for certification of Hg calibration standards.
3.2.1 Zero Gas means a calibration gas with a concentration that is below the level detectable by the measurement system.
3.2.2 Low-Level Gas means a calibration gas with a concentration that is 10 to 30 percent of the calibration span.
3.2.3 Mid-Level Gas means a calibration gas with a concentration that is 40 to 60 percent of the calibration span.
3.2.4 High-Level Gas means a calibration gas whose concentration is equal to the calibration span.
3.3 Converter means a device that reduces oxidized mercury (Hg 2 ) to elemental mercury (Hg 0 ).
3.4 Calibration Span means the upper limit of valid instrument response during sampling. To the extent practicable the measured emissions are to be between 10 and 100 percent of the selected calibration span ( i.e. , the measured emissions should be within the calibrated range determined by the Low- and High-Level gas standards). It is recommended that the calibration span be at least twice the native concentration to accommodate the dynamic spiking procedure.
3.5 Centroidal Area means the central area that has the same shape as the stack or duct cross section and is no greater than one percent of the stack or duct total cross-sectional area.
3.6 Data Recorder means the equipment that permanently records the concentrations reported by the analyzer.
3.7 Drift Check means the test to determine the difference between the measurement system readings obtained in a post-run system integrity check and the prior pre-run system integrity check at a specific calibration gas concentration level ( i.e. , zero, mid-level, or high-level).
3.8 Dynamic Spiking means a procedure in which a known mass or concentration of vapor phase HgCl 2 is injected into the probe sample gas stream at a known flow rate, in order to assess the effects of the flue gas matrix on the accuracy of the measurement system.
3.9 Gas Analyzer means the equipment that detects the total vapor phase Hg being measured and generates an output proportional to its concentration.
3.10 Interference Test means the test to detect analyzer responses to compounds other than Hg, usually gases present in the measured gas stream, that are not adequately accounted for in the calibration procedure and may cause measurement bias.
3.11 Measurement System means all of the equipment used to determine the Hg concentration. The measurement system may generally include the following major subsystems: sample acquisition, Hg 2 to Hg 0 converter, sample transport, sample conditioning, flow control/gas manifold, gas analyzer, and data recorder.
3.12 Native Concentration means the total vapor phase Hg concentration in the effluent gas stream.
3.13 NIST means the National Institute of Standards and Technology, located in Gaithersburg, Maryland.
3.14 Response Time means the time it takes for the measurement system, while operating normally at its target sample flow rate or dilution ratio, to respond to a known step change in gas concentration (from a low-level to a high-level gas) and to read within 5 percent of the stable high-level gas response.
3.15 Run means a series of gas samples taken successively from the stack or duct. A test normally consists of a specific number of runs.
3.16 System Calibration Error means the difference between the measured concentration of a low-, mid-, or high-level Hg 0 calibration gas and the certified concentration of the gas when it is introduced in system calibration mode.
3.17 System Calibration Mode means introducing the calibration gases into the measurement system at the probe, upstream of all sample conditioning components.
3.18 Test refers to the series of runs required by the applicable regulation.
4.0 Interferences
Interferences will vary among instruments and potential instrument-specific spectral and matrix interferences must be evaluated through the interference test and the dynamic spiking tests.
5.0 Safety
What safety measures should I consider when using this method?
This method may require you to work with hazardous materials and in hazardous conditions. You are encouraged to establish safety procedures before using the method. Among other precautions, you should become familiar with the safety recommendations in the gas analyzer user's manual. Occupational Safety and Health Administration (OSHA) regulations concerning use of compressed gas cylinders and noxious gases may apply.
6.0 Equipment and Supplies
6.1 What do I need for the measurement system? This method is intended to be applicable to multiple instrumental technologies. You may use any equipment and supplies that meet the following specifications.
6.1.1 All wetted sampling system components, including probe components prior to the point at which the calibration gas is introduced, must be chemically inert to all Hg species. Materials such as perfluoroalkoxy (PFA) Teflon TM , quartz, treated stainless steel (SS) are examples of such materials. [ Note: These materials of construction are required because components prior to the calibration gas injection point are not included in the system calibration error, system integrity, and interference tests.]
6.1.2 The interference, system calibration error, system integrity, drift and dynamic spiking test criteria must all be met by the system used.
6.1.3 The system must be capable of measuring and controlling sample flow rate.
6.1.4 All system components prior to the Hg 2 to Hg 0 converter must be maintained at a sample temperature above the acid gas dew point.
6.2 Measurement System Components. Figure 30A-1 in section 17.0 is an example schematic of a Method 30A measurement system.
6.2.1 Sample Probe. The probe must be made of the appropriate materials as noted in section 6.1.1, heated when necessary (see section 6.1.4), configured with ports for introduction of calibration and spiking gases, and of sufficient length to traverse all of the sample points.
6.2.2 Filter or Other Particulate Removal Device. The filter or other particulate removal device is considered to be a part of the measurement system, must be made of appropriate materials as noted in section 6.1.1, and must be included in all system tests.
6.2.3 Sample Line. The sample line that connects the probe to the converter, conditioning system and analyzer must be made of appropriate materials as noted in section 6.1.1.
6.2.4 Conditioning Equipment. For dry basis measurements, a condenser, dryer or other suitable device is required to remove moisture continuously from the sample gas. Any equipment needed to heat the probe, or sample line to avoid condensation prior to the moisture removal component is also required. For wet basis systems, you must keep the sample above its dew point either by: (1) Heating the sample line and all sample transport components up to the inlet of the analyzer (and, for hot-wet extractive systems, also heating the analyzer) or (2) by diluting the sample prior to analysis using a dilution probe system. The components required to do either of the above are considered to be conditioning equipment.
6.2.5 Sampling Pump. A pump is needed to push or pull the sample gas through the system at a flow rate sufficient to minimize the response time of the measurement system. If a mechanical sample pump is used and its surfaces are in contact with the sample gas prior to detection, the pump must be leak free and must be constructed of a material that is non-reactive to the gas being sampled (see section 6.1.1). For dilution-type measurement systems, an ejector pump (eductor) may be used to create a sufficient vacuum that sample gas will be drawn through a critical orifice at a constant rate. The ejector pump may be constructed of any material that is non-reactive to the gas being sampled.
6.2.6 Calibration Gas System(s). One or more systems may be needed to introduce calibration gases into the measurement system. A system should be able to flood the sampling probe sufficiently to prevent entry of gas from the effluent stream.
6.2.7 Dynamic Spiking Port. For the purposes of the dynamic spiking procedure described in section 8.2.7, the measurement system must be equipped with a port to allow introduction of the dynamic spike gas stream with the sample gas stream, at a point as close as possible to the inlet of the probe so as to ensure adequate mixing. The same port used for system calibrations and calibration error checks may be used for dynamic spiking purposes.
6.2.8 Sample Gas Delivery. The sample line may feed directly to a converter, to a by-pass valve (for speciating systems), or to a sample manifold. All valve and/or manifold components must be made of material that is non-reactive to the gas sampled and the calibration gas, and must be configured to safely discharge any excess gas.
6.2.9 Hg Analyzer. An instrument is required that continuously measures the total vapor phase Hg in the gas stream and meets the applicable specifications in section 13.0.
6.2.10 Data Recorder. A recorder, such as a computerized data acquisition and handling system (DAHS), digital recorder, strip chart, or data logger, is required for recording measurement data.
6.3 Moisture Measurement System. If correction of the measured Hg emissions for moisture is required (see section 8.5), either Method 4 in appendix A-3 to this part or other moisture measurement methods approved by the Administrator will be needed to measure stack gas moisture content.
7.0 Reagents and Standards
7.1 Calibration Gases. What calibration gases do I need? You will need calibration gases of known concentrations of Hg 0 and HgCl 2 . Special reagents and equipment may be required to prepare the HgCl 2 gas standards (e.g., a NIST-traceable solution of HgCl 2 and a gas generator equipped with mass flow controllers).
The following calibration gas concentrations are required:
7.1.1 High-Level Gas. Equal to the selected calibration span.
7.1.2 Mid-Level Gas. 40 to 60 percent of the calibration span.
7.1.3 Low-Level Gas. 10 to 30 percent of the calibration span.
7.1.4 Zero Gas. No detectable Hg.
7.1.5 Dynamic Spike Gas. The exact concentration of the HgCl 2 calibration gas used to perform the pre-test dynamic spiking procedure described in section 8.2.7 depends on the native Hg concentration in the stack The spike gas must produce a spiked sample concentration above the native concentration, as specified in section 8.2.7.2.2.
7.2 Interference Test. What reagents do I need for the interference test? Use the appropriate test gases listed in Table 30A-3 in section 17.0 (i.e., the potential interferents for the source to be tested, as identified by the instrument manufacturer) to conduct the interference check. These gases need not be of protocol gas quality.
8.0 Sample Collection
Emission Test Procedure
Figure 30A-2 in section 17.0 presents an overview of the test procedures required by this method. Since you may choose different options to comply with certain performance criteria, you must identify the specific options and associated frequencies you select and document your results in regard to the performance criteria.
8.1 Selection of Sampling Sites and Sampling Points. What sampling site and sampling points do I select?
8.1.1 When this method is used solely for Hg emission testing (e.g., to determine compliance with an emission standard or limit), use twelve sampling points located according to Table 1-1 or Table 1-2 of Method 1 in appendix A-1 to this part. Alternatively, you may conduct a stratification test as described in section 8.1.3 to determine the number and location of the sampling points.
8.1.2 When this method is used for relative accuracy testing of a Hg CEMS or sorbent trap monitoring system, follow the sampling site selection and sampling point layout procedures for gas monitor RATA testing described in the appropriate performance specification or applicable regulation (e.g., Performance Specification 2, section 8.1.3 of appendix B to this part or section 6.5.6 of appendix A to part 75 of this chapter), with one exception. If you elect to perform stratification testing as part of the sampling point selection process, perform the testing in accordance with section 8.1.3 of this method (see also “Summary Table of QA/QC Requirements” in section 9.0).
8.1.3 Determination of Stratification. If you elect to perform stratification testing as part of the sampling point selection process and the test results show your effluent gas stream to be unstratified or minimally stratified, you may be allowed to sample at fewer points or at different points than would otherwise be required.
8.1.3.1 Test Procedure. To test for stratification, use a probe of appropriate length to measure the total vapor phase Hg concentration at twelve traverse points located according to Table 1-1 or Table 1-2 of Method 1 in appendix A-1 to this part. Alternatively, for a sampling location where stratification is expected (e.g., after a wet scrubber or at a point where dissimilar gas streams are combined together), if a 12-point Hg stratification test has been previously performed at that location and the results of the test showed the location to be minimally stratified or unstratified according to the criteria in section 8.1.3.2, you may perform an abbreviated 3-point or 6-point Hg stratification test at the points specified in section 6.5.6.2(a) of appendix A to part 75 of this chapter in lieu of performing the 12-point test. Sample for a minimum of twice the system response time (see section 8.2.6) at each traverse point. Calculate the individual point and mean Hg concentrations.
8.1.3.2 Acceptance Criteria and Sampling Point Location.
8.1.3.2.1 If the Hg concentration at each traverse point differs from the mean concentration for all traverse points by no more than: (a) ±5 percent of the mean concentration; or (b) ±0.2 µg/m 3 (whichever is less restrictive), the gas stream is considered to be unstratified and you may collect samples from a single point that most closely matches the mean.
8.1.3.2.2 If the 5 percent or 0.2 µg/m 3 criterion in section 8.1.3.2.1 is not met, but the Hg concentration at each traverse point differs from the mean concentration for all traverse points by no more than: (a)±10 percent of the mean; or (b)±0.5 µg/m 3 (whichever is less restrictive), the gas stream is considered to be minimally stratified, and you may take samples from three points, provided the points are located on the measurement line exhibiting the highest average Hg concentration during the stratification test. If the stack diameter (or equivalent diameter, for a rectangular stack or duct) is greater than 2.4 meters (7.8 ft), locate the three sampling points at 0.4, 1.0, and 2.0 meters from the stack or duct wall. Alternatively, if a RATA required by part 75 of this chapter is being conducted, you may locate the three points at 4.4, 14.6, and 29.6 percent of the duct diameter, in accordance with Method 1 in appendix A-1 to this part. For stack or duct diameters of 2.4 meters (7.8 ft) or less, locate the three sampling points at 16.7, 50.0, and 83.3 percent of the measurement line.
8.1.3.2.3 If the gas stream is found to be stratified because the 10 percent or 0.5 µg/m 3 criterion in section 8.1.3.2.2 is not met, then either locate three sampling points at 16.7, 50.0, and 83.3 percent of the measurement line that exhibited the highest average Hg concentration during the stratification test, or locate twelve traverse points for the test in accordance with Table 1-1 or Table 1-2 of Method 1 in appendix A-1 to this part; or, if a RATA required by part 75 of this chapter is being conducted, locate six Method 1 points along the measurement line that exhibited the highest average Hg concentration.
8.1.3.3 Temporal Variations. Temporal variations in the source Hg concentration during a stratification test may complicate the determination of stratification. If temporal variations are a concern, you may use the following procedure to normalize the stratification test data. A second Hg measurement system, i.e., either an installed Hg CEMS or another Method 30A system, is required to perform this procedure. Position the sampling probe of the second Hg measurement system at a fixed point in the stack or duct, at least one meter from the stack or duct wall. Then, each time that the Hg concentration is measured at one of the stratification test points, make a concurrent measurement of Hg concentration at the fixed point. Normalize the Hg concentration measured at each traverse point, by multiplying it by the ratio of C F,avg to C F , where C F is the corresponding fixed-point Hg concentration measurement, and C F,avg is the average of all of the fixed-point measurements over the duration of the stratification test. Evaluate the results of the stratification test according to section 8.1.3.2, using the normalized Hg concentrations.
8.1.3.4 Stratification Testing Exemption. Stratification testing need not be performed at a test location where it would otherwise be required to justify using fewer sample points or different sample points, if the owner or operator documents that the Hg concentration in the stack gas is expected to be 3 µg/m 3 or less at the time of a Hg monitoring system RATA or an Hg emissions test. To demonstrate that a particular test location qualifies for the stratification testing exemption, representative Hg emissions data must be collected just prior to the RATA or emissions test. At least one hour of Hg concentration data is required for the demonstration. The data used for the demonstration shall be recorded at process operating conditions that closely approximate the operating conditions that will exist during the RATA or emissions test. It is recommended that collection of the demonstration data be integrated with the on-site pretest procedures required by the reference method being used for the RATA or emissions test (whether this method or another approved Hg reference method is used). Quality-assured data from an installed Hg monitoring system may also be used for the demonstration. If a particular test location qualifies for the stratification testing exemption, sampling shall be performed at three points, as described in section 8.1.3.2.2 of this method. The owner or operator shall fully document the method used to collect the demonstration data and shall keep this documentation on file with the data from the associated RATA or Hg emissions test.
8.1.3.5 Interim Alternative Stratification Test Procedures. In the time period between the effective date of this method and January 1, 2009, you may follow one of the following two procedures. Substitute a stratification test for sulfur dioxide (SO 2 ) for the Hg stratification test described in section 8.1.3.1. If this option is chosen, follow the test procedures in section 6.5.6.1 of appendix A to part 75 of this chapter. Evaluate the test results and determine the sampling point locations according to section 6.5.6.3 of appendix A to part 75 of this chapter. If the sampling location is found to be minimally stratified or unstratified for SO 2 , it shall be considered minimally stratified or unstratified for Hg. Alternatively, you may forgo stratification testing, assume the gas stream is minimally stratified, and sample at three points as described in section 8.1.3.2.2 of this method.
8.2 Initial Measurement System Performance Tests. What initial performance criteria must my system meet before I begin sampling? Before measuring emissions, perform the following procedures:
(a) Interference Test;
(b) Calibration Gas Verification;
(c) Measurement System Preparation;
(d) 3-Point System Calibration Error Test;
(e) System Integrity Check;
(f) Measurement System Response Time Test; and
(g) Dynamic Spiking Test.
8.2.1 Interference Test (Optional). Your measurement system should be free of known interferences. It is recommended that you conduct this interference test of your measurement system prior to its initial use in the field to verify that the candidate test instrument is free from inherent biases or interferences resulting from common combustion emission constituents. If you have multiple measurement systems with components of the same make and model numbers, you need only perform this interference check on one system and you may also rely on an interference test conducted by the manufacturer on a system having components of the same make and model(s) of the system that you use. The interference test procedure is found in section 8.6 of this method.
8.2.2 Calibration Gas Verification. How must I verify the concentrations of my calibration gases?
8.2.2.1 Cylinder Gas Standards. When cylinder gas standards are used for Hg 0 , obtain a certificate from the gas manufacturer and confirm that the documentation includes all information required by an EPA traceability protocol (see section 16). Confirm that the manufacturer certification is complete and current. Ensure that the calibration gas certifications have not expired.
8.2.2.2 Other Calibration Standards. All other calibration standards for HgCl 2 and Hg 0 , such as gas generators, must meet the requirements of an EPA traceability protocol (see section 16), and the certification procedures must be fully documented in the test report.
8.2.2.3 Calibration Span. Select the calibration span (i.e., high-level gas concentration) so that the measured source emissions are 10 to 100 percent of the calibration span. This requirement is waived for applications in which the Hg concentrations are consistently below 1 µg/m 3 ; however, the calibration span for these low-concentration applications shall not exceed 5 µg/m 3 .
8.2.3 Measurement System Preparation. How do I prepare my measurement system for use? Assemble, prepare, and precondition the measurement system according to your standard operating procedure. Adjust the system to achieve the correct sampling rate or dilution ratio (as applicable). Then, conduct a 3-point system calibration error test using Hg 0 as described in section 8.2.4, an initial system integrity check using HgCl 2 and a zero gas as described in section 8.2.5, and a pre-test dynamic spiking test as described in section 8.2.7.
8.2.4 System Calibration Error Test. Conduct a 3-point system calibration error test before the first test run. Use Hg 0 standards for this test. Introduce the low-, mid-, and high-level calibration gases in any order, in system calibration mode, unless you desire to determine the system response time during this test, in which case, inject the gases such that the high-level injection directly follows the low-level injection. For non-dilution systems, you may adjust the system to maintain the correct flow rate at the analyzer during the test, but you may not make adjustments for any other purpose. For dilution systems, you must operate the measurement system at the appropriate dilution ratio during all system calibration error checks, and you may make only the adjustments necessary to maintain the proper ratio. After each gas injection, wait until a stable response has been obtained. Record the analyzer's final, stable response to each calibration gas on a form similar to Table 30A-1 in section 17.0. For each calibration gas, calculate the system calibration error using Equation 30A-1 in section 12.2. The calibration error specification in section 13.1 must be met for the low-, mid-, and high-level gases. If the calibration error specification is not met for all three gases, take corrective action and repeat the test until an acceptable 3-point calibration is achieved.
8.2.5 System Integrity Check. Perform a two-point system integrity check before the first test run. Use the zero gas and either the mid- or high-level HgCl 2 calibration gas for the check, whichever one best represents the total vapor phase Hg concentration levels in the stack. Record the data on a form similar to Table 30A-2 in section 17.0. The system integrity check specification in section 13.2 must be met for both the zero gas and the mid- or high-level gas. If the system integrity specification is not met for both gases, take corrective action and repeat the test until an acceptable system integrity check is achieved.
8.2.6 Measurement System Response Time. The measurement system response time is used to determine the minimum sampling time for each sampling point and is equal to the time that is required for the measured Hg concentration to increase from the stable low-level calibration gas response to a value within 5 percent of the stable high-level calibration gas response during the system calibration error test in section 8.2.4. Round off the measured system response time to the nearest minute.
8.2.7 Dynamic Spiking Test. You must perform dynamic spiking prior to the first test run to validate your test data. The purpose of this procedure is to demonstrate that the site-specific flue gas matrix does not adversely affect the accuracy of the measurement system. The specifications in section 13.5 must be met to validate your data. If these specifications are not met for the pre-test dynamic spiking, you may not proceed with the test until satisfactory results are obtained. For the time period between the effective date of this method and January 1, 2009, the dynamic spiking requirement is waived.
8.2.7.1 How do I perform dynamic spiking? Dynamic spiking is a gas phase application of the method of standard additions, which involves injecting a known quantity of Hg into the measurement system upstream of all sample conditioning components, similar to system calibration mode, except the probe is not flooded and the resulting sample stream includes both effluent gas and the spike gas. You must follow a written procedure that details how the spike is added to the system, how the spike dilution factor (DF) is measured, and how the Hg concentration data are collected and processed.
8.2.7.2 Spiking Procedure Requirements.
8.2.7.2.1 Spiking Gas Requirements. The spike gas must also be a HgCl 2 calibration gas certified by an EPA traceability protocol. You must choose concentrations that can produce the target levels while being injected at a volumetric flow rate that is ≤20 percent of the total volumetric flow rate through the measurement system (i.e., sample flow rate plus spike gas flow rate).
8.2.7.2.2 Target Spiking Level. The target level for spiking must be 150 to 200 percent of the native Hg concentration; however, if the native Hg concentration is <1 µg/m 3 , set the target level to add between 1 and 4 µg/m 3 Hg 2 to the native concentration. Use Equation 30A-5 in section 12.5 to calculate the acceptable range of spike gas concentrations at the target level. Then select a spike gas concentration in that range.
8.2.7.2.3 Spike Injections. You must inject spikes in such a manner that the spiking does not alter the total volumetric sample system flow rate and dilution ratio (if applicable). You must collect at least 3 data points, and the relative standard deviation (RSD) specification in section 13.5 must be met. Each data point represents a single spike injection, and pre- and post-injection measurements of the native Hg concentration (or diluted native concentration, as applicable) are required for each spike injection.
8.2.7.2.4 Spike Dilution Factor (DF). For each spike injection, DF, the dilution factor must be determined. DF is the ratio of the total volumetric flow rate of gas through the measurement system to the spike gas flow rate. This factor must be ≥5. The spiking mass balance calculation is directly dependent on the accuracy of the DF determination. As a result, high accuracy total volumetric flow rate and spike gas flowrate measurements are required. These flow rates may be determined by direct or indirect measurement. Calibrated flow meters, venturies, orifices or tracer gas measurements are examples of potential flow measurement techniques.
8.2.7.2.5 Concentrations. The measurement system must record total vapor phase Hg concentrations continuously during the dynamic spiking procedure. It is possible that dynamic spiking at a level close to 200 percent of the native Hg concentration may cause the measured Hg concentration to exceed the calibration span value. Avoid this by choosing a lower spiking level or by recalibration at a higher span. The measurements shall not exceed 120 percent of the calibration span. The “baseline” measurements made between spikes may represent the native Hg concentration (if spike gas flow is stopped between injections) or the native Hg concentration diluted by blank or carrier gas flowing at the same rate as the spike gas (if gas flow cannot be stopped between injections). Each baseline measurement must include at least 4 readings or 1 minute (whichever is greater) of stable responses. Use Equation 30A-10 or 30A-11 in section 12.10 (as applicable) to convert baseline measurements to native concentration.
8.2.7.2.6 Recovery. Calculate spike recoveries using Equation 30A-7 in section 12.7. Mass recoveries may be calculated from stable responses based on injected mass flows or from integrated response peaks based on total mass injected. Calculate the mean and RSD for the three (or more) spike injections and compare to the specifications in section 13.5.
8.2.7.2.7 Error Adjustment Option. You may adjust the measurement data collected during dynamic spiking for the system calibration error using Equation 30A-3 in section 12. To do this, perform the initial system integrity check prior to the dynamic spiking test, and perform another system integrity check following the dynamic spiking test and before the first test run. If you choose this option, you must apply Equation 30A-3 to both the spiked sample concentration measurement (C ss ) and the baseline or native concentration measurement (C native ), each substituted in place of C avg in the equation.
8.2.7.3 Example Spiking Procedure Using a Hot Vapor Calibration Source Generator.
(a) Introduce the spike gas into the probe using a hot vapor calibration source generator and a solution of HgCl 2 in dilute HC1 and HNO 3 . The calibrator uses a mass flow controller (accurate within 2 percent) to measure the gas flow, and the solution feed is measured using a top-loading balance accurate to §0.01g. The challenges of injecting oxidized Hg may make it impractical to stop the flow of gas between spike injections. In this case, operate the hot vapor calibration source generator continuously during the spiking procedure, swapping blank solutions for HgCl 2 solutions when switching between spiking and baseline measurements.
(b) If applicable, monitor the measurement system to make sure the total sampling system flow rate and the sample dilution ratio do not change during this procedure. Record all data on a data sheet similar to Table 30A-5 in section 17.0. If the Hg measurement system design makes it impractical to measure the total volumetric flow rate through the system, use a spike gas that includes a tracer for measuring the dilution factor, DF (see Equation 30A-9 in section 12.9). Allow the measurements to stabilize between each spike injection, average the pre- and post-injection baseline measurements, and calculate the native concentration. If this measurement shifts by more than 5 percent during any injection, it may be necessary to discard that data point and repeat the injection to achieve the required RSD among the injections. If the spikes persistently show poor repeatability, or if the recoveries are not within the range specified in section 13.5, take corrective action.
8.2.8 Run Validation. How do I confirm that each run I conduct is valid?
8.2.8.1 System Integrity Checks.
(a) Before and after each test run, perform a two-point system integrity check using the same procedure as the initial system integrity check described in section 8.2.5. You may use data from that initial system integrity check as the pre-run data for the first test run, provided it is the most recent system integrity check done before the first run. You may also use the results of a successful post-run system integrity check as the pre-run data for the next test run. Do not make any adjustments to the measurement system during these checks, other than to maintain the target calibration gas flow rate and the proper dilution ratio.
(b) As a time-saving alternative, you may, at the risk of invalidating multiple test runs, skip one or more integrity checks during a test day. Provided there have been no auto-calibrations or other instrument alterations, a single integrity check may suffice as a post-run check to validate (or invalidate) as many consecutive test runs as can be completed during a single test day. All subsequent test days must begin with a pre-run system integrity check subject to the same performance criteria and corrective action requirements as a post-run system integrity check.
(c) Each system integrity check must meet the criteria for system integrity checks in section 13.2. If a post-run system integrity check is failed, all test runs since the last passed system integrity check are invalid. If a post-run or a pre-run system integrity check is failed, you must take corrective action and pass another 3-point Hg 0 system calibration error test (Section 8.2.4) followed by another system integrity check before conducting any additional test runs. Record the results of the pre- and post-run system integrity checks on a form similar to Table 30A-2 in section 17.0.
8.2.8.2 Drift Check. Using the data from the successful pre- and post-run system integrity checks, calculate the zero and upscale drift, using Equation 30A-2 in section 12.3. Exceeding the section 13.3 specification does not invalidate the run, but corrective action must be taken and a new 3-point Hg 0 system calibration error test and a system integrity check must be passed before any more runs are made.
8.3 Dilution-Type Systems - Special Considerations. When a dilution-type measurement system is used, there are three important considerations that must be taken into account to ensure the quality of the emissions data. First, the critical orifice size and dilution ratio must be selected properly so that the sample dew point will be below the sample line and analyzer temperatures. Second, a high-quality, accurate dilution controller must be used to maintain the correct dilution ratio during sampling. The dilution controller should be capable of monitoring the dilution air pressure, orifice upstream pressure, eductor vacuum, and sample flow rates. Third, differences between the molecular weight of calibration gas mixtures, dilution air, and the stack gas molecular weight must be considered because these can affect the dilution ratio and introduce measurement bias.
8.4 Sampling.
(a) Position the probe at the first sampling point. Allow the system to flush and equilibrate for at least two times the measurement system response time before recording any data. Then, traverse and record measurements at all required sampling points. Sample at each traverse point for an equal length of time, maintaining the appropriate sample flow rate or dilution ratio (as applicable). For all Hg instrumental method systems, the minimum sampling time at each sampling point must be at least two times the system response time, but not less than 10 minutes. For concentrating systems, the minimum sampling time must also include at least 4 concentration measurement cycles.
(b) After recording data for the appropriate period of time at the first traverse point, you may move the sample probe to the next point and continue recording, omitting the requirement to allow the system to equilibrate for two times the system response time before recording data at the subsequent traverse points. You must, however, sample at this and all subsequent traverse points for the required minimum amount of time specified in this section. If you must remove the probe from the stack for any reason, you must again allow the sampling system to equilibrate for at least two times the system response time prior to resuming data recording.
(c) If at any point the measured Hg concentration exceeds the calibration span value, you must at a minimum identify and report this as a deviation from the method. Depending on the data quality objectives of the test, thisevent may require corrective action before proceeding. If the average Hg concentration for any run exceeds the calibration span value, the run is invalidated.
8.5 Moisture Correction. If the moisture basis (wet or dry) of the measurements made with this method is different from the moisture basis of either: (1) The applicable emission limit; or (2) a Hg CEMS or sorbent trap monitoring system being evaluated for relative accuracy, you must determine the moisture content of the flue gas and correct the measured gas concentrations to a dry basis using Method 4 in appendix A-3 of this part or other appropriate methods, subject to the approval of the Administrator.
8.6 Optional Interference Test Procedure.
(a) Select an appropriate calibration span that reflects the source(s) to be tested and perform the interference check at 40 percent of the lowest calibration span value anticipated, e.g., 10 µg/m 3 . Alternatively, successfully conducting the interference test at an absolute Hg concentration of 2 µg/m 3 will demonstrate performance for an equivalent calibration span of 5 µg/m 3 , the lowest calibration span allowed for Method 30A testing. Therefore, performing the interference test at the 2 µ/m 3 level will serve to demonstrate acceptable performance for all calibration spans greater than or equal to 5 µg/m 3 .
(b) Introduce the interference test gases listed in Table 30A-3 in section 17.0 into the measurement system separately or as a mixture. The interference test gases HCl and NO must be introduced as a mixture. The interference test gases must be introduced into the sampling system at the probe such that the interference gas mixtures pass through all filters, scrubbers, conditioners, and other components as would be configured for normal sampling.
(c) The interference test must be performed using HgCl 2 , and each interference test gas (or gas mixture) must be evaluated in triplicate. This is accomplished by measuring the Hg response first with only the HgCl 2 gas present and then when adding the interference test gas(es) while maintaining the HgCl 2 concentration of the test stream constant. It is important that the equipment used to conduct the interference test be of sufficient quality so as to be capable of blending the HgCl 2 and interference gases while maintaining the Hg concentration constant. Gas blending system or manifolds may be used.
(d) The duration of each test should be for a sufficient period of time to ensure the Hg measurement system surfaces are conditioned and a stable output is obtained. Measure the Hg response of the analyzer to these gases in µg/m3. Record the responses and determine the overall interference response using Table 30A-4 in section 17.0 and the equations presented in section 12.11. The specification in section 13.4 must be met.
(e) A copy of these data, including the date completed and a signed certification, must be included with each test report. The intent of this test is that the interference test results are intended to be valid for the life of the system. As a result, the Hg measurement system should be operated and tested in a configuration consistent with the configuration that will be used for field applications. However, if the system used for field testing is not consistent with the system that was interference-tested, the interference test must be repeated before it is used for any field applications. Examples of such conditions include, but are not limited to: major changes in dilution ratio (for dilution based systems), changes in catalyst materials, changes in filtering device design or materials, changes in probe design or configuration, and changes in gas conditioning materials or approaches.
9.0 Quality Control
What quality control measures must I take?
The table which follows is a summary of the mandatory, suggested, and alternative quality assurance and quality control measures and the associated frequency and acceptance criteria. All of the QC data, along with the run data, must be documented and included in the test report.
| Status 1 | Process or element | QA/QC specification | Acceptance criteria | Checking frequency |
|---|---|---|---|---|
| S | Identify Data User | Regulatory Agency or other primary end user of data | Before designing test. | |
| M | Analyzer Design | Analyzer range | Sufficiently >high-level gas to allow determination of system calibration error | |
| S | Analyzer resolution or sensitivity | <2.0 % of full-scale range | Manufacturer design. | |
| S | Interference response | Overall response ≤3% of calibration span Alternatively, overall response ≤0.3 µg/m 3 | ||
| M | Calibration Gases | Traceability protocol | Validation of concentration required | |
| M | High-level Hg 0 gas | Equal to the calibration span | Each calibration error test. | |
| M | Mid-level Hg 0 gas | 40 to 60% of calibration span | Each calibration error test. | |
| M | Low-level Hg 0 gas | 10 to 30% of calibration span | Each calibration error test. | |
| M | High-level HgCl 2 gas | Equal to the calibration span | Each system integrity check (if it better represents C native than the mid level gas). | |
| M | Mid-level HgCl 2 | 40 to 60% of calibration span | Each system gas integrity check (if it better represents C native than the high level gas). | |
| M | Zero gas | Each system integrity check. | ||
| M | Dynamic spike gas (C native ≥1 µg/m 3 ) | A high-concentration HgCl 2 gas, used to produce a spiked sample concentration that is 150 to 200% of the native concentration | Pre-test; dynamic spiking not required until 1/1/09. | |
| M | Dynamic spike gas (C native <1 µg/m 3 ) | A high-concentration HgCl 2 gas, used to produce a spiked sample concentration that is 1 to 2 µg/m 3 above the native concentration | Pre-test; dynamic spiking not required until 1/1/09. | |
| S | Data Recorder Design | Data resolution | ≤0.5% of full-scale | Manufacturer design. |
| M | Sample Extraction | Probe material | Inert to sample constituents (e.g., PFA Teflon, or quartz if stack >500°F) | Each run. |
| M | Sample Extraction | Probe, filter and sample line temperature | For dry-basis analyzers, keep sample above the dew point, by heating prior to moisture removal For wet-basis analyzers, keep sample above dew point at all times, by heating or dilution | Each run. |
| M | Sample Extraction | Calibration valve material | Inert to sample constituents (e.g., PFA Teflon or PFA Teflon coated) | Each test. |
| S | Sample Extraction | Sample pump material | Inert to sample constituents | Each test. |
| M | Sample Extraction | Manifold material | Inert to sample constituents | Each test. |
| M | Particulate Removal | Filter inertness | Pass calibration error check | Each calibration error check. |
| M | System Calibration Performance | System calibration error (CE) test | CE ≤5.0 % of the calibration span for the low-, mid-or high-level Hg 0 calibration gas Alternative specification: ≤0.5 µg/m 3 absolute difference between system response and reference value | Before initial run and after a failed system integrity check or drift test. |
| M | System Calibration Performance | System integrity check | Error ≤5.0% of the calibration span for the zero and mid- or high-level HgCl 2 calibration gas Alternative specification: ≤0.5 µg/m 3 absolute difference between system response and reference value | Before initial run, after each run, at the beginning of subsequent test days, and after a failed system integrity check or drift test. |
| M | System Performance | System response time | Used to determine minimum sampling time per point | During initial 3-point system calibration error test. |
| M | System Performance | Drift | ≤3.0% of calibration span for the zero and mid- or high-level gas Alternative specification: ≤0.3 µg/m 3 absolute difference between pre- and post-run system calibration error percentages. | At least once per test day. |
| M | System Performance | Minimum sampling time | The greater of two times the system response time or 10 minutes. Concentrating systems must also include at least 4 cycles | Each sampling point. |
| M | System Performance | Percentage spike recovery and relative standard deviation | Percentage spike recovery, at the target level: 100 ±10% Relative standard deviation: ≤5 percent Alternative specification: absolute difference between calculated and measured spike values ≤0.5 µg/m 3 | Before initial run; dynamic spiking not required until 1/1/09. |
| M | Sample Point Selection | Number and Location of Sample Points | For emission testing applications, use 12 points, located according to Method 1 in appendix A-1 to this part, unless the results of a stratification test allow fewer points to be used | Prior to first run. |
| For Part 60 RATAs, follow the procedures in Performance Specification 2, section 8.1.3, and for Part 75 RATAs, follow the procedures in section 6.5.6 of appendix A to Part 75. That is: | ||||
| At any test location, you may use 3 sample points located at 16.7, 50.0, and 83.3% of a “long” measurement line passing through the centroidal area; or | ||||
| At any test location, you may use 6 sample points along a diameter, located according to Method 1 (Part 75 RATAs, only); or | ||||
| At a location where stratification is not expected and the measurement line is >2.4 m (7.8 ft), you may use 3 sample points located along a “short” measurement line at 0.4, 1.0, and 2.0 m from the stack or duct wall or, for Part 75 only, sample points may be located at 4.4, 14.6, and 29.6% of the measurement line; or | ||||
| After a wet scrubber or at a point where dissimilar gas streams are combined, either locate 3 sample points along the “long” measurement line or locate 6 Method 1 points along a diameter (Part 75, only), unless the results of a stratification test allow you to use a “short” 3-point measurement line or to sample at a single point | ||||
| If it can be demonstrated that stack gas concentration is ≤3 µg/m 3 , then the test site is exempted from stratification testing. Use the 3-point “short” measurement line if the stack diameter is >2.4 m (7.8 ft) and the 3-point “long” line for stack diameters ≤2.4 m (7.8 ft) | ||||
| A | Sample Point Selection | Stratification Test (See section 8.1.3). | If the Hg concentration 2 at each traverse point during the stratification test is: Within ±5% of mean, use 1-point sampling (at the point closest to the mean); or Not within ±5% of mean, but is within ±10% of mean, use 3-point sampling. Locate points according to section 8.1.3.2.2 of this method. Alternatively, if the Hg concentration at each point is: Within ±0.2 µg/m 3 of mean, use 1-point sampling (at the point closest to the mean); or Not within ±0.2 µg/m 3 of mean, but is within ±0.5 µg/m 3 of mean, use 3-point sampling. Locate points according to section 8.1.3.2.2 of this method. | Prior to first run. Prior to 1/1/09, you may (1) forgo stratification testing and use 3 sampling points (as per section 8.1.3.2.2) or (2) perform a SO 2 stratification test (see sections 6.5.6.1 and 6.5.6.3 of appendix A to part 75), in lieu of a Hg stratification test. If the test location is unstratified or minimally stratified for SO 2 , it can be considered unstratified or minimally stratified for Hg also. |
| M | Data Recording | Frequency | Once per cycle | During run. |
| S | Data Parameters | Sample concentration and calibration span | All analyzer readings during each run within calibration span | Each run. |
| M | Data Parameters | Sample concentration and calibration span | All analyzer readings during dynamic spiking tests within 120% of calibration span | Each spike injection. |
| M | Data Parameters | Sample concentration and calibration span | Average Hg concentration for the run ≤calibration span | Each run. |
| 1 M = Mandatory; S = Suggested; A = Alternative. 2 These may either be the unadjusted Hg concentrations or concentrations normalized to account for temporal variations. | ||||
10.0 Calibration and Standardization
What measurement system calibrations are required?
Your analyzer must be calibrated with Hg° standards. The initial 3-point system calibration error test described in section 8.2.4 is required before you start the test. Also, prior to and following test runs, the two-point system integrity checks described in sections 8.2.5 and 8.2.8 are required. On and after January 1, 2009, the pre-test dynamic spiking procedure described in section 8.2.7 is also required to verify that the accuracy of the measurement system is suitable and not adversely affected by the flue gas matrix.
11.0 Analytical Procedures
Because sample collection and analysis are performed together (see section 8), additional discussion of the analytical procedure is not necessary.
12.0 Calculations and Data Analysis
You must follow the procedures for calculations and data analysis listed in this section.
12.1 Nomenclature. The terms used in the equations are defined as follows:
B ws = Moisture content of sample gas as measured by Method 4 in Appendix A-3 to this part, percent/100.
C avg = Average unadjusted Hg concentration for the test run, as indicated by the data recorder µg/m 3 .
C͞ baseline = Average Hg concentration measured before and after dynamic spiking injections, µg/m 3 .
C d = Hg concentration, dry basis, µg/m 3 .
C dif = Absolute value of the difference between the measured Hg concentrations of the reference HgCl 2 calibration gas, with and without the individual or combined interference gases, µg/m 3 .
C dif avg = Average of the 3 absolute values of the difference between the measured Hg concentrations of the reference HgCl 2 calibration gas, with and without the individual or combined interference gases, µg/m 3 .
C gas = Average Hg concentration in the effluent gas for the test run, adjusted for system calibration error, µg/m 3 .
C int = Measured Hg concentration of the reference HgCl 2 calibration gas plus the individual or combined interference gases, µg/m 3 .
C m = Average of pre- and post-run system integrity check responses for the upscale (i.e., mid- or high-level) calibration gas, µg/m 3 .
C ma = Actual concentration of the upscale (i.e., mid- or high-level) calibration gas used for the system integrity checks, µg/m 3 .
C 0 = Average of pre- and post-run system integrity check responses from the zero gas, µg/m 3 .
C native = Vapor phase Hg concentration in the source effluent, µg/m 3 .
C ref = Measured Hg concentration of the reference HgCl 2 calibration gas alone, in the interference test, µg/m 3 .
C s = Measured concentration of a calibration gas (zero-, low-, mid-, or high-level), when introduced in system calibration mode, µg/m 3 .
C spike = Actual Hg concentration of the spike gas, µg/m 3 .
C* spike = Hg concentration of the spike gas required to achieve a certain target value for the spiked sample Hg concentration, µg/m 3 .
C ss = Measured Hg concentration of the spiked sample at the target level, µg/m 3 .
C* ss = Expected Hg concentration of the spiked sample at the target level, µg/m 3 .
C target = Target Hg concentration of the spiked sample, µg/m 3 .
C Tnative = Measured tracer gas concentration present in native effluent gas, ppm.
C Tdir = Tracer gas concentration injected with spike gas, ppm.
C Tv = Diluted tracer gas concentration measured in a spiked sample, ppm.
C v = Certified Hg° or HgCl 2 concentration of a calibration gas (zero, low, mid, or high), µg/m 3 .
C w = Hg concentration measured under moist sample conditions, wet basis, µg/m 3 .
CS = Calibration span, µg/m 3 .
D = Zero or upscale drift, percent of calibration span.
DF = Dilution factor of the spike gas, dimensionless.
I = Interference response, percent of calibration span.
Q probe = Total flow rate of the stack gas sample plus the spike gas, liters/min.
Q spike = Flow rate of the spike gas, liters/min.
R i = Individual injection spike recovery, %.
R͞ = Mean value of spike recoveries at a particular target level, %.
RSD = Relative standard deviation, %;.
SCE = System calibration error, percent of calibration span.
SCE i = Pre-run system calibration error during the two-point system integrity check, percent of calibration span.
SCE f = Post-run system calibration error during the two-point system integrity check, percent of calibration span.
12.2 System Calibration Error. Use Equation 30A-1 to calculate the system calibration error. Equation 30A-1 applies to: 3-point system calibration error tests performed with Hg° standards; and pre- and post-run two-point system integrity checks performed with HgCl 2 .
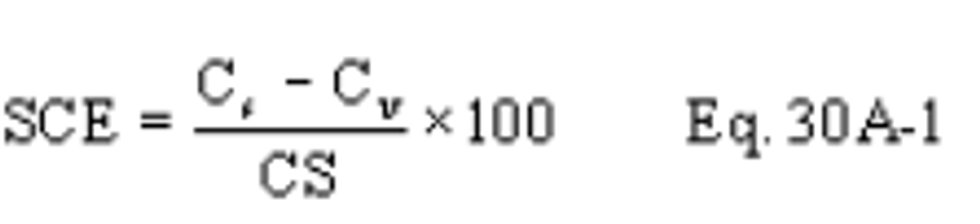
12.3 Drift Assessment. Use Equation 30A-2 to separately calculate the zero and upscale drift for each test run.
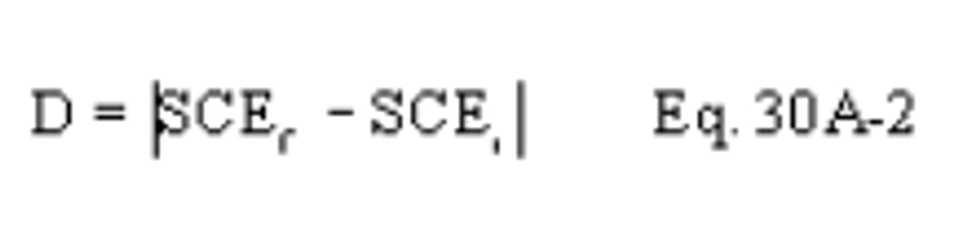
12.3 Effluent Hg Concentration. For each test run, calculate C avg , the arithmetic average of all valid Hg concentration values recorded during the run. Then, adjust the value of C avg for system calibration error, using Equation 30A-3.
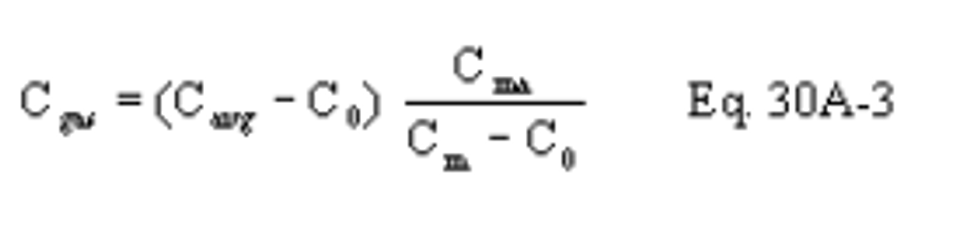
12.4 Moisture Correction. Use Equation 30A-4a if your measurements need to be corrected to a dry basis.
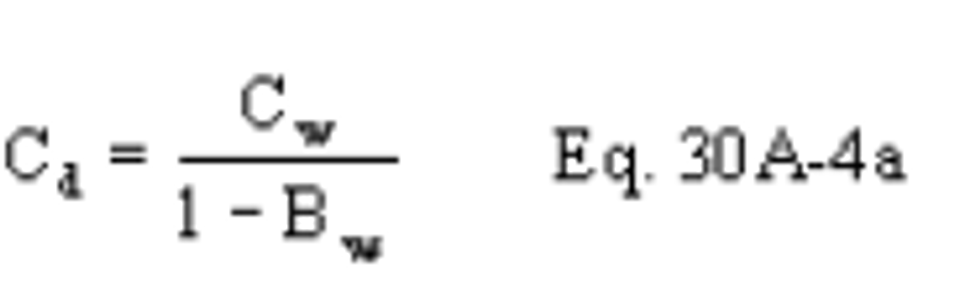
Use Equation 30A-4b if your measurements need to be corrected to a wet basis.
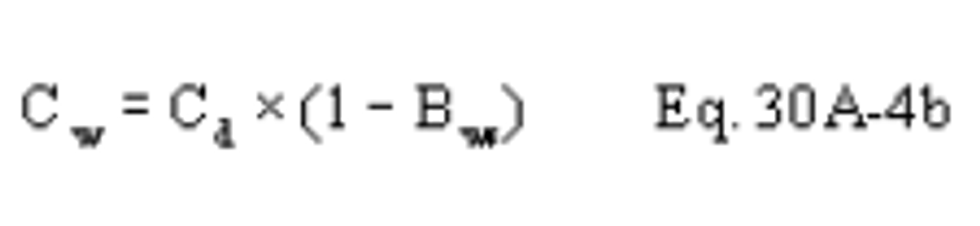
12.5 Dynamic Spike Gas Concentrations. Use Equation 30A-5 to determine the spike gas concentration needed to produce a spiked sample with a certain “target” Hg concentration.
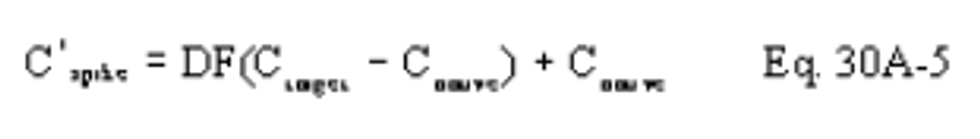
12.6 Spiked Sample Concentration. Use Equation 30A-6 to determine the expected or theoretical Hg concentration of a spiked sample.
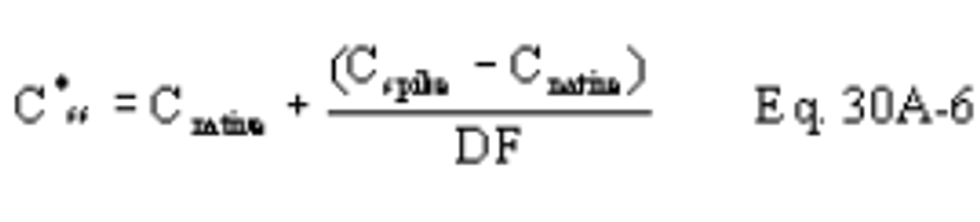
12.7 Spike Recovery. Use Equation 30A-7 to calculate the percentage recovery of each spike.
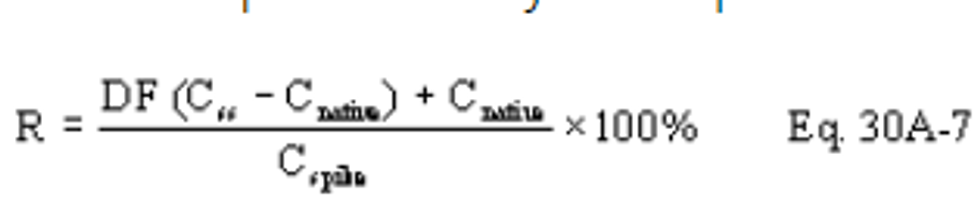
12.8 Relative Standard Deviation. Use Equation 30A-8 to calculate the relative standard deviation of the individual percentage spike recovery values from the mean.
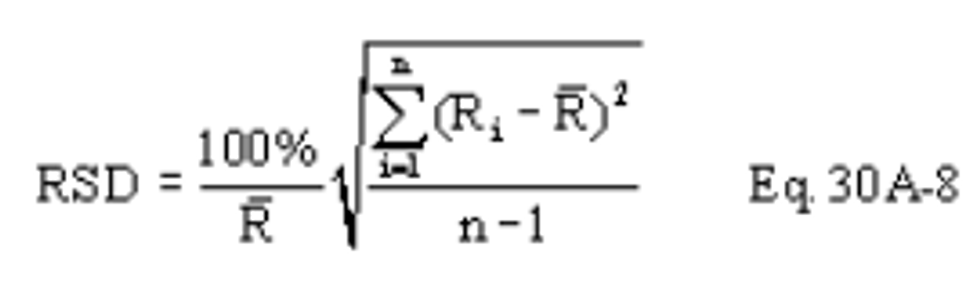
12.9 Spike Dilution Factor. Use Equation 30A-9 to calculate the spike dilution factor, using either direct flow measurements or tracer gas measurements.
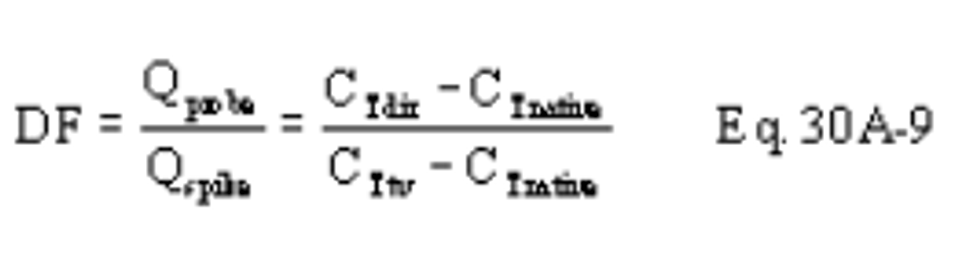
12.10 Native Concentration. For spiking procedures that inject blank or carrier gases (at the spiking flow rate, Q spike ) between spikes, use Equation 30A-10 to calculate the native concentration.
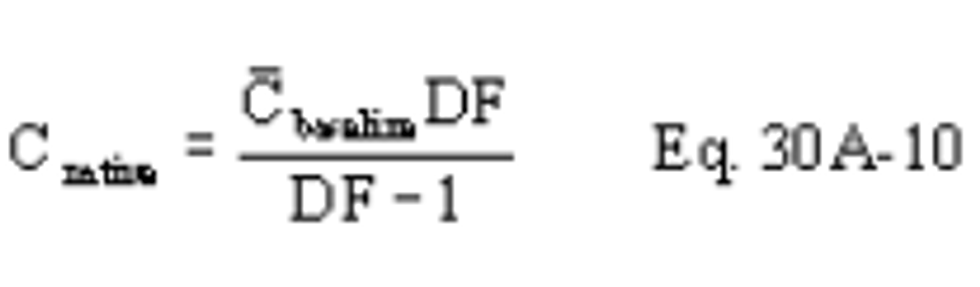
For spiking procedures that halt all injections between spikes, the native concentration equals the average baseline concentration (see Equation 30A-11).
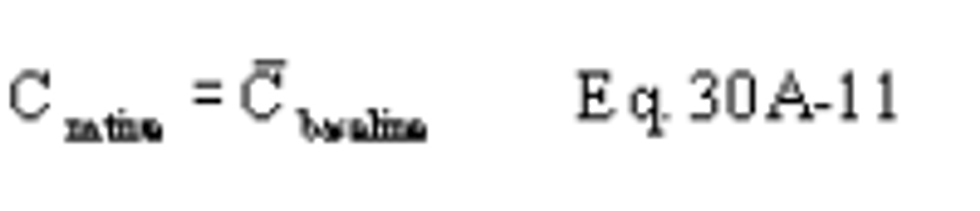
12.11 Overall Interference Response. Use equation 30A-12 to calculate the overall interference response.
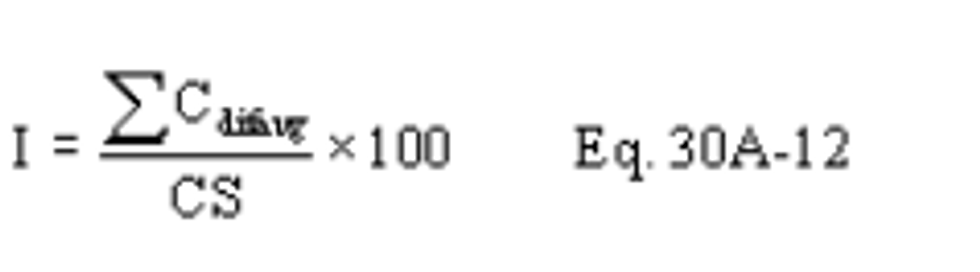
Where, for each interference gas (or mixture):

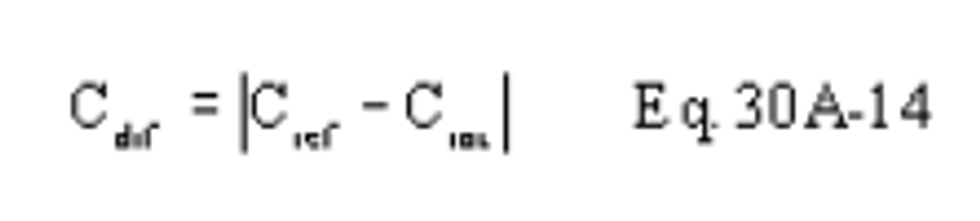
13.0 Method Performance
13.1 System Calibration Error Test. This specification applies to the 3-point system calibration error tests using Hg 0 . At each calibration gas level tested (low-, mid-, or high-level), the calibration error must be within ±5.0 percent of the calibration span. Alternatively, the results are acceptable if | C s − C v | ≤0.5 µg/m 3 .
13.2 System Integrity Checks. This specification applies to all pre- and post-run 2-point system integrity checks using HgCl 2 and zero gas. At each calibration gas level tested (zero and mid- or high-level), the error must be within ±5.0 percent of the calibration span. Alternatively, the results are acceptable if | C s − C v | ≤0.5 µg/m 3 .
13.3 Drift. For each run, the low-level and upscale drift must be less than or equal to 3.0 percent of the calibration span. The drift is also acceptable if the pre- and post-run system integrity check responses do not differ by more than 0.3 µg/m 3 (i.e., | C s post-run − C s pre-run | ≤0.3 µg/m 3 ).
13.4 Interference Test. Summarize the results following the format contained in Table 30A-4. For each interference gas (or mixture), calculate the mean difference between the measurement system responses with and without the interference test gas(es). The overall interference response for the analyzer that was used for the test (calculated according to Equation 30A-12), must not be greater than 3.0 percent of the calibration span used for the test (see section 8.6). The results of the interference test are also acceptable if the sum of the absolute average differences for all interference gases (i.e., Σ C dif avg ) does not exceed 0.3 µg/m 3 .
13.5 Dynamic Spiking Test. For the pre-test dynamic spiking, the mean value of the percentage spike recovery must be 100 ±10 percent. In addition, the relative standard deviation (RSD) of the individual percentage spike recovery values from the mean must be ≤5.0 percent. Alternatively, if the mean percentage recovery is not met, the results are acceptable if the absolute difference between the theoretical spiked sample concentration (see section 12.6) and the actual average value of the spiked sample concentration is ≤0.5 µg/m 3 .
14.0 Pollution Prevention [Reserved]
15.0 Waste Management [Reserved]
16.0 References
1. EPA Traceability Protocol for Qualification and Certification of Elemental Mercury Gas Generators, expected publication date December 2008, see www.epa.gov/ttn/emc.
2. EPA Traceability Protocol for Qualification and Certification of Oxidized Mercury Gas Generators, expected publication date December 2008, see www.epa.gov/ttn/emc.
3. EPA Traceability Protocol for Assay and Certification of Gaseous Calibration Standards, expected revision publication date December 2008, see www.epa.gov/ttn/emc.
17.0 Figures and Tables
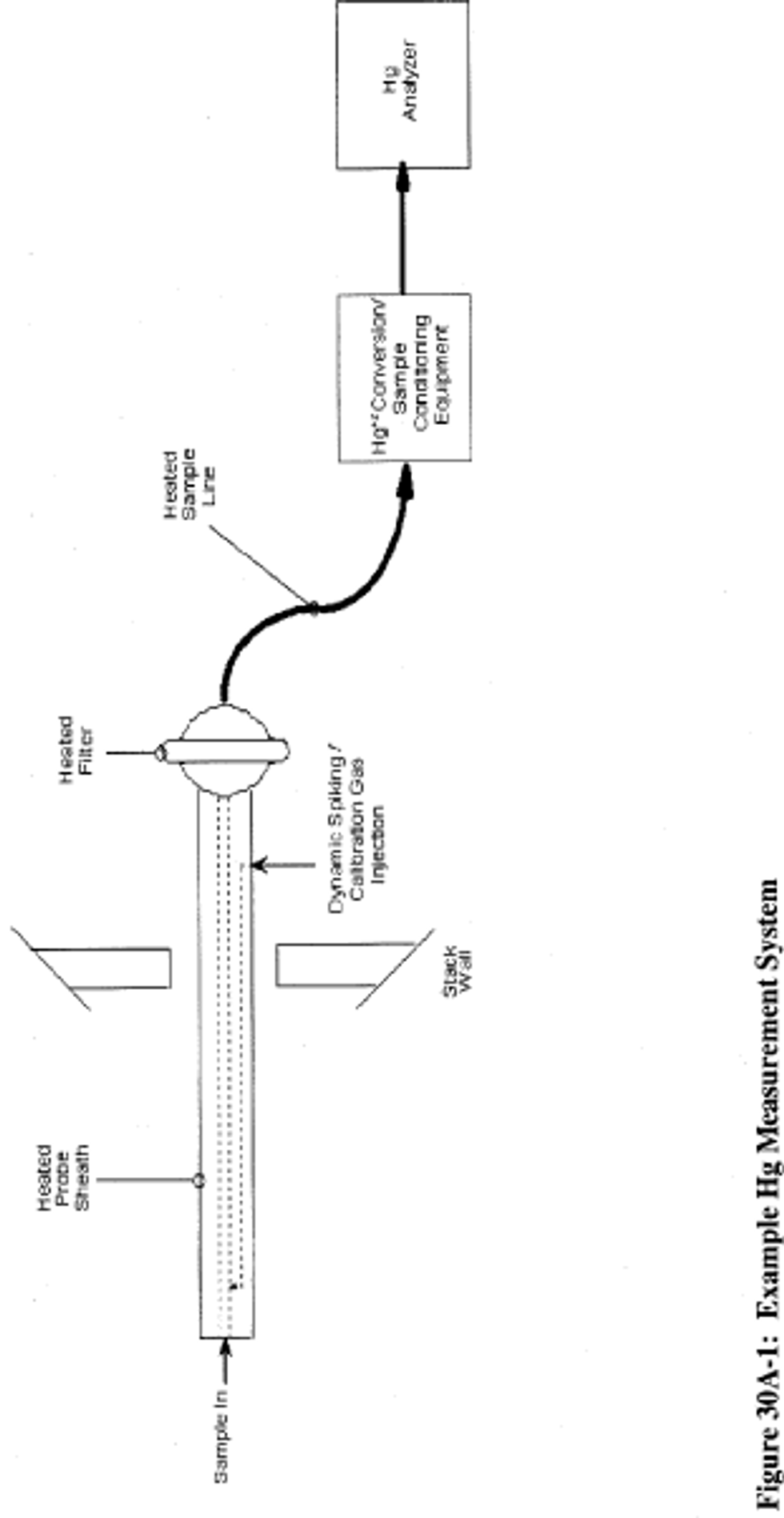
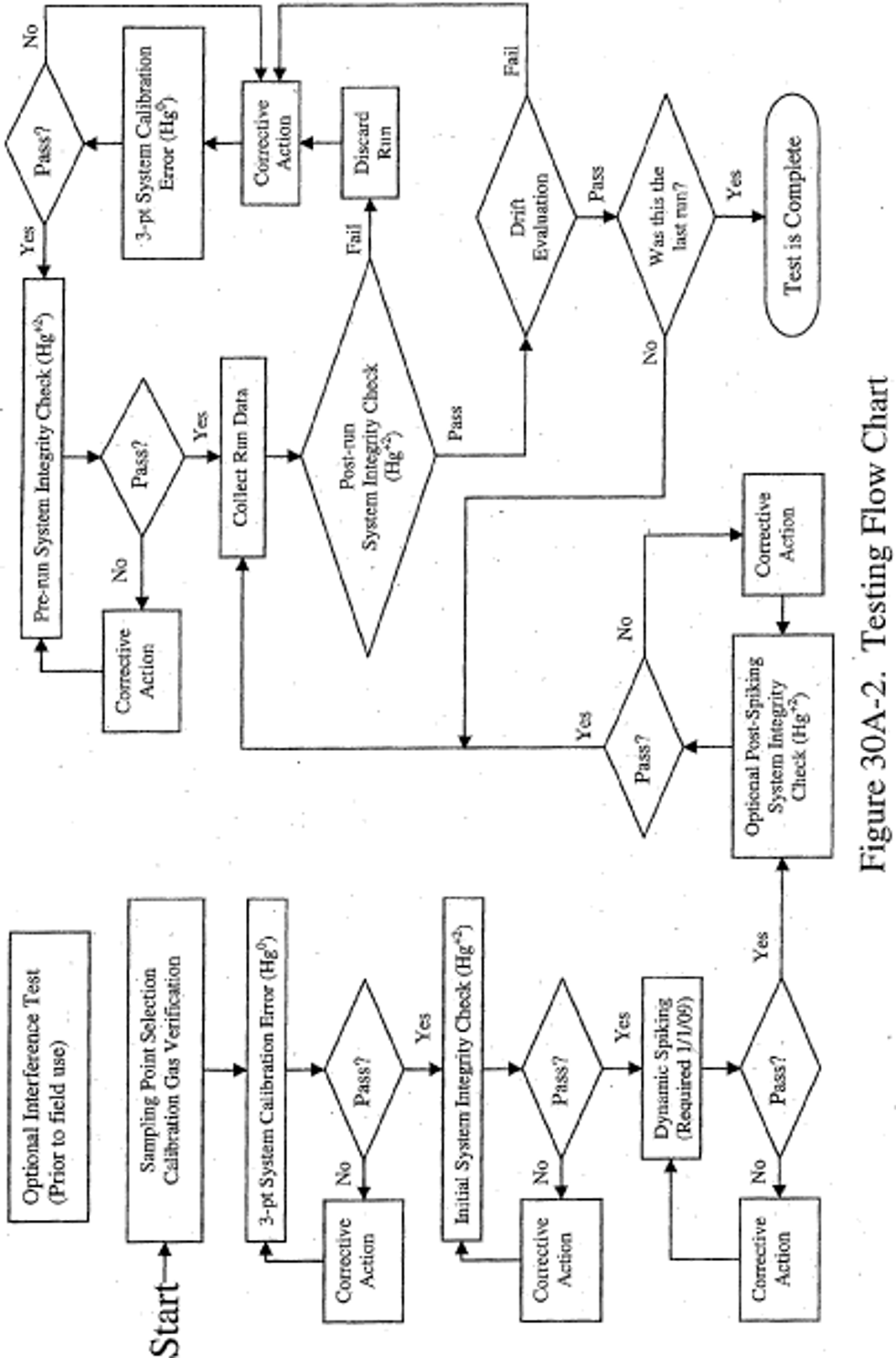
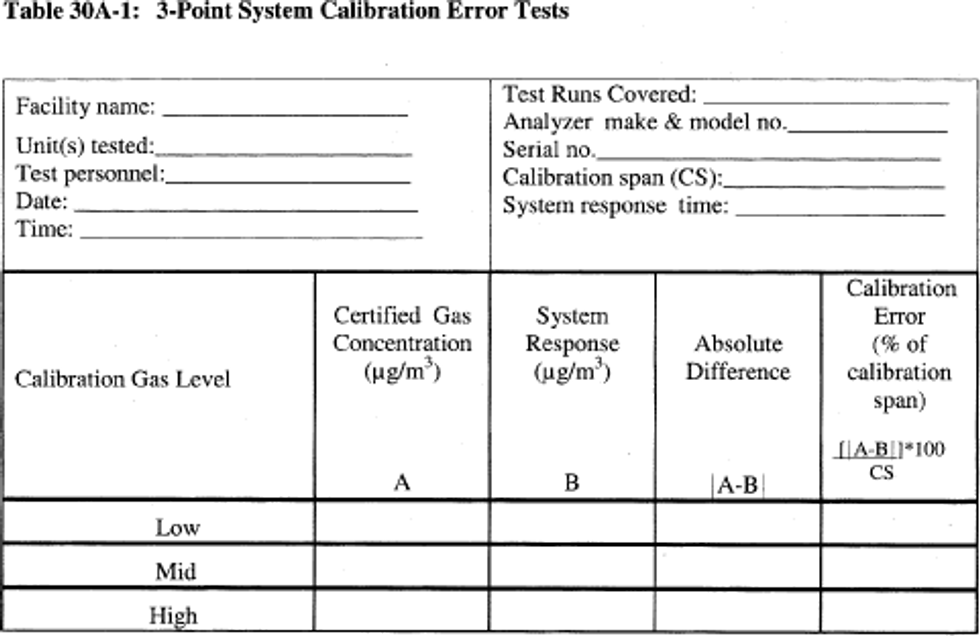
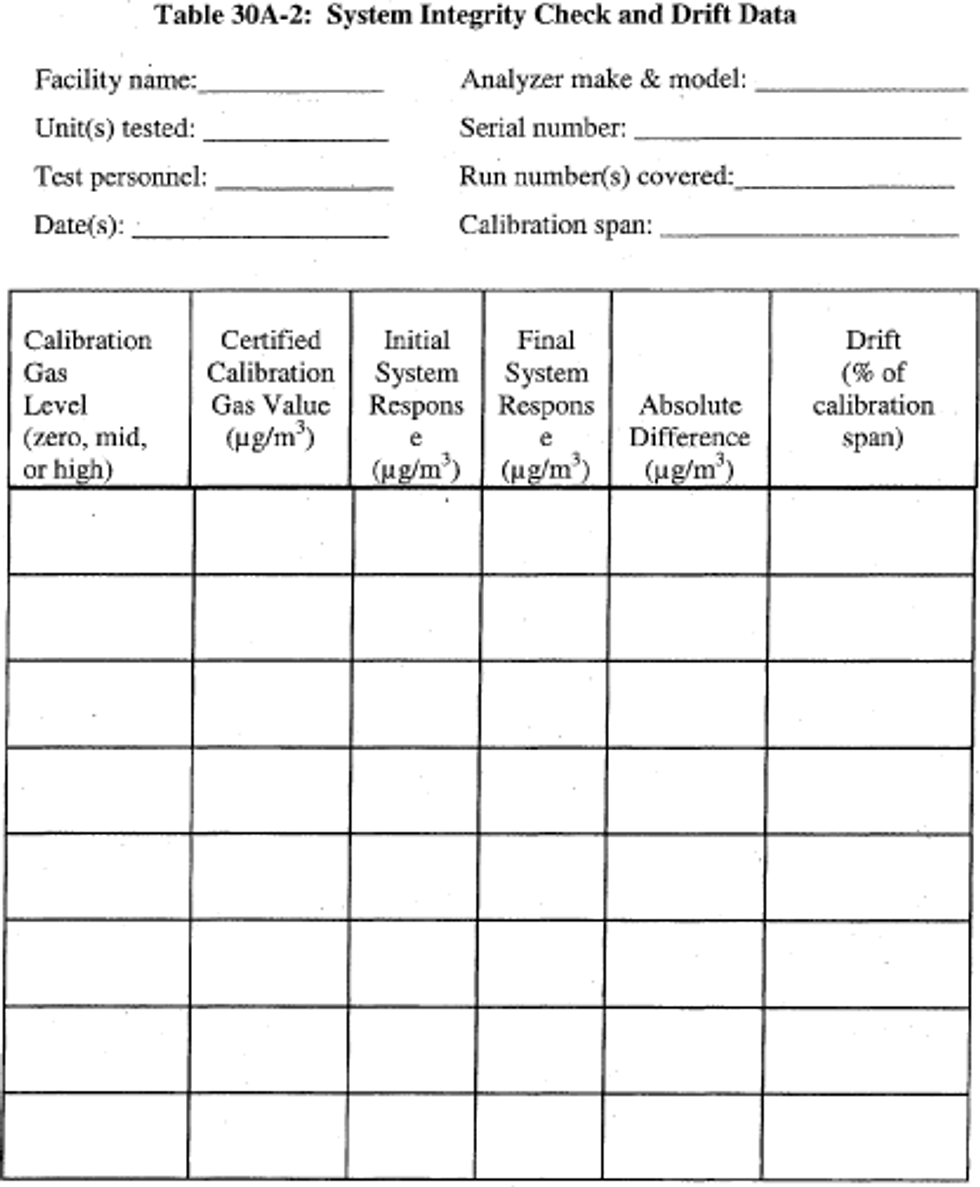
| Potential interferent gas 1 | Concentration, tentative - (balance N 2 ) |
|---|---|
| CO 2 | 15% ±1% CO 2 |
| CO | 100 ±20 ppm |
| HCl 2 | 100 ±20 ppm |
| NO 2 | 250 ±50 ppm |
| SO 2 | 200 ±20 ppm |
| O 2 | 3% ±1% O 2 |
| H 2 O | 10% ±1% H 2 O |
| Nitrogen | Balance |
| Other | |
| 1 Any of these specific gases can be tested at a lower level if the manufacturer has provided reliable means for limiting or scrubbing that gas to a specified level. 2 HCl and NO must be tested as a mixture. | |


Method 30B - Determination of Total Vapor Phase Mercury Emissions From Coal-Fired Combustion Sources Using Carbon Sorbent Traps
1.0 Scope and Application
What is Method 30B?
Method 30B is a procedure for measuring total vapor phase mercury (Hg) emissions from coal-fired combustion sources using sorbent trap sampling and an extractive or thermal analytical technique. This method is only intended for use only under relatively low particulate conditions (e.g., sampling after all pollution control devices). Quality assurance and quality control requirements are included to assure that you, the tester, collect data of known and acceptable quality for each testing program. This method does not completely describe all equipment, supplies, and sampling and analytical procedures you will need, but instead refers to other test methods for some of the details. Therefore, to obtain reliable results, you should also have a thorough knowledge of these additional methods which are found in Appendices A-1 and A-3 to this part:
(a) Method 1 - Sample and Velocity Traverses for Stationary Sources.
(b) Method 4 - Determination of Moisture Content in Stack Gases.
(c) Method 5 - Determination of Particulate Matter Emissions from Stationary Sources
1.1 Analytes. What does this method determine? This method is designed to measure the mass concentration of total vapor phase Hg in flue gas, including elemental Hg (Hg 0 ) and oxidized forms of Hg (Hg 2 ), in micrograms per dry standard cubic meter (µg/dscm).
| Analyte | CAS No. | Analytical range and sensitivity |
|---|---|---|
| Elemental Hg (Hg 0 ) | 7439-97-6 | Typically 0.1 µg/dscm to >50 µg/dscm. |
| Oxidized Hg (Hg 2 ) | (Same) |
1.2 Applicability. When is this method required? Method 30B is a reference method for relative accuracy test audits (RATAs) of vapor phase Hg CEMS and sorbent trap monitoring systems installed at coal-fired boilers and is also appropriate for Hg emissions testing at such boilers. It is intended for use only under relatively low particulate conditions (i.e., sampling after all pollution control devices); in cases where significant amounts of particle-bound Hg may be present, an isokinetic sampling method for Hg should be used. Method 30B may also be specified by New Source Performance Standards (NSPS), National Emission Standards for Hazardous Air Pollutants (NESHAP), emissions trading programs, State Implementation Plans (SIPs), and operating permits that require measurement of Hg concentrations in stationary source emissions, either to determine compliance with an applicable emission standard or limit, or to conduct RATAs of Hg CEMS and sorbent trap monitoring systems.
1.3 Data Quality Objectives (DQO). How good must my collected data be? Method 30B has been designed to provide data of high and known quality for Hg emissions testing and for RATA testing of Hg monitoring systems, including CEMS and sorbent trap monitors. In these and other applications, the principal objective is to ensure the accuracy of the data at the actual emissions levels and in the actual emissions matrix encountered. To meet this objective, NIST-traceable calibration standards must be used and method performance tests are required.
2.0 Summary of Method
Known volumes of flue gas are extracted from a stack or duct through paired, in-stack sorbent media traps at an appropriate flow rate. Collection of mercury on the sorbent media in the stack mitigates potential loss of mercury during transport through a probe/sample line. For each test run, paired train sampling is required to determine measurement precision and verify acceptability of the measured emissions data. A field recovery test which assesses recovery of an elemental Hg spike to determine measurement bias is also used to verify data acceptability. The sorbent traps are recovered from the sampling system, prepared for analysis as needed, and analyzed by any suitable determinative technique that can meet the performance criteria.
3.0 Definitions
3.1 Analytical System is the combined equipment and apparatus used to perform sample analyses. This includes any associated sample preparation apparatus e.g., digestion equipment, spiking systems, reduction devices, etc., as well as analytical instrumentation such as UV AA and UV AF cold vapor analyzers.
3.2 Calibration Standards are the Hg containing solutions prepared from NIST traceable standards and are used to directly calibrate analytical systems.
3.3 Independent Calibration Standard is a NIST traceable standard obtained from a source or supplier independent of that for the calibration standards and is used to confirm the integrity of the calibration standards used.
3.4 Method Detection Limit (MDL) is the lowest mass of Hg greater than zero that can be estimated and reported by your candidate analytical technique. The MDL is statistically derived from replicate low level measurements near your analytical instrument's detection level.
3.5 NIST means the National Institute of Standards and Technology, located in Gaithersburg, Maryland.
3.6 Run means a series of gas samples taken successively from the stack or duct. A test normally consists of a specific number of runs.
3.7 Sorbent Trap means a cartridge or sleeve containing a sorbent media (typically activated carbon treated with iodine or some other halogen) with multiple sections separated by an inert material such as glass wool. These sorbent traps are optimized for the quantitative capture of elemental and oxidized forms of Hg and can be analyzed by multiple techniques.
3.8 Test refers to the series of runs required by the applicable regulation.
3.9 Thermal Analysis means an analytical technique where the contents of the sorbent traps are analyzed using a thermal technique (desorption or combustion) to release the captured Hg in a detectable form for quantification.
3.10 Wet Analysis means an analytical technique where the contents of the sorbent tube are first leached or digested to quantitatively transfer the captured Hg to liquid solution for subsequent analysis.
4.0 Interferences
Interferences may result from the sorbent trap material used as well as from the measurement environment itself. The iodine present on some sorbent traps may impart a negative measurement bias. High levels of sulfur trioxide (SO 3 ) are also suspected to compromise the performance of sorbent trap Hg capture. These, and other, potential interferences are assessed by performing the analytical matrix interference, Hg 0 and HgCl 2 analytical bias and field recovery tests.
5.0 Safety
What safety measures should I consider when using this method? This method may require you to work with hazardous materials and in hazardous conditions. You are encouraged to establish safety procedures before using the method. Among other precautions, you should become familiar with the safety recommendations in the gas analyzer user's manual. Occupational Safety and Health Administration (OSHA) regulations concerning use of compressed gas cylinders and noxious gases may apply.
5.1 Site Hazards. Prior to applying these procedures/specifications in the field, the potential hazards at the test site should be considered; advance coordination with the site is critical to understand the conditions and applicable safety policies. At a minimum, portions of the sampling system will be hot, requiring appropriate gloves, long sleeves, and caution in handling this equipment.
5.2 Laboratory Safety. Policies should be in place to minimize risk of chemical exposure and to properly handle waste disposal in the laboratory. Personnel shall wear appropriate laboratory attire according to a Chemical Hygiene Plan established by the laboratory.
5.3 Reagent Toxicity/Carcinogenicity. The toxicity and carcinogenicity of any reagents used must be considered. Depending upon the sampling and analytical technologies selected, this measurement may involve hazardous materials, operations, and equipment and this method does not address all of the safety problems associated with implementing this approach. It is the responsibility of the user to establish appropriate safety and health practices and determine the applicable regulatory limitations prior to performance. Any chemical should be regarded as a potential health hazard and exposure to these compounds should be minimized. Chemists should refer to the Material Safety Data Sheet (MSDS) for each chemical used.
5.4 Waste Disposal. Any waste generated by this procedure must be disposed of according to a hazardous materials management plan that details and tracks various waste streams and disposal procedures.
6.0 Equipment and Supplies
The following list is presented as an example of key equipment and supplies likely required to measure vapor-phase Hg using a sorbent trap sampling system. It is recognized that additional equipment and supplies may be needed. Collection of paired samples is required.
6.1 Sorbent Trap Sampling System. A typical sorbent trap sampling system is shown in Figure 30B-1 in section 17.0. The sorbent trap sampling system shall include the following components:
6.1.1 Sorbent Traps. The sorbent media used to collect Hg must be configured in a trap with at least two distinct segments or sections, connected in series, that are amenable to separate analyses. section 1 is designated for primary capture of gaseous Hg. section 2 is designated as a backup section for determination of vapor phase Hg breakthrough. Each sorbent trap must be inscribed or otherwise permanently marked with a unique identification number, for tracking purposes. The sorbent media may be any collection material (e.g., carbon, chemically-treated filter, etc.) capable of quantitatively capturing and recovering for subsequent analysis, all gaseous forms of Hg in the emissions from the intended application. Selection of the sorbent media shall be based on the material's ability to achieve the performance criteria contained in this method as well as the sorbent's vapor phase Hg capture efficiency for the emissions matrix and the expected sampling duration at the test site. The sorbent media must be obtained from a source that can demonstrate their quality assurance and quality control (see section 7.2). The paired sorbent traps are supported on a probe (or probes) and inserted directly into the flue gas stream.
6.1.2 Sampling Probe Assembly. Each probe assembly shall have a leak-free attachment to the sorbent trap(s). Each sorbent trap must be mounted at the entrance of or within the probe such that the gas sampled enters the trap directly. Each probe/sorbent trap assembly must be heated to a temperature sufficient to prevent liquid condensation in the sorbent trap(s). Auxiliary heating is required only where the stack temperature is too low to prevent condensation. Use a calibrated thermocouple to monitor the stack temperature. A single probe capable of operating the paired sorbent traps may be used. Alternatively, individual probe/sorbent trap assemblies may be used, provided that the individual sorbent traps are co-located to ensure representative Hg monitoring.
6.1.3 Moisture Removal Device. A moisture removal device or system shall be used to remove water vapor from the gas stream prior to entering dry gas flow metering devices.
6.1.4 Vacuum Pump. Use a leak-tight, vacuum pump capable of operating within the system's flow range.
6.1.5 Gas Flow Meter. A gas flow meter (such as a dry gas meter, thermal mass flow meter, or other suitable measurement device) shall be used to determine the total sample volume on a dry basis, in units of standard cubic meters. The meter must be sufficiently accurate to measure the total sample volume to within 2 percent and must be calibrated at selected flow rates across the range of sample flow rates at which the sampling train will be operated. The gas flow meter shall be equipped with any necessary auxiliary measurement devices (e.g., temperature sensors, pressure measurement devices) needed to correct the sample volume to standard conditions.
6.1.6 Sample Flow Rate Meter and Controller. Use a flow rate indicator and controller for maintaining necessary sampling flow rates.
6.1.7 Temperature Sensor. Same as section 6.1.1.7 of Method 5 in Appendix A-3 to this part.
6.1.8 Barometer. Same as section 6.1.2 of Method 5 in Appendix A-3 to this part.
6.1.9 Data Logger (optional). Device for recording associated and necessary ancillary information (e.g., temperatures, pressures, flow, time, etc.).
6.2 Gaseous Hg 0 Sorbent Trap Spiking System. A known mass of gaseous Hg 0 must be either present on or spiked onto the first section of sorbent traps in order to perform the Hg 0 and HgCl 2 analytical bias test and the field recovery study. Any approach capable of quantitatively delivering known masses of Hg 0 onto sorbent traps is acceptable. Several spiking technologies or devices are available to meet this objective. Their practicality is a function of Hg mass spike levels. For low levels, NIST-certified or NIST-traceable gas generators or tanks may be suitable. An alternative system, capable of delivering almost any mass required, makes use of NIST-certified or NIST-traceable Hg salt solutions (e.g., HgCl 2 , Hg(NO 3 ) 2 ). With this system, an aliquot of known volume and concentration is added to a reaction vessel containing a reducing agent (e.g., stannous chloride); the Hg salt solution is reduced to Hg 0 and purged onto the sorbent trap using an impinger sparging system. When available, information on example spiking systems will be posted at http://www.epa.gov/ttn/emc.
6.3 Sample Analysis Equipment. Any analytical system capable of quantitatively recovering and quantifying total Hg from the sorbent media selected is acceptable provided that the analysis can meet the performance criteria described in this method. Example recovery techniques include acid leaching, digestion, and thermal desorption/direct combustion. Example analytical techniques include, but are not limited to, ultraviolet atomic fluorescence (UV AF), ultraviolet atomic absorption (UV AA) with and without gold trapping, and X-ray fluorescence (XRF) analysis.
6.3 Moisture Measurement System. If correction of the measured Hg emissions for moisture is required (see section 8.3.3.7), either Method 4 in Appendix A-3 to this part or other moisture measurement methods approved by the Administrator will be needed to measure stack gas moisture content.
7.0 Reagents and Standards
7.1 Reagents and Standards. Only NIST-certified or NIST-traceable calibration standards, standard reference materials, and reagents shall be used for the tests and procedures required by this method.
7.2 Sorbent Trap Media. The sorbent trap media shall be prepared such that the material used for testing is of known and acceptable quality. Sorbent supplier quality assurance/quality control measures to ensure appropriate and consistent performance such as sorptive capacity, uniformity of preparation treatments, and background levels shall be considered.
8.0 Sample Collection and Handling
This section presents the sample collection and handling procedures along with the pretest and on-site performance tests required by this method. Since you may choose different options to comply with certain performance criteria, each test report must identify the specific options selected and document the results with respect to the performance criteria of this method.
8.1 Selection of Sampling Sites and Sampling Points. What sampling site and sampling points do I select? Same as section 8.1 of Method 30A of this appendix.
8.2 Measurement System Performance Tests. What performance criteria must my measurement system meet? The following laboratory and field procedures and associated criteria of this section are designed to ensure (1) selection of a sorbent and analytical technique combination capable of quantitative collection and analysis of gaseous Hg, (2) collection of an adequate amount of Hg on each sorbent trap during field tests, and (3) adequate performance of the method for each test program: The primary objectives of these performance tests are to characterize and verify the performance of your intended analytical system and associated sampling and analytical procedures, and to define the minimum amount of Hg (as the sample collection target) that can be quantified reliably.
(a) Analytical Matrix Interference Test;
(b) Determination of Minimum Sample Mass;
(c) Hg 0 and HgCl 2 Analytical Bias Test;
(d) Determination of Nominal Sample Volume;
(e) Field Recovery Test.
8.2.1 Analytical Matrix Interference Test and Minimum Sample Dilution.
(a) The analytical matrix interference test is a laboratory procedure. It is required only if you elect to use a liquid digestion analytical approach and needs to be performed only once for each sorbent material used. The purpose of the test is to verify the presence or absence of known and potential analytical matrix interferences, including the potential negative bias associated with iodine common to many sorbent trap materials. The analytical matrix interference test determines the minimum dilution (if any) necessary to mitigate matrix effects on the sample digestate solutions.
(b) The result of the analytical matrix interference test, i.e., the minimum sample dilution required (if any) for all sample analyses, is used to establish the minimum sample mass needed for the Hg 0 and HgCl 2 analytical bias test and to determine the nominal sample volume for a test run. The analytical matrix interference test is sorbent material-specific and shall be performed for each sorbent material you intend to use for field sampling and analysis. The test shall be performed using a mass of sorbent material comparable to the sorbent mass typically used in the first section of the trap for sampling. Similar sorbent materials from different sources of supply are considered to be different materials and must be tested individually. You must conduct the analytical matrix interference test for each sorbent material prior to the analysis of field samples.
8.2.1.1 Analytical Matrix Interference Test Procedures. Digest and prepare for analysis a representative mass of sorbent material (unsampled) according to your intended laboratory techniques for field samples. Analyze the digestate according to your intended analytical conditions at the least diluted level you intend to use for sample analysis (e.g., undiluted, 1 in 10 dilution, etc.). Determine the Hg concentration of the undiluted digestate solution. Prepare a series of solutions with a fixed final volume containing graduated aliquots of the sample digestate and, a fixed aliquot of a calibration standard (with the balance being Hg-free reagent or H 2 0) to establish solutions of varied digestate dilution ratio (e.g., 1:2, 1:5, 1:10, 1:100, etc. - see example in section 8.2.1.3, below). One of these solutions should contain only the aliquot of the calibration standard in Hg-free reagent or H 2 O. This will result in a series of solutions where the amount of Hg is held relatively constant and only the volume of digestate diluted is varied. Analyze each of these solutions following intended sample analytical procedures and conditions, determining the concentration for each solution.
8.2.1.2 Analytical Matrix Interference Test Acceptance Criteria. Compare the measured concentration of each solution containing digestate to the measured concentration of the digestate-free solution. The lowest dilution ratio of any solution having a Hg concentration within ±5 percent of the digestate-free solution is the minimum dilution ratio required for analysis of all samples. If you desire to measure the digestate without dilution, the ±5 percent criterion must be met at a dilution ratio of at least 9:10 (i.e., ≥90% digestate).
8.2.1.3 Example Analytical Matrix Interference Test. An example analytical matrix interference test is presented below. Additional information on the conduct of the analytical matrix interference test will be posted at http://www.epa.gov/ttn/emc. Determine the most sensitive working range for the analyzer to be used. This will be a narrow range of concentrations. Digest and prepare for analysis a representative mass of sorbent material (unsampled) according to your intended laboratory techniques for sample preparation and analysis. Prepare a calibration curve for the most sensitive analytical region, e.g., 0.0, 0.5, 1.0, 3.0, 5.0, 10 ppb. Using the highest calibration standard, e.g., 10.0 ppb, prepare a series of solutions by adding successively smaller increments of the digestate to a fixed volume of the calibration standard and bringing each solution to a final fixed volume with mercury-free deionized water (diH 2 O). To 2.0 ml of the calibration standard add 18.0, 10.0, 4.0, 2.0, 1.0, 0.2, and 0.0 ml of the digestate. Bring the final volume of each solution to a total volume of 20 ml by adding 0.0, 8.0, 14.0, 16.0, 17.0, 17.8, and 18.0 ml of diH 2 O. This will yield solutions with dilution ratios of 9:10, 1:2, 1:5, 1:10, 1:20, 1:100, and 0:10, respectively. Determine the Hg concentration of each solution. The dilution ratio of any solution having a concentration that is within ±5 percent of the concentration of the solution containing 0.0 ml of digestate is an acceptable dilution ratio for analyzing field samples. If more than one solution meets this criterion, the one with the lowest dilution ratio is the minimum dilution required for analysis of field samples. If the 9:10 dilution meets this criterion, then no sample dilution is required.
8.2.2 Determination of Minimum Sample Mass. The minimum mass of Hg that must be collected per sample must be determined. This information is necessary in order to effectively perform the Hg 0 and HgCl 2 Analytical Bias Test, to estimate target sample volumes/sample times for test runs, and to ensure the quality of the measurements. The determination of minimum sample mass is a direct function of analytical technique, measurement sensitivity, dilutions, etc. This determination is required for all analytical techniques. Based on the analytical approach you employ, you should determine the most sensitive calibration range. Based on a calibration point within that range, you must consider all sample treatments (e.g., dilutions) to determine the mass of sample that needs to be collected to ensure that all sample analyses fall within your calibration curve.
8.2.2.1 Determination of Minimum Calibration Concentration or Mass. Based on your instrument's sensitivity and linearity, determine the calibration concentrations or masses that make up a representative low level calibration range. Verify that you are able to meet the multipoint calibration performance criteria in section 11.0 of this method. Select a calibration concentration or mass that is no less than 2 times the lowest concentration or mass in your calibration curve. The lowest point in your calibration curve must be at least 5, and preferably 10, times the Method Detection Limit (MDL), which is the minimum amount of the analyte that can be detected and reported. The MDL must be determined at least once for the analytical system using an MDL study such as that found in section 15.0 to Method 301 of appendix A to part 63 of this chapter.
Note to section 8.2.2.1:
While it might be desirable to base the minimum calibration concentration or mass on the lowest point in the calibration curve, selecting a higher concentration or mass is necessary to ensure that all analyses of the field samples will fall within the calibration curve. Therefore, it is strongly recommended that you select a minimum calibration concentration or mass that is sufficiently above the lowest point of the calibration curve (see examples in sections 8.2.2.2.1 and 8.2.2.2.2 below).
8.2.2.2 Determination of Minimum Sample Mass. Based on your minimum calibration concentration or mass and other sample treatments including, but not limited to, final digestate volume and minimum sample dilution, determine the minimum sample mass. Consideration should also be given to the Hg levels expected to be measured in section 2 of the sorbent traps and to the breakthrough criteria presented in Table 9-1.
8.2.2.2.1 Example Determination of Minimum Sample Mass for Thermal Desorption Analysis. A thermal analysis system has been calibrated at five Hg mass levels: 10 ng, 20 ng, 50 ng, 100 ng, 200 ng, and shown to meet the calibration performance criteria in this method. Based on 2 times the lowest point in the calibration curve, 20 ng is selected as the minimum calibration mass. Because the entire sample is analyzed and there are no dilutions involved, the minimum sample mass is also 20 ng.
Note:
In this example, if the typical background (blank) Hg levels in section 2 were relatively high (e.g., 3 to 5 ng), a sample mass of 20 ng might not have been sufficient to ensure that the breakthrough criteria in Table 9-1 would be met, thereby necessitating the use of a higher point on the calibration curve (e.g., 50 ng) as the minimum calibration and sample mass.
8.2.2.2.2 Example Determination of Minimum Sample Mass for Acid Leachate/Digestate Analysis. A cold vapor analysis system has been calibrated at four Hg concentration levels: 2 ng/L, 5 ng, 10 ng/L, 20 ng/L, and shown to meet the calibration performance criteria in this method. Based on 2 times the lowest point in the calibration curve, 4 ng/L was selected as the minimum calibration concentration. The final sample volume of a digestate is nominally 50 ml (0.05 L) and the minimum dilution necessary was determined to be 1:100 by the Analytical Matrix Interference Test of section 8.2.1. The following calculation would be used to determine the minimum sample mass.
Minimum sample mass = (4 ng/L) × (0.05 L) × (100) = 20 ng
Note:
In this example, if the typical background (blank) Hg levels in section 2 were relatively high (e.g., 3 to 5 ng), a sample mass of 20 ng might not have been sufficient to ensure that the breakthrough criterion in Table 9-1 would be met, thereby necessitating the use of a higher point on the calibration curve (e.g., 10 ng/L) as the minimum calibration concentration.
8.2.3 Hg 0 and HgCl 2 Analytical Bias Test. Before analyzing any field samples, the laboratory must demonstrate the ability to recover and accurately quantify Hg 0 and HgCl 2 from the chosen sorbent media by performing the following analytical bias test for sorbent traps spiked with Hg 0 and HgCl 2 . The analytical bias test is performed at a minimum of two distinct sorbent trap Hg loadings that will: (1) Represent the lower and upper bound of sample Hg loadings for application of the analytical technique to the field samples, and (2) be used for data validation.
8.2.3.1 Hg 0 and HgCl 2 Analytical Bias Test Procedures. Determine the lower and upper bound mass loadings. The minimum sample mass established in section 8.2.2.2 can be used for the lower bound Hg mass loading although lower Hg loading levels are acceptable. The upper bound Hg loading level should be an estimate of the greatest mass loading that may result as a function of stack concentration and volume sampled. As previously noted, this test defines the bounds that actual field samples must be within in order to be valid.
8.2.3.1.1 Hg 0 Analytical Bias Test. Analyze the front section of three sorbent traps containing Hg 0 at the lower bound mass loading level and the front section of three sorbent traps containing Hg 0 at the upper bound mass loading level. In other words, analyze each mass loading level in triplicate. You may refer to section 6.2 for spiking guidance. Prepare and analyze each spiked trap, using the same techniques that will be used to prepare and analyze the field samples. The average recovery for the three traps at each mass loading level must be between 90 and 110 percent. If multiple types of sorbent media are to be analyzed, a separate analytical bias test is required for each sorbent material.
8.2.3.1.2 HgCl 2 Analytical Bias Test. Analyze the front section of three sorbent traps containing HgCl 2 at the lower bound mass loading level and the front section of three traps containing HgCl 2 at the upper bound mass loading level. HgCl 2 can be spiked as a gas, or as a liquid solution containing HgCl 2 . However the liquid volume spiked must be <100 µL. Prepare and analyze each spiked trap, using the techniques that will be used to prepare and analyze the field samples. The average recovery for three traps at each spike concentration must be between 90 and 110 percent. Again, if multiple types of sorbent media are to be analyzed, a separate analytical bias test is required for each sorbent material.
8.2.4 Determination of Target Sample Volume. The target sample volume is an estimate of the sample volume needed to ensure that valid emissions data are collected (i.e., that sample mass Hg loadings fall within the analytical calibration curve and are within the upper and lower bounds set by the analytical bias tests). The target sample volume and minimum sample mass can also be determined by performing a diagnostic test run prior to initiation of formal testing.
Example:
If the minimum sample mass is 50 ng and the concentration of mercury in the stack gas is estimated to be 2 µg/m 3 (ng/L) then the following calculation would be used to determine the target sample volume:
Target Sample Volume = (50 ng) / (2 ng/L) = 25 L
Note to section 8.2.4:
For the purposes of relative accuracy testing of Hg monitoring systems under subpart UUUUU of part 63 of this chapter and Performance Specifications 12A and 12B in appendix B to this part, when the stack gas Hg concentration is expected to be very low (<0.5 µg/dscm), you may estimate the Hg concentration at 0.5 µg/dscm.
8.2.5 Determination of Sample Run Time. Sample run time will be a function of minimum sample mass (see section 8.2.2), target sample volume and nominal equipment sample flow rate. The minimum sample run time for conducting relative accuracy test audits of Hg monitoring systems is 30 minutes and for emissions testing to characterize an emission source is 1 hour. The target sample run time can be calculated using the following example.
Example:
If the target sample volume has been determined to be 25 L, then the following formula would be used to determine the sampling time necessary to acquire 25 L of gas when sampling at a rate of 0.4 L/min.
Sampling time (min) = 25 L / 0.4 L/min = 63 minutes
8.2.6 Field Recovery Test. The field recovery test provides a test program-specific verification of the performance of the combined sampling and analytical approach. Three sets of paired samples, one of each pair which is spiked with a known level of Hg, are collected and analyzed and the average recovery of the spiked samples is used to verify performance of the measurement system under field conditions during that test program. The conduct of this test requires an estimate or confirmation of the stack Hg concentrations at the time of testing.
8.2.6.1 Calculation of Pre-sampling Spiking Level. Determine the sorbent trap spiking level for the field recovery test using estimates of the stack Hg concentration, the target sample flow rate, and the planned sample duration. First, determine the Hg mass expected to be collected in section 1 of the sorbent trap. The pre-sampling spike must be within 50 to 150 percent of this expected mass.
Example calculation:
For an expected stack Hg concentration of 5 ug/m 3 (ng/L) a target sample rate of 0.40 liters/min, and a sample duration of 1 hour:
(0.40 L/min) * (60 min) * (5ng/L) = 120 ng
A Hg spike of 60 to 180 ng (50-150% of 120 ng) would be appropriate.
8.2.6.2 Procedures. Set up two identical sampling trains. One of the sampling trains shall be designated the spiked train and the other the unspiked train. Spike Hg 0 onto the front section of the sorbent trap in the spiked train before sampling. The mass of Hg spiked shall be 50 to 150 percent of the mass expected to be collected with the unspiked train. Sample the stack gas with the two trains simultaneously using the same procedures as for the field samples (see section 8.3). The total sample volume must be within ±20 percent of the target sample volume for the field sample test runs. Analyze the sorbent traps from the two trains utilizing the same analytical procedures and instrumentation as for the field samples (see section 11.0). Determine the fraction of spiked Hg recovered (R) using the equations in section 12.7. Repeat this procedure for a total of three runs. Report the individual R values in the test report; the average of the three R values must be between 85 and 115 percent.
Note to section 8.2.6.2:
It is acceptable to perform the field recovery test concurrent with actual test runs (e.g., through the use of a quad probe). It is also acceptable to use the field recovery test runs as test runs for emissions testing or for the RATA of a Hg monitoring system under subpart UUUUU of part 63 of this chapter and Performance Specifications 12A and 12B in appendix B to this part, if certain conditions are met. To determine whether a particular field recovery test run may be used as a RATA run, subtract the mass of the Hg 0 spike from the total Hg mass collected in sections 1 and 2 of the spiked trap. The difference represents the mass of Hg in the stack gas sample. Divide this mass by the sample volume to obtain the Hg concentration in the effluent gas stream, as measured with the spiked trap. Compare this concentration to the corresponding Hg concentration measured with the unspiked trap. If the paired trains meet the relative deviation and other applicable data validation criteria in Table 9-1, then the average of the two Hg concentrations may be used as an emissions test run value or as the reference method value for a RATA run.
8.3 Sampling. This section describes the procedures and criteria for collecting the field samples for analysis. As noted in section 8.2.6, the field recovery test samples are also collected using these procedures.
8.3.1 Pre-test leak check. Perform a leak check of the sampling system with the sorbent traps in place. For each of the paired sampling trains, draw a vacuum in the train, and adjust the vacuum to ∼15″ Hg; and, using the gas flow meter, determine leak rate. The leak rate for an individual train must not exceed 4 percent of the target sampling rate. Once the leak check passes this criterion, carefully release the vacuum in the sample train, then seal the sorbent trap inlet until the probe is ready for insertion into the stack or duct.
8.3.2 Determination of Flue Gas Characteristics. Determine or measure the flue gas measurement environment characteristics (gas temperature, static pressure, gas velocity, stack moisture, etc.) in order to determine ancillary requirements such as probe heating requirements (if any), initial sampling rate, moisture management, etc.
8.3.3 Sample Collection
8.3.3.1 Remove the plug from the end of each sorbent trap and store each plug in a clean sorbent trap storage container. Remove the stack or duct port cap and insert the probe(s). Secure the probe(s) and ensure that no leakage occurs between the duct and environment.
8.3.3.2 Record initial data including the sorbent trap ID, date, and the run start time.
8.3.3.3 Record the initial gas flow meter reading, stack temperature, meter temperatures (if needed), and any other appropriate information, before beginning sampling. Begin sampling and target a sampling flow rate similar to that for the field recovery test. Then, at regular intervals (≤5 minutes) during the sampling period, record the date and time, the sample flow rate, the gas meter reading, the stack temperature, the flow meter temperatures (if using a dry gas meter), temperatures of heated equipment such as the vacuum lines and the probes (if heated), and the sampling system vacuum readings. Adjust the sampling flow rate as necessary to maintain the initial sample flow rate. Ensure that the total volume sampled for each run is within 20 percent of the total volume sampled for the field recovery test.
8.3.3.4 Data Recording. Obtain and record any essential operating data for the facility during the test period, e.g., the barometric pressure must be obtained for correcting sample volume to standard conditions when using a dry gas meter. At the end of the data collection period, record the final gas flow meter reading and the final values of all other essential parameters.
8.3.3.5 Post-Test Leak Check. When sampling is completed, turn off the sample pump, remove the probe(s) with sorbent traps from the port, and carefully seal the end of each sorbent trap. Perform another leak check of each sampling train with the sorbent trap in place, at the maximum vacuum reached during the sampling period. Record the leakage rates and vacuums. The leakage rate for each train must not exceed 4 percent of the average sampling rate for the data collection period. Following each leak check, carefully release the vacuum in the sample train.
8.3.3.6 Sample Recovery. Recover each sampled sorbent trap by removing it from the probe and sealing both ends. Wipe any deposited material from the outside of the sorbent trap. Place the sorbent trap into an appropriate sample storage container and store/preserve in appropriate manner (see section 8.3.3.8).
8.3.3.7 Stack Gas Moisture Determination. If the moisture basis of the measurements made with this method (dry) is different from the moisture basis of either: (1) the applicable emission limit; or (2) a Hg CEMS being evaluated for relative accuracy, you must determine the moisture content of the flue gas and correct for moisture using Method 4 in appendix A-3 to this part. If correction of the measured Hg concentrations for moisture is required, at least one Method 4 moisture determination shall be made during each test run.
8.3.3.8 Sample Handling, Preservation, Storage, and Transport. While the performance criteria of this approach provides for verification of appropriate sample handling, it is still important that the user consider, determine and plan for suitable sample preservation, storage, transport, and holding times for these measurements. Therefore, procedures in ASTM D6911-15 “Standard Guide for Packaging and Shipping Environmental Samples for Laboratory Analysis” (incorporated by reference-see 40 CFR 60.17) shall be followed for all samples, where appropriate. To avoid Hg contamination of the samples, special attention should be paid to cleanliness during transport, field handling, sampling, recovery, and laboratory analysis, as well as during preparation of the sorbent cartridges. Collection and analysis of blank samples ( e.g., reagent, sorbent, field, etc.) is useful in verifying the absence or source of contaminant Hg.
8.3.3.9 Sample Custody. Proper procedures and documentation for sample chain of custody are critical to ensuring data integrity. The chain of custody procedures in ASTM D4840-99 “Standard Guide for Sampling Chain-of-Custody Procedures” shall be followed for all samples (including field samples and blanks).
9.0 Quality Assurance and Quality Control
Table 9-1 summarizes the QA/QC performance criteria that are used to validate the Hg emissions data from Method 30B sorbent trap measurement systems.
| QA/QC test or specification | Acceptance criteria | Frequency | Consequences if not met |
|---|---|---|---|
| Gas flow meter calibration (At 3 settings or points) | Calibration factor (Y i ) at each flow rate must be within ±2% of the average value (Y) | Prior to initial use and when post-test check is not within ±5% of Y | Recalibrate at 3 points until the acceptance criteria are met. |
| Gas flow meter post-test calibration check (Single-point) | Calibration factor (Y i ) must be within ±5% of the Y value from the most recent 3-point calibration | After each field test. For mass flow meters, must be done on-site, using stack gas | Recalibrate gas flow meter at 3 points to determine a new value of Y. For mass flow meters, must be done on-site, using stack gas. Apply the new Y value to the field test data. |
| Temperature sensor calibration | Absolute temperature measures by sensor within ±1.5% of a reference sensor | Prior to initial use and before each test thereafter | Recalibrate; sensor may not be used until specification is met. |
| Barometer calibration | Absolute pressure measured by instrument within ±10 mm Hg of reading with a mercury barometer or NIST traceable barometer | Prior to initial use and before each test thereafter | Recalibrate; instrument may not be used until specification is met. |
| Pre-test leak check | ≤4% of target sampling rate | Prior to sampling | Sampling shall not commence until the leak check is passed. |
| Post-test leak check | ≤4% of average sampling rate | After sampling | Sample invalidated.* |
| Analytical matrix interference test (wet chemical analysis, only) | Establish minimum dilution (if any) needed to eliminate sorbent matrix interferences | Prior to analyzing any field samples; repeat for each type of sorbent used | Field sample results not validated. |
| Analytical bias test | Average recovery between 90% and 110% for Hg 0 and HgCl 2 at each of the 2 spike concentration levels | Prior to analyzing field samples and prior to use of new sorbent media | Field samples shall not be analyzed until the percent recovery criteria has been met. |
| Multipoint analyzer calibration | Each analyzer reading within ±10% of true value and r 2 ≥0.99 | On the day of analysis, before analyzing any samples | Recalibrate until successful. |
| Analysis of independent calibration standard | Within ±10% of true value | Following daily calibration, prior to analyzing field samples | Recalibrate and repeat independent standard analysis until successful. |
| Analysis of continuing calibration verification standard (CCVS) | Within ±10% of true value | Following daily calibration, after analyzing ≤10 field samples, and at end of each set of analyses | Recalibrate and repeat independent standard analysis, reanalyze samples until successful, if possible; for destructive techniques, samples invalidated. |
| Test run total sample volume | Within ±20% of total volume sampled during field recovery test | Each individual sample | Sample invalidated. |
| Sorbent trap section 2 breakthrough | For compliance/emissions testing: | Every sample | Sample invalidated.* |
| ≤10% of section 1 Hg mass for Hg concentrations >1 µg/dscm; | |||
| ≤20% of section 1 Hg mass for Hg concentrations ≤1 µg/dscm | |||
| ≤50% of section 1 Hg mass if the stack Hg concentration is ≤30% of the Hg concentration that is equivalent to the applicable emission limit | |||
| For relative accuracy testing: | |||
| ≤10% of section 1 Hg mass for Hg concentrations >1 µg/dscm; | |||
| ≤20% of section 1 Hg mass for Hg concentrations ≤1 µg/dscm and >0.5 µg/dscm; | |||
| ≤50% of section 1 Hg mass for Hg concentrations ≤0.5 µg/dscm >0.1 µg/dscm; | |||
| no criterion for Hg concentrations ≤0.1 µg/dscm (must meet all other QA/QC specifications) | |||
| Paired sorbent trap agreement | ≤10% Relative Deviation (RD) mass for Hg concentrations >1 µg/dscm; | Every run | Run invalidated.* |
| ≤20% RD or ≤0.2 µg/dscm absolute difference for Hg concentrations ≤1 µg/dscm | |||
| Sample analysis | Within valid calibration range (within calibration curve) | All Section 1 samples where stack Hg concentration is ≥0.02 µg/dscm except in case where stack Hg concentration is ≤30% of the applicable emission limit | Reanalyze at more concentrated level if possible, samples invalidated if not within calibrated range. |
| Sample analysis | Within bounds of Hg 0 and HgCl 2 Analytical Bias Test | All Section 1 samples where stack Hg concentration is ≥0.5 µg/dscm | Expand bounds of Hg 0 and HgCl 2 Analytical Bias Test; if not successful, samples invalidated. |
| Field recovery test | Average recovery between 85% and 115% for Hg 0 | Once per field test | Field sample runs not validated without successful field recovery test. |
| * And data from the pair of sorbent traps are also invalidated. | |||
10.0 Calibration and Standardization
10.1 Only NIST-certified and NIST-traceable calibration standards (i.e., calibration gases, solutions, etc.) shall be used for the spiking and analytical procedures in this method.
10.2 Gas Flow Meter Calibration.
10.2.1 Preliminaries. The manufacturer or equipment supplier of the gas flow meter should perform all necessary set-up, testing, programming, etc., and should provide the end user with any necessary instructions, to ensure that the meter will give an accurate readout of dry gas volume in standard cubic meters for this method.
10.2.2 Initial Calibration. Prior to its initial use, a calibration of the gas flow meter shall be performed. The initial calibration may be done by the manufacturer, by the equipment supplier, or by the end user. If the flow meter is volumetric in nature ( e.g. , a dry gas meter), the manufacturer or end user may perform a direct volumetric calibration using any gas. For a mass flow meter, the manufacturer, equipment supplier, or end user may calibrate the meter using either: (1) A bottled gas mixture containing 12 ±0.5% CO 2 , 7 ±0.5% O 2 , and balance N 2 (when this method is applied to coal-fired boilers); (2) a bottled gas mixture containing CO 2 , O 2 , and N 2 in proportions representative of the expected stack gas composition; or (3) the actual stack gas.
10.2.2.1 Initial Calibration Procedures. Determine an average calibration factor (Y) for the gas flow meter by calibrating it at three sample flow rate settings covering the range of sample flow rates at which the sampling system will be operated. You may either follow the procedures in section 10.3.1 of Method 5 in appendix A-3 to this part or in section 16 of Method 5 in appendix A-3 to this part. If a dry gas meter is being calibrated, use at least five revolutions of the meter at each flow rate.
10.2.2.2 Alternative Initial Calibration Procedures. Alternatively, you may perform the initial calibration of the gas flow meter using a reference gas flow meter (RGFM). The RGFM may be: (1) A wet test meter calibrated according to section 10.3.1 of Method 5 in appendix A-3 to this part; (2) a gas flow metering device calibrated at multiple flow rates using the procedures in section 16 of Method 5 in appendix A-3 to this part; or (3) a NIST-traceable calibration device capable of measuring volumetric flow to an accuracy of 1 percent. To calibrate the gas flow meter using the RGFM, proceed as follows: While the Method 30B sampling system is sampling the actual stack gas or a compressed gas mixture that simulates the stack gas composition (as applicable), connect the RGFM to the discharge of the system. Care should be taken to minimize the dead volume between the gas flow meter being tested and the RGFM. Concurrently measure dry stack gas volume with the RGFM and the flow meter being calibrated for at least 10 minutes at each of three flow rates covering the typical range of operation of the sampling system. For each set of concurrent measurements, record the total sample volume, in units of dry standard cubic meters (dscm), measured by the RGFM and the gas flow meter being tested.
10.2.2.3 Initial Calibration Factor. Calculate an individual calibration factor Y i at each tested flow rate from section 10.2.2.1 or 10.2.2.2 of this method (as applicable) by taking the ratio of the reference sample volume to the sample volume recorded by the gas flow meter. Average the three Y i values, to determine Y, the calibration factor for the flow meter. Each of the three individual values of Y i must be within ±0.02 of Y. Except as otherwise provided in sections 10.2.2.4 and 10.2.2.5 of this method, use the average Y value from the initial 3-point calibration to adjust subsequent gas volume measurements made with the gas flow meter.
10.2.2.4 Pretest On-Site Calibration Check (Optional). For a mass flow meter, if the most recent 3-point calibration of the flow meter was performed using a compressed gas mixture, you may want to conduct the following on-site calibration check prior to testing, to ensure that the flow meter will accurately measure the volume of the stack gas: While sampling stack gas, check the calibration of the flow meter at one intermediate flow rate setting representative of normal operation of the sampling system. If the pretest calibration check shows that the value of Y i , the calibration factor at the tested flow rate, differs from the current value of Y by more than 5 percent, perform a full 3-point recalibration of the meter using stack gas to determine a new value of Y, and (except as otherwise provided in section 10.2.2.5 of this method) apply the new Y value to the data recorded during the field test.
10.2.2.5 Post-Test Calibration Check. Check the calibration of the gas flow meter following each field test at one intermediate flow rate setting, either at, or in close proximity to, the average sample flow rate during the field test. For dry gas meters, ensure at least three revolutions of the meter during the calibration check. For mass flow meters, this check must be performed before leaving the test site, while sampling stack gas. If a one-point calibration check shows that the value of Y i at the tested flow rate differs by more than 5 percent from the current value of Y, repeat the full 3-point calibration procedure to determine a new value of Y, and apply the new Y value to the gas volume measurements made with the gas flow meter during the field test that was just completed. For mass flow meters, perform the 3-point recalibration while sampling stack gas.
10.3 Thermocouples and Other Temperature Sensors. Use the procedures and criteria in Section 10.3 of Method 2 in appendix A-1 to this part to calibrate in-stack temperature sensors and thermocouples. Dial thermometers shall be calibrated against mercury-in-glass thermometers or equivalent. Calibrations must be performed prior to initial use and before each field test thereafter. At each calibration point, the absolute temperature measured by the temperature sensor must agree to within ±1.5 percent of the temperature measured with the reference sensor, otherwise the sensor may not continue to be used.
10.4 Barometer. Calibrate against a mercury barometer or other NIST-traceable barometer as per Section 10.6 of Method 5 in appendix A-3 to this part. Calibration must be performed prior to initial use and before each test program, and the absolute pressure measured by the barometer must agree to within ±10 mm Hg of the pressure measured by the mercury or other NIST-traceable barometer, otherwise the barometer may not continue to be used.
10.5 Other Sensors and Gauges. Calibrate all other sensors and gauges according to the procedures specified by the instrument manufacturer(s).
10.6 Analytical System Calibration. See section 11.1 of this method.
11.0 Analytical Procedures
The analysis of Hg in the field and quality control samples may be conducted using any instrument or technology capable of quantifying total Hg from the sorbent media and meeting the performance criteria in this method. Because multiple analytical approaches, equipment and techniques are appropriate for the analysis of sorbent traps, it is not possible to provide detailed, technique-specific analytical procedures. As they become available, detailed procedures for a variety of candidate analytical approaches will be posted at http://www.epa.gov/ttn/emc.
11.1 Analytical System Calibration. Perform a multipoint calibration of the analyzer at three or more upscale points over the desired quantitative range (multiple calibration ranges shall be calibrated, if necessary). The field samples analyzed must fall within a calibrated, quantitative range and meet the performance criteria specified below. For samples suitable for aliquotting, a series of dilutions may be needed to ensure that the samples fall within a calibrated range. However, for sorbent media samples consumed during analysis (e.g., when using thermal desorption techniques), extra care must be taken to ensure that the analytical system is appropriately calibrated prior to sample analysis. The calibration curve range(s) should be determined such that the levels of Hg mass expected to be collected and measured will fall within the calibrated range. The calibration curve may be generated by directly introducing standard solutions into the analyzer or by spiking the standards onto the sorbent media and then introducing into the analyzer after preparing the sorbent/standard according to the particular analytical technique. For each calibration curve, the value of the square of the linear correlation coefficient, i.e., r 2 , must be ≥0.99, and the analyzer response must be within ±10 percent of the reference value at each upscale calibration point. Calibrations must be performed on the day of the analysis, before analyzing any of the samples. Following calibration, an independent standard shall be analyzed. The measured value of the independently prepared standard must be within ±10 percent of the expected value.
11.2 Sample Preparation. Carefully separate the sections of each sorbent trap. Combine for analysis all materials associated with each section; any supporting substrate that the sample gas passes through prior to entering a media section (e.g., glass wool separators, acid gas traps, etc.) must be analyzed with that segment.
11.3 Field Sample Analyses. Analyze the sorbent trap samples following the same procedures that were used for conducting the Hg 0 and HgCl 2 analytical bias tests. The individual sections of the sorbent trap and their respective components must be analyzed separately ( i.e. , section 1 and its components, then section 2 and its components). All sorbent trap section 1 sample analyses must be within the calibrated range of the analytical system as specified in Table 9-1. For wet analyses, the sample can simply be diluted to fall within the calibrated range. However, for the destructive thermal analyses, samples that are not within the calibrated range cannot be re-analyzed. As a result, the sample cannot be validated, and another sample must be collected. It is strongly suggested that the analytical system be calibrated over multiple ranges so that thermally analyzed samples fall within the calibrated range. The total mass of Hg measured in each sorbent trap section 1 must also fall within the lower and upper mass limits established during the initial Hg 0 and HgCl 2 analytical bias test. If a sample is analyzed and found to fall outside of these limits, it is acceptable for an additional Hg 0 and HgCl 2 analytical bias test to be performed that now includes this level. However, some samples (e.g., the mass collected in trap section 2), may have Hg levels so low that it may not be possible to quantify them in the analytical system's calibrated range. Because a reliable estimate of these low-level Hg measurements is necessary to fully validate the emissions data, the MDL (see section 8.2.2.1 of this method) is used to establish the minimum amount that can be detected and reported. If the measured mass or concentration is below the lowest point in the calibration curve and above the MDL, the analyst must estimate the mass or concentration of the sample based on the analytical instrument response relative to an additional calibration standard at a concentration or mass between the MDL and the lowest point in the calibration curve. This is accomplished by establishing a response factor (e.g., area counts per Hg mass or concentration) and estimating the amount of Hg present in the sample based on the analytical response and this response factor.
Example:
The analysis of a particular sample results in a measured mass above the MDL, but below the lowest point in the calibration curve which is 10 ng. An MDL of 1.3 ng Hg has been established by the MDL study. A calibration standard containing 5 ng of Hg is analyzed and gives an analytical response of 6,170 area counts, which equates to a response factor of 1,234 area counts/ng Hg. The analytical response for the sample is 4,840 area counts. Dividing the analytical response for the sample (4,840 area counts) by the response factor gives 3.9 ng Hg, which is the estimated mass of Hg in the sample.
11.4 Analysis of Continuing Calibration Verification Standard (CCVS). After no more than 10 samples and at the end of each set of analyses, a continuing calibration verification standard must be analyzed. The measured value of the continuing calibration standard must be within ±10 percent of the expected value.
11.5 Blanks. The analysis of blanks is optional. The analysis of blanks is useful to verify the absence of, or an acceptable level of, Hg contamination. Blank levels should be considered when quantifying low Hg levels and their potential contribution to meeting the sorbent trap section 2 breakthrough requirements; however, correcting sorbent trap results for blank levels is prohibited.
12.0 Calculations and Data Analysis
You must follow the procedures for calculation and data analysis listed in this section.
12.1 Nomenclature. The terms used in the equations are defined as follows:
B = Breakthrough (%).
B ws = Moisture content of sample gas as measured by Method 4, percent/100.
C a = Concentration of Hg for the sample collection period, for sorbent trap “a” (µg/dscm).
C b = Concentration of Hg for the sample collection period, for sorbent trap “b” (µg/dscm).
C d = Hg concentration, dry basis (µg/dscm).
C rec = Concentration of spiked compound measured (µg/m 3 ).
C w = Hg concentration, wet basis (µg/m 3 ).
m 1 = Mass of Hg measured on sorbent trap section 1 (µg).
m 2 = Mass of Hg measured on sorbent trap section 2 (µg).
m recovered = Mass of spiked Hg recovered in Analytical Bias or Field Recovery Test (µg).
m s = Total mass of Hg measured on spiked trap in Field Recovery Test (µg).
m spiked = Mass of Hg spiked in Analytical Bias or Field Recovery Test (µg).
m u = Total mass of Hg measured on unspiked trap in Field Recovery Test (µg).
R = Percentage of spiked mass recovered (%).
RD = Relative deviation between the Hg concentrations from traps “a” and “b” (%).
v s = Volume of gas sampled, spiked trap in Field Recovery Test (dscm).
V t = Total volume of dry gas metered during the collection period (dscm); for the purposes of this method, standard temperature and pressure are defined as 20°C and 760 mm Hg, respectively.
v u = Volume of gas sampled, unspiked trap in Field Recovery Test (dscm).
12.2 Calculation of Spike Recovery (Analytical Bias Test). Calculate the percent recovery of Hg 0 and HgCl 2 using Equation 30B-1.
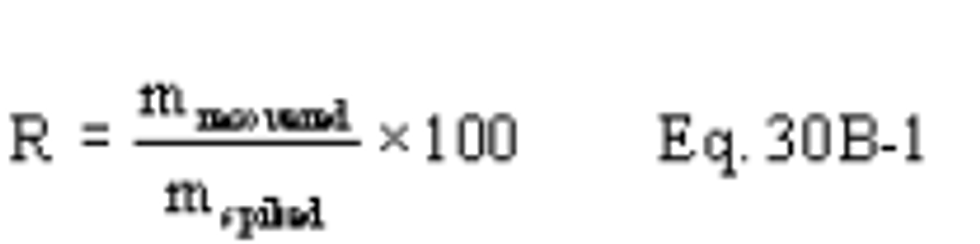
12.3 Calculation of Breakthrough. Use Equation 30B-2 to calculate the percent breakthrough to the second section of the sorbent trap.
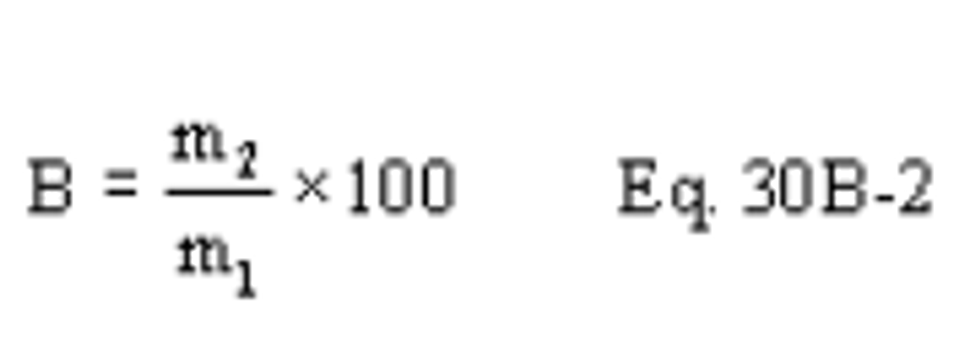
12.4 Calculation of Hg Concentration. Calculate the Hg concentration measured with sorbent trap “a”, using Equation 30B-3.
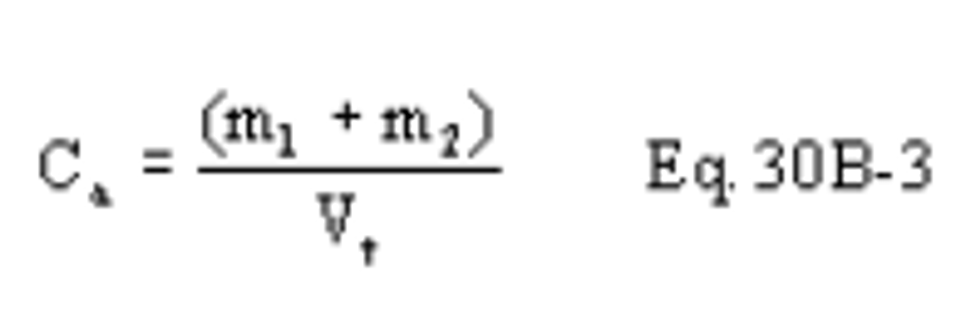
For sorbent trap “b”, replace “C a ” with “C b ” in Equation 30B-3. Report the average concentration, i.e., 1/2 (C a C b ).
12.5 Moisture Correction. Use Equation 30B-4 if your measurements need to be corrected to a wet basis.
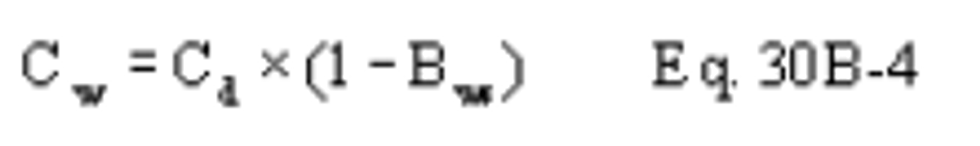
12.6 Calculation of Paired Trap Agreement. Calculate the relative deviation (RD) between the Hg concentrations measured with the paired sorbent traps using Equation 30B-5.
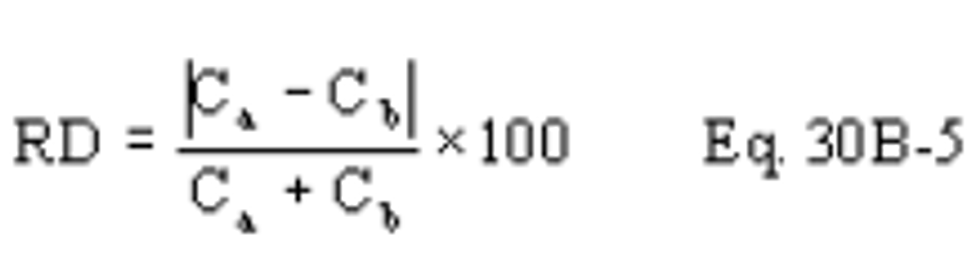
12.7 Calculation of Measured Spike Hg Concentration (Field Recovery Test). Calculate the measured spike concentration using Equation 30B-6.
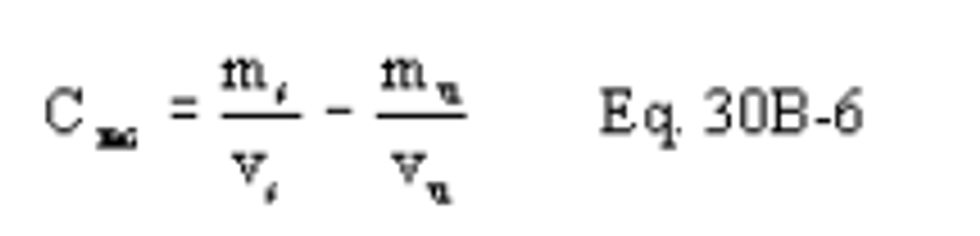
Then calculate the spiked Hg recovery, R, using Equation 30B-7.
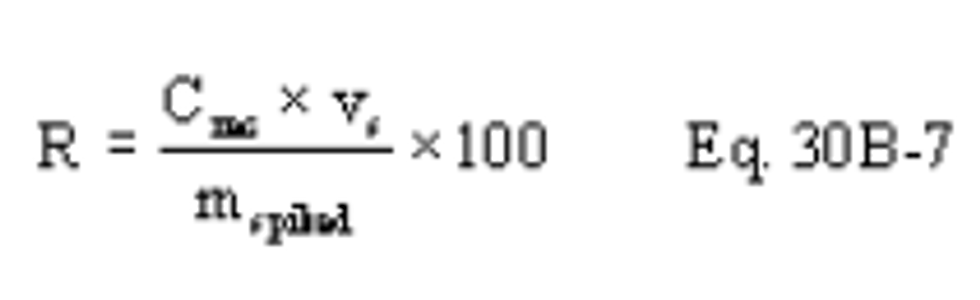
13.0 Method Performance
How do I validate my data? Measurement data are validated using initial, one-time laboratory tests coupled with test program-specific tests and procedures. The analytical matrix interference test and the Hg 0 and HgCl 2 analytical bias test described in section 8.2 are used to verify the appropriateness of the selected analytical approach(es) as well as define the valid working ranges for sample analysis. The field recovery test serves to verify the performance of the combined sampling and analysis as applied for each test program. Field test samples are validated by meeting the above requirements as well as meeting specific sampling requirements (i.e., leak checks, paired train agreement, total sample volume agreement with field recovery test samples) and analytical requirements (i.e., valid calibration curve, continuing calibration performance, sample results within calibration curve and bounds of Hg 0 and HgCl 2 analytical bias test). Complete data validation requirements are summarized in Table 9-1.
14.0 Pollution Prevention [Reserved]
15.0 Waste Management [Reserved]
16.0 References
1. EPA Traceability Protocol for Qualification and Certification of Elemental Mercury Gas Generators, expected publication date December 2008, see www.epa.gov/ttn/emc.
2. EPA Traceability Protocol for Qualification and Certification of Oxidized Mercury Gas Generators, expected publication date December 2008, see www.epa.gov/ttn/emc.
3. EPA Traceability Protocol for Assay and Certification of Gaseous Calibration Standards, expected revision publication date December 2008, see www.epa.gov/ttn/emc.
17.0 Figures and Tables
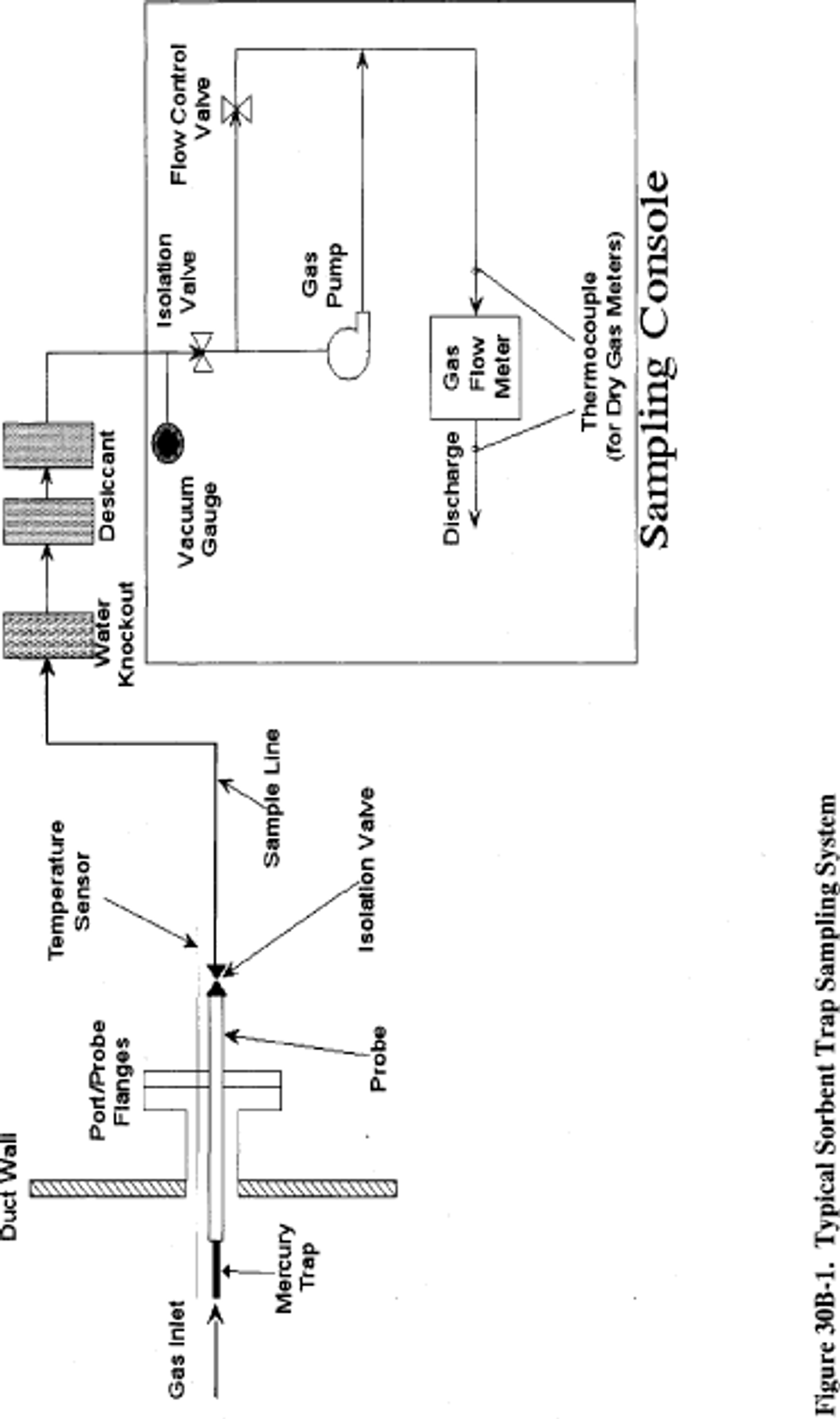
Editorial Note: For Federal Register citations affecting appendix A-8 to part 60, see the List of CFR sections Affected, which appears in the Finding Aids section of the printed volume and at www.govinfo.gov.
[36 FR 24877, Dec. 23, 1971; 85 FR 63416, Oct. 7, 2020; 88 FR 18406, March 29, 2023]
['Air Programs']
['Air Quality']
UPGRADE TO CONTINUE READING