Compliance Just Got Easier: Stay ahead of regulatory changes with instant notifications on updates that matter.
['Air Programs']
['Air Quality']
11/20/2023

Copyright 2026 J. J. Keller & Associate, Inc. For re-use options please contact copyright@jjkeller.com or call 800-558-5011.
Method 6 - Determination of sulfur dioxide emissions from stationary sources
Method 6A - Determination of sulfur dioxide, moisture, and carbon dioxide emissions from fossil fuel combustion sources
Method 6B - Determination of sulfur dioxide and carbon dioxide daily average emissions from fossil fuel combustion sources
Method 6C - Determination of Sulfur Dioxide Emissions From Stationary Sources (Instrumental Analyzer Procedure)
Method 7 - Determination of nitrogen oxide emissions from stationary sources
Method 7A - Determination of nitrogen oxide emissions from stationary sources - Ion chromatographic method
Method 7B - Determination of nitrogen oxide emissions from stationary sources (Ultraviolet spectrophotometry)
Method 7C - Determination of nitrogen oxide emissions from stationary sources - Alkaline-permanganate/colorimetric method
Method 7D - Determination of nitrogen oxide emissions from stationary sources - Alkaline-permanganate/ion chromatographic method
Method 7E - Determination of Nitrogen Oxides Emissions From Stationary Sources (Instrumental Analyzer Procedure)
Method 8 - Determination of sulfuric acid mist and sulfur dioxide emissions from stationary sources
Method 9 - Visual determination of the opacity of emissions from stationary sources
Alternate method 1 - Determination of the opacity of emissions from stationary sources remotely by lidar
Method 10 - Determination of carbon monoxide emissions from stationary sources
Method 10A - Determination of carbon monoxide emissions in certifying continuous emission monitoring systems at petroleum refineries
Method 10B - Determination of carbon monoxide emissions from stationary sources
The test methods in this appendix are referred to in §60.8 (Performance Tests) and §60.11 (Compliance With Standards and Maintenance Requirements) of 40 CFR part 60, subpart A (General Provisions). Specific uses of these test methods are described in the standards of performance contained in the subparts, beginning with Subpart D.
Within each standard of performance, a section title “Test Methods and Procedures” is provided to: (1) Identify the test methods to be used as reference methods to the facility subject to the respective standard and (2) identify any special instructions or conditions to be followed when applying a method to the respective facility. Such instructions (for example, establish sampling rates, volumes, or temperatures) are to be used either in addition to, or as a substitute for procedures in a test method. Similarly, for sources subject to emission monitoring requirements, specific instructions pertaining to any use of a test method as a reference method are provided in the subpart or in Appendix B.
Inclusion of methods in this appendix is not intended as an endorsement or denial of their applicability to sources that are not subject to standards of performance. The methods are potentially applicable to other sources; however, applicability should be confirmed by careful and appropriate evaluation of the conditions prevalent at such sources.
The approach followed in the formulation of the test methods involves specifications for equipment, procedures, and performance. In concept, a performance specification approach would be preferable in all methods because this allows the greatest flexibility to the user. In practice, however, this approach is impractical in most cases because performance specifications cannot be established. Most of the methods described herein, therefore, involve specific equipment specifications and procedures, and only a few methods in this appendix rely on performance criteria.
Minor changes in the test methods should not necessarily affect the validity of the results and it is recognized that alternative and equivalent methods exist. section 60.8 provides authority for the Administrator to specify or approve (1) equivalent methods, (2) alternative methods, and (3) minor changes in the methodology of the test methods. It should be clearly understood that unless otherwise identified all such methods and changes must have prior approval of the Administrator. An owner employing such methods or deviations from the test methods without obtaining prior approval does so at the risk of subsequent disapproval and retesting with approved methods.
Within the test methods, certain specific equipment or procedures are recognized as being acceptable or potentially acceptable and are specifically identified in the methods. The items identified as acceptable options may be used without approval but must be identified in the test report. The potentially approvable options are cited as “subject to the approval of the Administrator” or as “or equivalent.” Such potentially approvable techniques or alternatives may be used at the discretion of the owner without prior approval. However, detailed descriptions for applying these potentially approvable techniques or alternatives are not provided in the test methods. Also, the potentially approvable options are not necessarily acceptable in all applications. Therefore, an owner electing to use such potentially approvable techniques or alternatives is responsible for: (1) assuring that the techniques or alternatives are in fact applicable and are properly executed; (2) including a written description of the alternative method in the test report (the written method must be clear and must be capable of being performed without additional instruction, and the degree of detail should be similar to the detail contained in the test methods); and (3) providing any rationale or supporting data necessary to show the validity of the alternative in the particular application. Failure to meet these requirements can result in the Administrator's disapproval of the alternative.
Method 6 - Determination of Sulfur Dioxide Emissions From Stationary Sources
Note:
This method does not include all of the specifications (e.g., equipment and supplies) and procedures (e.g., sampling and analytical) essential to its performance. Some material is incorporated by reference from other methods in this part. Therefore, to obtain reliable results, persons using this method should have a thorough knowledge of at least the following additional test methods: Method 1, Method 2, Method 3, Method 5, and Method 8.
1.0 Scope and Application
1.1 Analytes.
| Analyte | CAS No. | Sensitivity |
|---|---|---|
| SO2 | 7449-09-5 | 3.4 mg SO2/m 3 (2.12 × 10)−7 lb/ft 3 |
1.2 Applicability. This method applies to the measurement of sulfur dioxide (SO2) emissions from stationary sources.
1.3 Data Quality Objectives. Adherence to the requirements of this method will enhance the quality of the data obtained from air pollutant sampling methods.
2.0 Summary of Method
2.1 A gas sample is extracted from the sampling point in the stack. The SO2 and the sulfur trioxide, including those fractions in any sulfur acid mist, are separated. The SO2 fraction is measured by the barium-thorin titration method.
3.0 Definitions [Reserved]
4.0 Interferences
4.1 Free Ammonia. Free ammonia interferes with this method by reacting with SO2 to form particulate sulfite and by reacting with the indicator. If free ammonia is present (this can be determined by knowledge of the process and/or noticing white particulate matter in the probe and isopropanol bubbler), alternative methods, subject to the approval of the Administrator are required. One approved alternative is listed in Reference 13 of section 17.0.
4.2 Water-Soluble Cations and Fluorides. The cations and fluorides are removed by a glass wool filter and an isopropanol bubbler; therefore, they do not affect the SO2 analysis. When samples are collected from a gas stream with high concentrations of metallic fumes (i.e., very fine cation aerosols) a high-efficiency glass fiber filter must be used in place of the glass wool plug (i.e., the one in the probe) to remove the cation interferent.
5.0 Safety
5.1 Disclaimer. This method may involve hazardous materials, operations, and equipment. This test method may not address all of the safety problems associated with its use. It is the responsibility of the user to establish appropriate safety and health practices and determine the applicability of regulatory limitations before performing this test method.
5.2 Corrosive reagents. The following reagents are hazardous. Personal protective equipment and safe procedures are useful in preventing chemical splashes. If contact occurs, immediately flush with copious amounts of water for at least 15 minutes. Remove clothing under shower and decontaminate. Treat residual chemical burns as thermal burns.
5.2.1 Hydrogen Peroxide (H2O2). Irritating to eyes, skin, nose, and lungs. 30% H2O2 is a strong oxidizing agent. Avoid contact with skin, eyes, and combustible material. Wear gloves when handling.
5.2.2 Sodium Hydroxide (NaOH). Causes severe damage to eyes and skin. Inhalation causes irritation to nose, throat, and lungs. Reacts exothermically with limited amounts of water.
5.2.3 Sulfuric Acid (H2SO4). Rapidly destructive to body tissue. Will cause third degree burns. Eye damage may result in blindness. Inhalation may be fatal from spasm of the larynx, usually within 30 minutes. May cause lung tissue damage with edema. 1 mg/m 3 for 8 hours will cause lung damage or, in higher concentrations, death. Provide ventilation to limit inhalation. Reacts violently with metals and organics.
6.0 Equipment and Supplies
6.1 Sample Collection. The following items are required for sample collection:
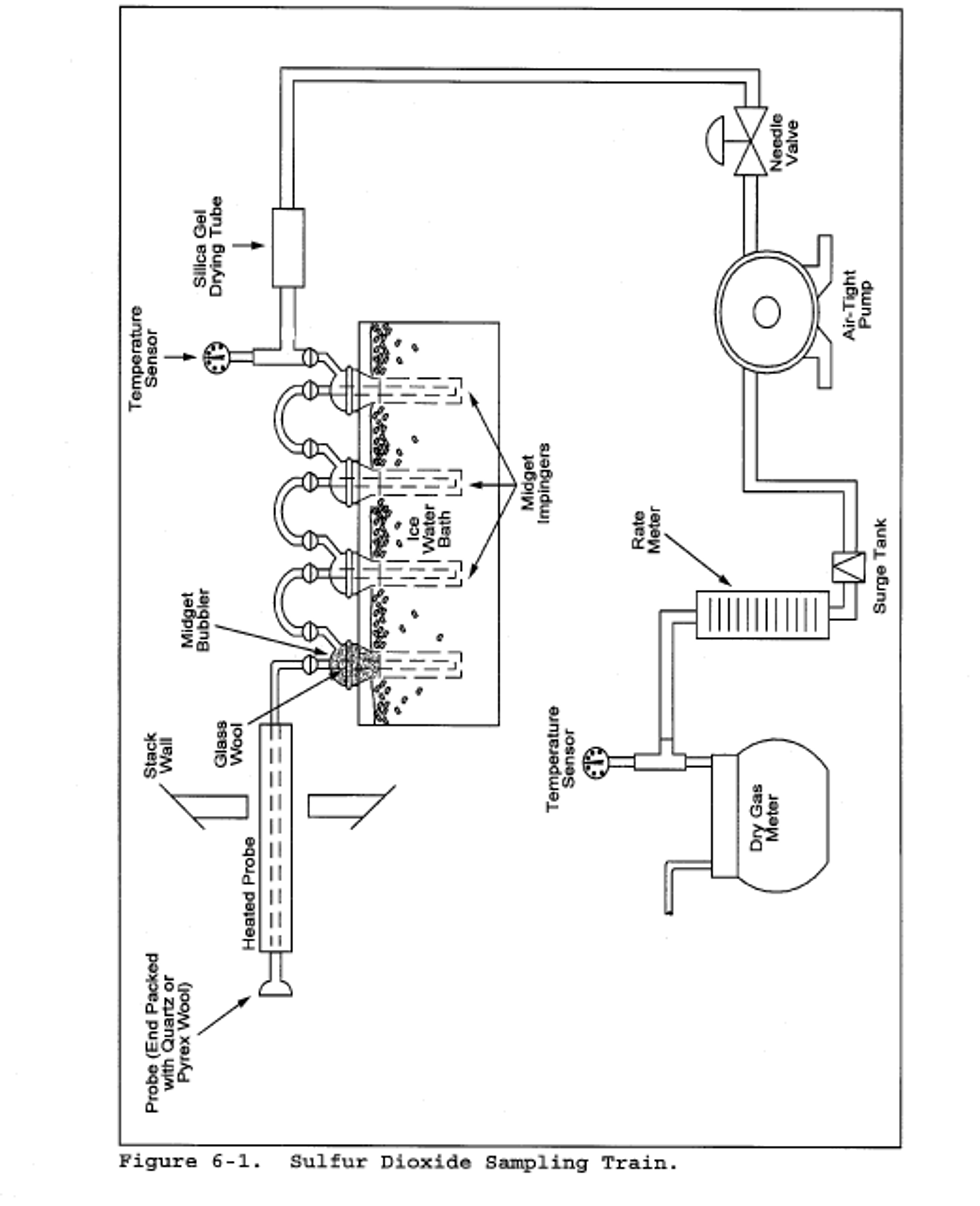
6.1.1 Sampling Train. A schematic of the sampling train is shown in Figure 6-1. The sampling equipment described in Method 8 may be substituted in place of the midget impinger equipment of Method 6. However, the Method 8 train must be modified to include a heated filter between the probe and isopropanol impinger, and the operation of the sampling train and sample analysis must be at the flow rates and solution volumes defined in Method 8. Alternatively, SO2 may be determined simultaneously with particulate matter and moisture determinations by either (1) replacing the water in a Method 5 impinger system with a 3 percent H2O2 solution, or (2) replacing the Method 5 water impinger system with a Method 8 isopropanol-filter-H2O2 system. The analysis for SO2 must be consistent with the procedure of Method 8. The Method 6 sampling train consists of the following components:
6.1.1.1 Probe. Borosilicate glass or stainless steel (other materials of construction may be used, subject to the approval of the Administrator), approximately 6 mm (0.25 in.) inside diameter, with a heating system to prevent water condensation and a filter (either in-stack or heated out-of-stack) to remove particulate matter, including sulfuric acid mist. A plug of glass wool is a satisfactory filter.
6.1.1.2 Bubbler and Impingers. One midget bubbler with medium-coarse glass frit and borosilicate or quartz glass wool packed in top (see Figure 6-1) to prevent sulfuric acid mist carryover, and three 30-ml midget impingers. The midget bubbler and midget impingers must be connected in series with leak-free glass connectors. Silicone grease may be used, if necessary, to prevent leakage. A midget impinger may be used in place of the midget bubbler.
Note:
Other collection absorbers and flow rates may be used, subject to the approval of the Administrator, but the collection efficiency must be shown to be at least 99 percent for each test run and must be documented in the report. If the efficiency is found to be acceptable after a series of three tests, further documentation is not required. To conduct the efficiency test, an extra absorber must be added and analyzed separately. This extra absorber must not contain more than 1 percent of the total SO2.
6.1.1.3 Glass Wool. Borosilicate or quartz.
6.1.1.4 Stopcock Grease. Acetone-insoluble, heat-stable silicone grease may be used, if necessary.
6.1.1.5 Temperature Sensor. Dial thermometer, or equivalent, to measure temperature of gas leaving impinger train to within 1°C (2°F).
6.1.1.6 Drying Tube. Tube packed with 6- to 16- mesh indicating-type silica gel, or equivalent, to dry the gas sample and to protect the meter and pump. If silica gel is previously used, dry at 177°C (350°F) for 2 hours. New silica gel may be used as received. Alternatively, other types of desiccants (equivalent or better) may be used, subject to the approval of the Administrator.
6.1.1.7 Valve. Needle valve, to regulate sample gas flow rate.
6.1.1.8 Pump. Leak-free diaphragm pump, or equivalent, to pull gas through the train. Install a small surge tank between the pump and rate meter to negate the pulsation effect of the diaphragm pump on the rate meter.
6.1.1.9 Rate Meter. Rotameter, or equivalent, capable of measuring flow rate to within 2 percent of the selected flow rate of about 1 liter/min (0.035 cfm).
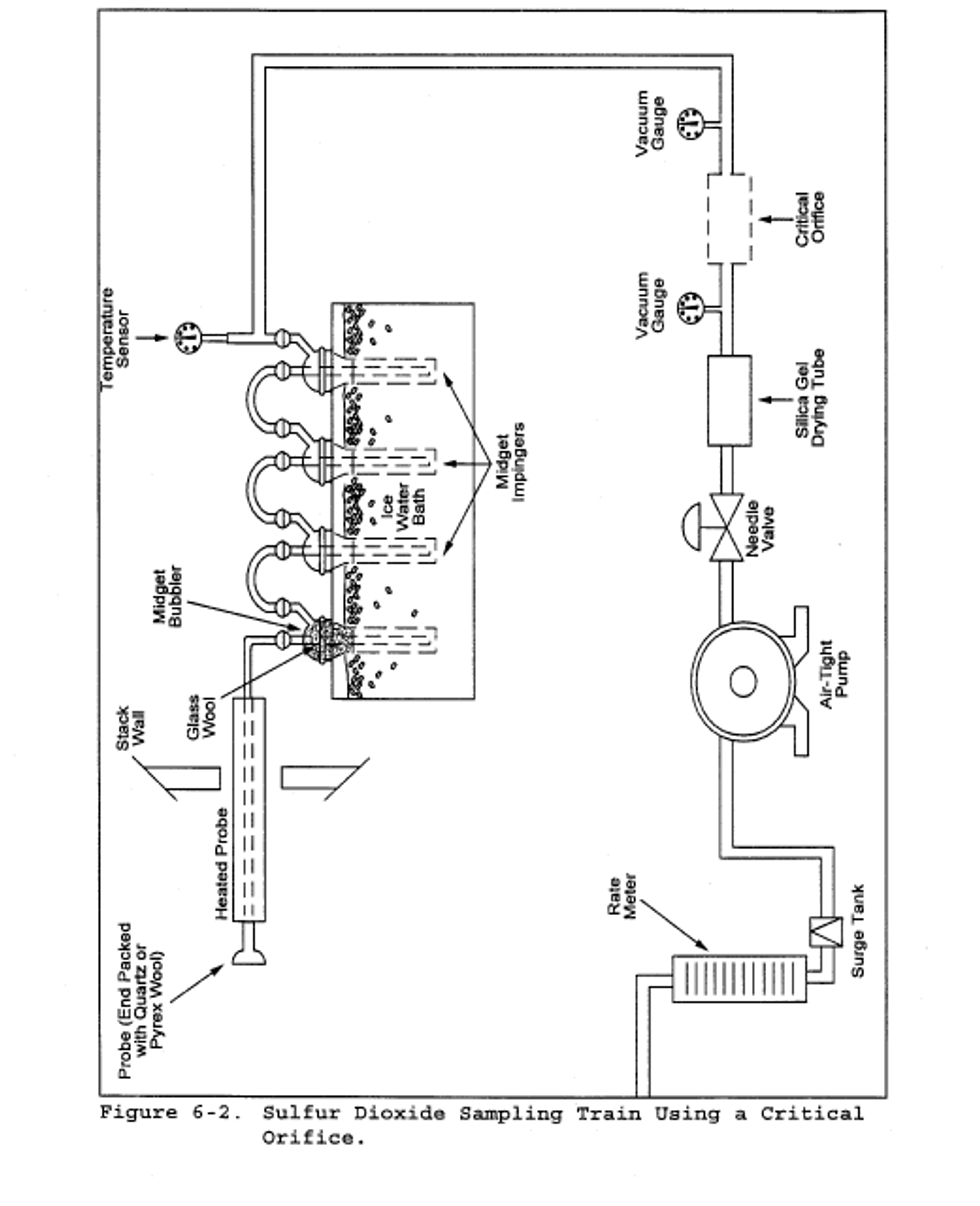
6.1.1.10 Volume Meter. Dry gas meter (DGM), sufficiently accurate to measure the sample volume to within 2 percent, calibrated at the selected flow rate and conditions actually encountered during sampling, and equipped with a temperature sensor (dial thermometer, or equivalent) capable of measuring temperature accurately to within 3°C (5.4°F). A critical orifice may be used in place of the DGM specified in this section provided that it is selected, calibrated, and used as specified in section 16.0.
6.1.2 Barometer. Mercury, aneroid, or other barometer capable of measuring atmospheric pressure to within 2.5 mm Hg (0.1 in. Hg). See the note in Method 5, section 6.1.2.
6.1.3 Vacuum Gauge and Rotameter. At least 760-mm Hg (30-in. Hg) gauge and 0- to 40-ml/min rotameter, to be used for leak-check of the sampling train.
6.2 Sample Recovery. The following items are needed for sample recovery:
6.2.1 Wash Bottles. Two polyethylene or glass bottles, 500-ml.
6.2.2 Storage Bottles. Polyethylene bottles, 100-ml, to store impinger samples (one per sample).
6.3 Sample Analysis. The following equipment is needed for sample analysis:
6.3.1 Pipettes. Volumetric type, 5-ml, 20-ml (one needed per sample), and 25-ml sizes.
6.3.2 Volumetric Flasks. 100-ml size (one per sample) and 1000-ml size.
6.3.3 Burettes. 5- and 50-ml sizes.
6.3.4 Erlenmeyer Flasks. 250-ml size (one for each sample, blank, and standard).
6.3.5 Dropping Bottle. 125-ml size, to add indicator.
6.3.6 Graduated Cylinder. 100-ml size.
6.3.7 Spectrophotometer. To measure absorbance at 352 nm.
7.0 Reagents and Standards
Note:
Unless otherwise indicated, all reagents must conform to the specifications established by the Committee on Analytical Reagents of the American Chemical Society. Where such specifications are not available, use the best available grade.
7.1 Sample Collection. The following reagents are required for sample collection:
7.1.1 Water. Deionized distilled to conform to ASTM Specification D 1193-77 or 91 Type 3 (incorporated by reference - see §60.17). The KMnO4 test for oxidizable organic matter may be omitted when high concentrations of organic matter are not expected to be present.
7.1.2 Isopropanol, 80 Percent by Volume. Mix 80 ml of isopropanol with 20 ml of water.
7.1.2.1 Check each lot of isopropanol for peroxide impurities as follows: Shake 10 ml of isopropanol with 10 ml of freshly prepared 10 percent potassium iodide solution. Prepare a blank by similarly treating 10 ml of water. After 1 minute, read the absorbance at 352 nm on a spectrophotometer using a 1-cm path length. If absorbance exceeds 0.1, reject alcohol for use.
7.1.2.2 Peroxides may be removed from isopropanol by redistilling or by passage through a column of activated alumina; however, reagent grade isopropanol with suitably low peroxide levels may be obtained from commercial sources. Rejection of contaminated lots may, therefore, be a more efficient procedure.
7.1.3 Hydrogen Peroxide (H2O2), 3 Percent by Volume. Add 10 ml of 30 percent H2O2 to 90 ml of water. Prepare fresh daily.
7.1.4 Potassium Iodide Solution, 10 Percent Weight by Volume (w/v). Dissolve 10.0 g of KI in water, and dilute to 100 ml. Prepare when needed.
7.2 Sample Recovery. The following reagents are required for sample recovery:
7.2.1 Water. Same as in section 7.1.1.
7.2.2 Isopropanol, 80 Percent by Volume. Same as in section 7.1.2.
7.3 Sample Analysis. The following reagents and standards are required for sample analysis:
7.3.1 Water. Same as in section 7.1.1.
7.3.2 Isopropanol, 100 Percent.
7.3.3 Thorin Indicator. 1-(o-arsonophenylazo)-2-naphthol-3,6-disulfonic acid, disodium salt, or equivalent. Dissolve 0.20 g in 100 ml of water.
7.3.4 Barium Standard Solution, 0.0100 N. Dissolve 1.95 g of barium perchlorate trihydrate [Ba(ClO4)2 3H2O] in 200 ml water, and dilute to 1 liter with isopropanol. Alternatively, 1.22 g of barium chloride dihydrate [BaCl2 2H2O] may be used instead of the barium perchlorate trihydrate. Standardize as in section 10.5.
7.3.5 Sulfuric Acid Standard, 0.0100 N. Purchase or standardize to ±0.0002 N against 0.0100 N NaOH which has previously been standardized against potassium acid phthalate (primary standard grade).
8.0 Sample Collection, Preservation, Storage and Transport
8.1 Preparation of Sampling Train. Measure 15 ml of 80 percent isopropanol into the midget bubbler and 15 ml of 3 percent H2O2 into each of the first two midget impingers. Leave the final midget impinger dry. Assemble the train as shown in Figure 6-1. Adjust the probe heater to a temperature sufficient to prevent water condensation. Place crushed ice and water around the impingers.
8.2 Sampling Train Leak-Check Procedure. A leak-check prior to the sampling run is recommended, but not required. A leak-check after the sampling run is mandatory. The leak-check procedure is as follows:
8.2.1 Temporarily attach a suitable (e.g., 0- to 40- ml/min) rotameter to the outlet of the DGM, and place a vacuum gauge at or near the probe inlet. Plug the probe inlet, pull a vacuum of at least 250 mm Hg (10 in. Hg), and note the flow rate as indicated by the rotameter. A leakage rate in excess of 2 percent of the average sampling rate is not acceptable.
Note:
Carefully (i.e., slowly) release the probe inlet plug before turning off the pump.
8.2.2 It is suggested (not mandatory) that the pump be leak-checked separately, either prior to or after the sampling run. To leak-check the pump, proceed as follows: Disconnect the drying tube from the probe-impinger assembly. Place a vacuum gauge at the inlet to either the drying tube or the pump, pull a vacuum of 250 mm Hg (10 in. Hg), plug or pinch off the outlet of the flow meter, and then turn off the pump. The vacuum should remain stable for at least 30 seconds.
If performed prior to the sampling run, the pump leak-check shall precede the leak-check of the sampling train described immediately above; if performed after the sampling run, the pump leak-check shall follow the sampling train leak-check.
8.2.3 Other leak-check procedures may be used, subject to the approval of the Administrator.
8.3 Sample Collection.
8.3.1 Record the initial DGM reading and barometric pressure. To begin sampling, position the tip of the probe at the sampling point, connect the probe to the bubbler, and start the pump. Adjust the sample flow to a constant rate of approximately 1.0 liter/min as indicated by the rate meter. Maintain this constant rate (±10 percent) during the entire sampling run.
8.3.2 Take readings (DGM volume, temperatures at DGM and at impinger outlet, and rate meter flow rate) at least every 5 minutes. Add more ice during the run to keep the temperature of the gases leaving the last impinger at 20°C (68°F) or less.
8.3.3 At the conclusion of each run, turn off the pump, remove the probe from the stack, and record the final readings. Conduct a leak-check as described in section 8.2. (This leak-check is mandatory.) If a leak is detected, void the test run or use procedures acceptable to the Administrator to adjust the sample volume for the leakage.
8.3.4 Drain the ice bath, and purge the remaining part of the train by drawing clean ambient air through the system for 15 minutes at the sampling rate. Clean ambient air can be provided by passing air through a charcoal filter or through an extra midget impinger containing 15 ml of 3 percent H2O2. Alternatively, ambient air without purification may be used.
8.4 Sample Recovery. Disconnect the impingers after purging. Discard the contents of the midget bubbler. Pour the contents of the midget impingers into a leak-free polyethylene bottle for shipment. Rinse the three midget impingers and the connecting tubes with water, and add the rinse to the same storage container. Mark the fluid level. Seal and identify the sample container.
9.0 Quality Control
| Section | Quality control measure | Effect |
|---|---|---|
| 7.1.2 | Isopropanol check | Ensure acceptable level of peroxide impurities in isopropanol. |
| 8.2, 10.1-10.4 | Sampling equipment leak-check and calibration | Ensure accurate measurement of stack gas flow rate, sample volume. |
| 10.5 | Barium standard solution standardization | Ensure precision of normality determination |
| 11.2.3 | Replicate titrations | Ensure precision of titration determinations. |
10.0 Calibration and Standardization
10.1 Volume Metering System.
10.1.1 Initial Calibration.
10.1.1.1 Before its initial use in the field, leak-check the metering system (drying tube, needle valve, pump, rate meter, and DGM) as follows: Place a vacuum gauge at the inlet to the drying tube and pull a vacuum of 250 mm Hg (10 in. Hg). Plug or pinch off the outlet of the flow meter, and then turn off the pump. The vacuum must remain stable for at least 30 seconds. Carefully release the vacuum gauge before releasing the flow meter end.
10.1.1.2 Remove the drying tube, and calibrate the metering system (at the sampling flow rate specified by the method) as follows: Connect an appropriately sized wet-test meter (e.g., 1 liter per revolution) to the inlet of the needle valve. Make three independent calibration runs, using at least five revolutions of the DGM per run. Calculate the calibration factor Y (wet-test meter calibration volume divided by the DGM volume, both volumes adjusted to the same reference temperature and pressure) for each run, and average the results (Yi). If any Y-value deviates by more than 2 percent from (Yi), the metering system is unacceptable for use. If the metering system is acceptable, use (Yi) as the calibration factor for subsequent test runs.
10.1.2 Post-Test Calibration Check. After each field test series, conduct a calibration check using the procedures outlined in section 10.1.1.2, except that three or more revolutions of the DGM may be used, and only two independent runs need be made. If the average of the two post-test calibration factors does not deviate by more than 5 percent from Yi, then Yi is accepted as the DGM calibration factor (Y), which is used in Equation 6-1 to calculate collected sample volume (see section 12.2). If the deviation is more than 5 percent, recalibrate the metering system as in section 10.1.1, and determine a post-test calibration factor (Yf). Compare Yi and Yf; the smaller of the two factors is accepted as the DGM calibration factor. If recalibration indicates that the metering system is unacceptable for use, either void the test run or use methods, subject to the approval of the Administrator, to determine an acceptable value for the collected sample volume.
10.1.3 DGM as a Calibration Standard. A DGM may be used as a calibration standard for volume measurements in place of the wet-test meter specified in section 10.1.1.2, provided that it is calibrated initially and recalibrated periodically according to the same procedures outlined in Method 5, section 10.3 with the following exceptions: (a) the DGM is calibrated against a wet-test meter having a capacity of 1 liter/rev (0.035 ft 3/rev) or 3 liters/rev (0.1 ft 3/rev) and having the capability of measuring volume to within 1 percent; (b) the DGM is calibrated at 1 liter/min (0.035 cfm); and (c) the meter box of the Method 6 sampling train is calibrated at the same flow rate.
10.2 Temperature Sensors. Calibrate against mercury-in-glass thermometers. An alternative mercury-free thermometer may be used if the thermometer is, at a minimum, equivalent in terms of performance or suitably effective for the specific temperature measurement application.
10.3 Rate Meter. The rate meter need not be calibrated, but should be cleaned and maintained according to the manufacturer's instructions.
10.4 Barometer. Calibrate against a mercury barometer or NIST-traceable barometer prior to the field test.
10.5 Barium Standard Solution. Standardize the barium perchlorate or chloride solution against 25 ml of standard sulfuric acid to which 100 ml of 100 percent isopropanol has been added. Run duplicate analyses. Calculate the normality using the average of duplicate analyses where the titrations agree within 1 percent or 0.2 ml, whichever is larger.
11.0 Analytical Procedure
11.1 Sample Loss Check. Note level of liquid in container and confirm whether any sample was lost during shipment; note this finding on the analytical data sheet. If a noticeable amount of leakage has occurred, either void the sample or use methods, subject to the approval of the Administrator, to correct the final results.
11.2 Sample Analysis.
11.2.1 Transfer the contents of the storage container to a 100-ml volumetric flask, dilute to exactly 100 ml with water, and mix the diluted sample.
11.2.2 Pipette a 20-ml aliquot of the diluted sample into a 250-ml Erlenmeyer flask and add 80 ml of 100 percent isopropanol plus two to four drops of thorin indicator. While stirring the solution, titrate to a pink endpoint using 0.0100 N barium standard solution.
11.2.3 Repeat the procedures in section 11.2.2, and average the titration volumes. Run a blank with each series of samples. Replicate titrations must agree within 1 percent or 0.2 ml, whichever is larger.
Note:
Protect the 0.0100 N barium standard solution from evaporation at all times.
12.0 Data Analysis and Calculations
Carry out calculations, retaining at least one extra significant figure beyond that of the acquired data. Round off figures after final calculation.
12.1 Nomenclature
CSO2 = Concentration of SO2, dry basis, corrected to standard conditions, mg/dscm (lb/dscf).
N = Normality of barium standard titrant, meq/ml.
Pbar = Barometric pressure, mm Hg (in. Hg).
Pstd = Standard absolute pressure, 760 mm Hg (29.92 in. Hg).
Tm = Average DGM absolute temperature,°K (°R).
Tstd = Standard absolute temperature, 293°K (528°R).
Va = Volume of sample aliquot titrated, ml.
Vm = Dry gas volume as measured by the DGM, dcm (dcf).
Vm(std) = Dry gas volume measured by the DGM, corrected to standard conditions, dscm (dscf).
Vsoln = Total volume of solution in which the SO2 sample is contained, 100 ml.
Vt = Volume of barium standard titrant used for the sample (average of replicate titration), ml.
Vtb = Volume of barium standard titrant used for the blank, ml.
Y = DGM calibration factor.
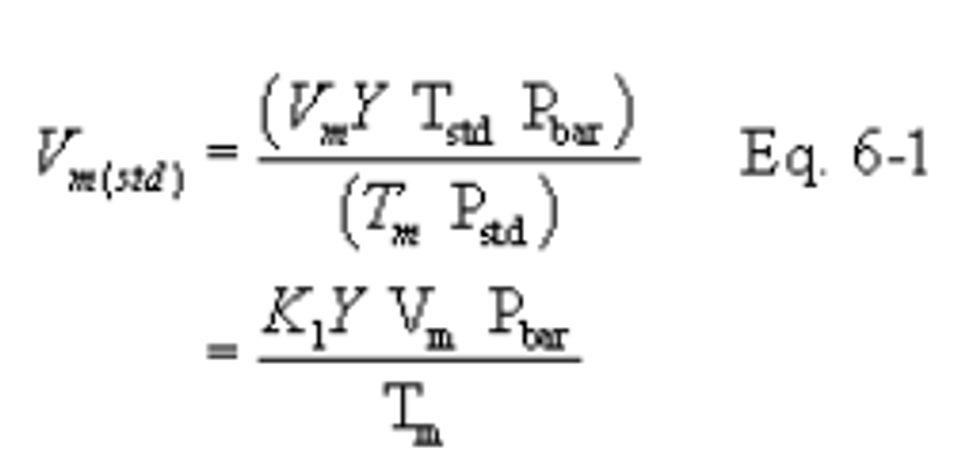
12.2 Dry Sample Gas Volume, Corrected to Standard Conditions.
Where:
K1 = 0.3855°K/mm Hg for metric units,
K1 = 17.65°R/in. Hg for English units.
12.3 SO2 Concentration.
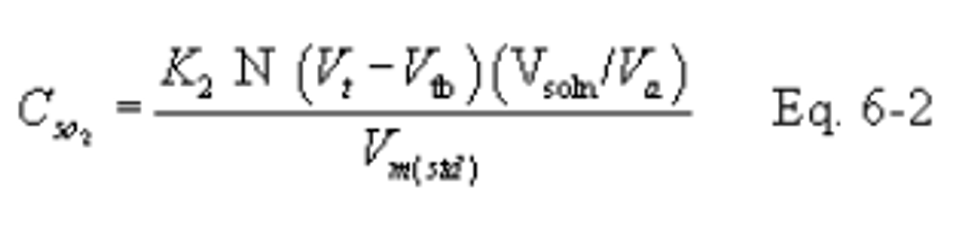
Where:
K2 = 32.03 mg SO2/meq for metric units,
K2 = 7.061 × 10−5 lb SO2/meq for English units.
13.0 Method Performance
13.1 Range. The minimum detectable limit of the method has been determined to be 3.4 mg SO2/m 3 (2.12 × 10−7 lb/ft 3). Although no upper limit has been established, tests have shown that concentrations as high as 80,000 mg/m 3 (0.005 lb/ft 3) of SO2 can be collected efficiently at a rate of 1.0 liter/min (0.035 cfm) for 20 minutes in two midget impingers, each containing 15 ml of 3 percent H2O2. Based on theoretical calculations, the upper concentration limit in a 20 liter (0.7 ft 3) sample is about 93,300 mg/m 3 (0.00583 lb/ft 3).
14.0 Pollution Prevention [Reserved]
15.0 Waste Management [Reserved]
16.0 Alternative Procedures
16.1 Nomenclature. Same as section 12.1, with the following additions:
Bwa = Water vapor in ambient air, proportion by volume.
Ma = Molecular weight of the ambient air saturated at impinger temperature, g/g-mole (lb/lb-mole).
Ms = Molecular weight of the sample gas saturated at impinger temperature, g/g-mole (lb/lb-mole).
Pc = Inlet vacuum reading obtained during the calibration run, mm Hg (in. Hg).
Psr = Inlet vacuum reading obtained during the sampling run, mm Hg (in. Hg).
Q͞ std = Volumetric flow rate through critical orifice, scm/min (scf/min).
Qstd = Average flow rate of pre-test and post-test calibration runs, scm/min (scf/min).
Tamb = Ambient absolute temperature of air,°K (°R).
Vsb = Volume of gas as measured by the soap bubble meter, m 3 (ft 3).
Vsb(std) = Volume of gas as measured by the soap bubble meter, corrected to standard conditions, scm (scf).
θ = Soap bubble travel time, min.
θs = Time, min.
16.2 Critical Orifices for Volume and Rate Measurements. A critical orifice may be used in place of the DGM specified in section 6.1.1.10, provided that it is selected, calibrated, and used as follows:
16.2.1 Preparation of Sampling Train. Assemble the sampling train as shown in Figure 6-2. The rate meter and surge tank are optional but are recommended in order to detect changes in the flow rate.
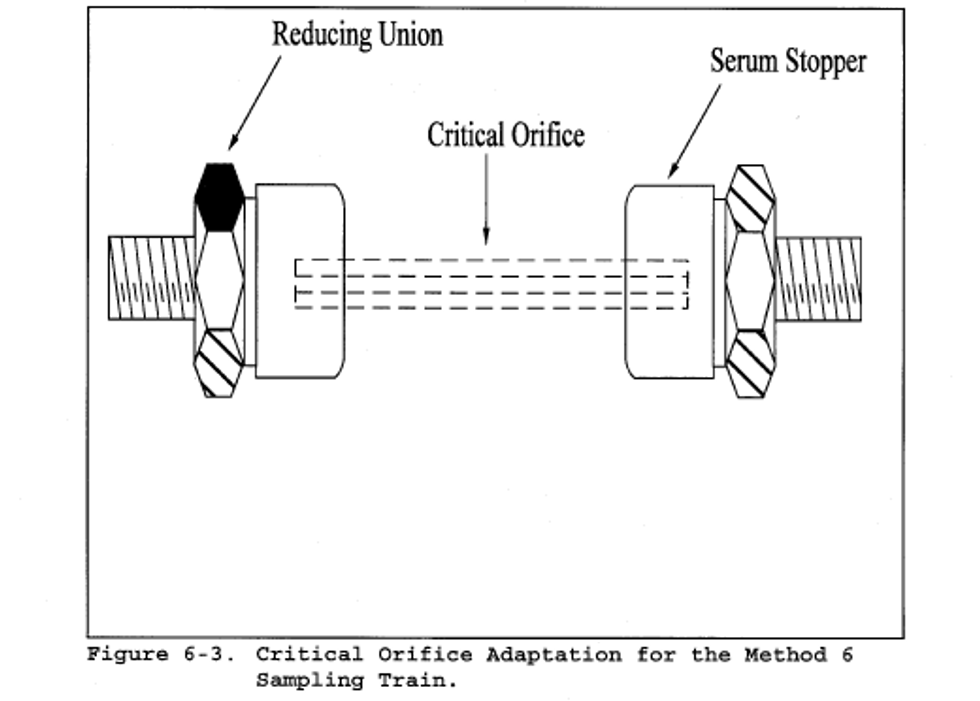
Note:
The critical orifices can be adapted to a Method 6 type sampling train as follows: Insert sleeve type, serum bottle stoppers into two reducing unions. Insert the needle into the stoppers as shown in Figure 6-3.
16.2.2 Selection of Critical Orifices.
16.2.2.1 The procedure that follows describes the use of hypodermic needles and stainless steel needle tubings, which have been found suitable for use as critical orifices. Other materials and critical orifice designs may be used provided the orifices act as true critical orifices, (i.e., a critical vacuum can be obtained) as described in this section. Select a critical orifice that is sized to operate at the desired flow rate. The needle sizes and tubing lengths shown in Table 6-1 give the following approximate flow rates.
16.2.2.2 Determine the suitability and the appropriate operating vacuum of the critical orifice as follows: If applicable, temporarily attach a rate meter and surge tank to the outlet of the sampling train, if said equipment is not present (see section 16.2.1). Turn on the pump and adjust the valve to give an outlet vacuum reading corresponding to about half of the atmospheric pressure. Observe the rate meter reading. Slowly increase the vacuum until a stable reading is obtained on the rate meter. Record the critical vacuum, which is the outlet vacuum when the rate meter first reaches a stable value. Orifices that do not reach a critical value must not be used.
16.2.3 Field Procedures.
16.2.3.1 Leak-Check Procedure. A leak-check before the sampling run is recommended, but not required. The leak-check procedure is as follows: Temporarily attach a suitable (e.g., 0-40 ml/min) rotameter and surge tank, or a soap bubble meter and surge tank to the outlet of the pump. Plug the probe inlet, pull an outlet vacuum of at least 250 mm Hg (10 in. Hg), and note the flow rate as indicated by the rotameter or bubble meter. A leakage rate in excess of 2 percent of the average sampling rate (Q͞ std) is not acceptable. Carefully release the probe inlet plug before turning off the pump.
16.2.3.2 Moisture Determination. At the sampling location, prior to testing, determine the percent moisture of the ambient air using the wet and dry bulb temperatures or, if appropriate, a relative humidity meter.
16.2.3.3 Critical Orifice Calibration. At the sampling location, prior to testing, calibrate the entire sampling train (i.e., determine the flow rate of the sampling train when operated at critical conditions). Attach a 500-ml soap bubble meter to the inlet of the probe, and operate the sampling train at an outlet vacuum of 25 to 50 mm Hg (1 to 2 in. Hg) above the critical vacuum. Record the information listed in Figure 6-4. Calculate the standard volume of air measured by the soap bubble meter and the volumetric flow rate using the equations below:
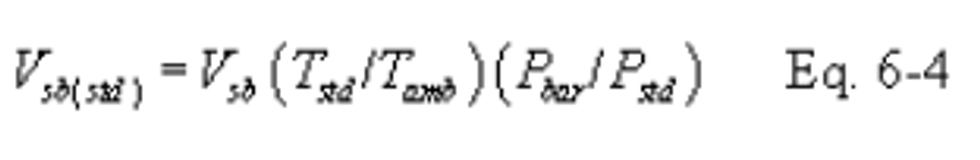
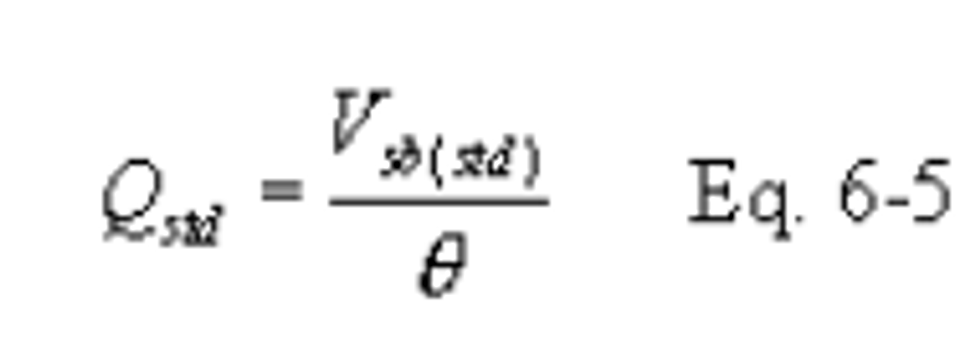
16.2.3.4 Sampling.
16.2.3.4.1 Operate the sampling train for sample collection at the same vacuum used during the calibration run. Start the watch and pump simultaneously. Take readings (temperature, rate meter, inlet vacuum, and outlet vacuum) at least every 5 minutes. At the end of the sampling run, stop the watch and pump simultaneously.
16.2.3.4.2 Conduct a post-test calibration run using the calibration procedure outlined in section 16.2.3.3. If the Qstd obtained before and after the test differ by more than 5 percent, void the test run; if not, calculate the volume of the gas measured with the critical orifice using Equation 6-6 as follows:
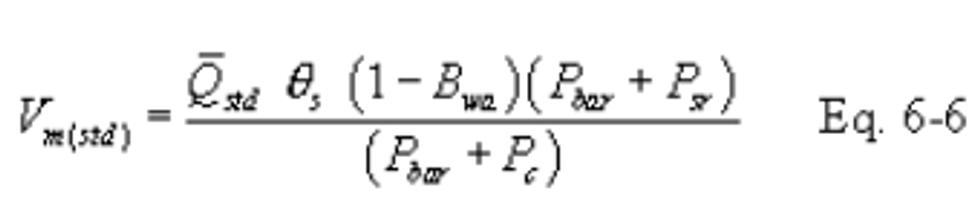
16.2.3.4.3 If the percent difference between the molecular weight of the ambient air at saturated conditions and the sample gas is more that ±3 percent, then the molecular weight of the gas sample must be considered in the calculations using the following equation:
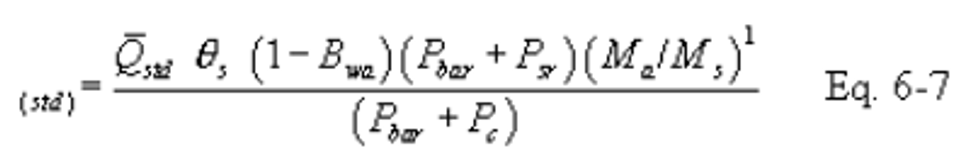
Note:
A post-test leak-check is not necessary because the post-test calibration run results will indicate whether there is any leakage.
16.2.3.4.4 Drain the ice bath, and purge the sampling train using the procedure described in section 8.3.4.
16.3 Elimination of Ammonia Interference. The following alternative procedures must be used in addition to those specified in the method when sampling at sources having ammonia emissions.
16.3.1 Sampling. The probe shall be maintained at 275°C (527°F) and equipped with a high-efficiency in-stack filter (glass fiber) to remove particulate matter. The filter material shall be unreactive to SO2. Whatman 934AH (formerly Reeve Angel 934AH) filters treated as described in Reference 10 in section 17.0 of Method 5 is an example of a filter that has been shown to work. Where alkaline particulate matter and condensed moisture are present in the gas stream, the filter shall be heated above the moisture dew point but below 225°C (437°F).
16.3.2 Sample Recovery. Recover the sample according to section 8.4 except for discarding the contents of the midget bubbler. Add the bubbler contents, including the rinsings of the bubbler with water, to a separate polyethylene bottle from the rest of the sample. Under normal testing conditions where sulfur trioxide will not be present significantly, the tester may opt to delete the midget bubbler from the sampling train. If an approximation of the sulfur trioxide concentration is desired, transfer the contents of the midget bubbler to a separate polyethylene bottle.
16.3.3 Sample Analysis. Follow the procedures in sections 11.1 and 11.2, except add 0.5 ml of 0.1 N HCl to the Erlenmeyer flask and mix before adding the indicator. The following analysis procedure may be used for an approximation of the sulfur trioxide concentration. The accuracy of the calculated concentration will depend upon the ammonia to SO2 ratio and the level of oxygen present in the gas stream. A fraction of the SO2 will be counted as sulfur trioxide as the ammonia to SO2 ratio and the sample oxygen content increases. Generally, when this ratio is 1 or less and the oxygen content is in the range of 5 percent, less than 10 percent of the SO2 will be counted as sulfur trioxide. Analyze the peroxide and isopropanol sample portions separately. Analyze the peroxide portion as described above. Sulfur trioxide is determined by difference using sequential titration of the isopropanol portion of the sample. Transfer the contents of the isopropanol storage container to a 100-ml volumetric flask, and dilute to exactly 100 ml with water. Pipette a 20-ml aliquot of this solution into a 250-ml Erlenmeyer flask, add 0.5 ml of 0.1 N HCl, 80 ml of 100 percent isopropanol, and two to four drops of thorin indicator. Titrate to a pink endpoint using 0.0100 N barium perchlorate. Repeat and average the titration volumes that agree within 1 percent or 0.2 ml, whichever is larger. Use this volume in Equation 6-2 to determine the sulfur trioxide concentration. From the flask containing the remainder of the isopropanol sample, determine the fraction of SO2 collected in the bubbler by pipetting 20-ml aliquots into 250-ml Erlenmeyer flasks. Add 5 ml of 3 percent H2O2, 100 ml of 100 percent isopropanol, and two to four drips of thorin indicator, and titrate as before. From this titration volume, subtract the titrant volume determined for sulfur trioxide, and add the titrant volume determined for the peroxide portion. This final volume constitutes Vt, the volume of barium perchlorate used for the SO2 sample.
17.0 References
1. Atmospheric Emissions from Sulfuric Acid Manufacturing Processes. U.S. DHEW, PHS, Division of Air Pollution. Public Health Service Publication No. 999-AP-13. Cincinnati, OH. 1965.
2. Corbett, P.F. The Determination of SO2 and SO3 in Flue Gases. Journal of the Institute of Fuel. 24:237-243. 1961.
3. Matty, R.E., and E.K. Diehl. Measuring Flue-Gas SO2 and SO3. Power. 101:94-97. November 1957.
4. Patton, W.F., and J.A. Brink, Jr. New Equipment and Techniques for Sampling Chemical Process Gases. J. Air Pollution Control Association. 13:162. 1963.
5. Rom, J.J. Maintenance, Calibration, and Operation of Isokinetic Source Sampling Equipment. Office of Air Programs, U.S. Environmental Protection Agency. Research Triangle Park, NC. APTD-0576. March 1972.
6. Hamil, H.F., and D.E. Camann. Collaborative Study of Method for the Determination of Sulfur Dioxide Emissions from Stationary Sources (Fossil-Fuel Fired Steam Generators). U.S. Environmental Protection Agency, Research Triangle Park, NC. EPA-650/4-74-024. December 1973.
7. Annual Book of ASTM Standards. Part 31; Water, Atmospheric Analysis. American Society for Testing and Materials. Philadelphia, PA. 1974. pp. 40-42.
8. Knoll, J.E., and M.R. Midgett. The Application of EPA Method 6 to High Sulfur Dioxide Concentrations. U.S. Environmental Protection Agency. Research Triangle Park, NC. EPA-600/4-76-038. July 1976.
9. Westlin, P.R., and R.T. Shigehara. Procedure for Calibrating and Using Dry Gas Volume Meters as Calibration Standards. Source Evaluation Society Newsletter. 3(1):17-30. February 1978.
10. Yu, K.K. Evaluation of Moisture Effect on Dry Gas Meter Calibration. Source Evaluation Society Newsletter. 5(1):24-28. February 1980.
11. Lodge, J.P., Jr., et al. The Use of Hypodermic Needles as Critical Orifices in Air Sampling. J. Air Pollution Control Association. 16:197-200. 1966.
12. Shigehara, R.T., and C.B. Sorrell. Using Critical Orifices as Method 5 CalibrationStandards. Source Evaluation Society Newsletter. 10:4-15. August 1985.
13. Curtis, F., Analysis of Method 6 Samples in the Presence of Ammonia. Source Evaluation Society Newsletter. 13(1):9-15 February 1988.
18.0 Tables, Diagrams, Flowcharts and Validation Data
| Needle size (gauge) | Needle length (cm) | Flow rate (ml/min) |
|---|---|---|
| 21 | 7.6 | 1,100 |
| 22 | 2.9 | 1,000 |
| 22 | 3.8 | 900 |
| 23 | 3.8 | 500 |
| 23 | 5.1 | 450 |
| 24 | 3.2 | 400 |

Method 6A - Determination of Sulfur Dioxide, Moisture, and Carbon Dioxide From Fossil Fuel Combustion Sources
Note:
This method does not include all of the specifications (e.g., equipment and supplies) and procedures (e.g., sampling and analytical) essential to its performance. Some material is incorporated by reference from other methods in this part. Therefore, to obtain reliable results, persons using this method should have a thorough knowledge of at least the following additional test methods: Method 1, Method 2, Method 3, Method 5, Method 6, and Method 19.
1.0 Scope and Application
1.1 Analytes.
| Analyte | CAS No. | Sensitivity |
|---|---|---|
| SO2 | 7449-09-05 | 3.4 mg SO2/m 3 (2.12 × 10−7 lb/ft 3) |
| CO2 | 124-38-9 | N/A |
| H2O | 7732-18-5 | N/A |
1.2 Applicability. This method is applicable for the determination of sulfur dioxide (SO2) emissions from fossil fuel combustion sources in terms of concentration (mg/dscm or lb/dscf) and in terms of emission rate (ng/J or lb/10 6 Btu) and for the determination of carbon dioxide (CO2) concentration (percent). Moisture content (percent), if desired, may also be determined by this method.
1.3 Data Quality Objectives. Adherence to the requirements of this method will enhance the quality of the data obtained from air pollutant sampling methods.
2.0 Summary of Method
2.1 A gas sample is extracted from a sampling point in the stack. The SO2 and the sulfur trioxide, including those fractions in any sulfur acid mist, are separated. The SO2 fraction is measured by the barium-thorin titration method. Moisture and CO2 fractions are collected in the same sampling train, and are determined gravimetrically.
3.0 Definitions [Reserved]
4.0 Interferences
Same as Method 6, section 4.0.
5.0 Safety
5.1 Disclaimer. This method may involve hazardous materials, operations, and equipment. This test method may not address all of the safety problems associated with its use. It is the responsibility of the user to establish appropriate safety and health practices and determine the applicability of regulatory limitations prior to performing this test method.
5.2 Corrosive reagents. Same as Method 6, section 5.2.
6.0 Equipment and Supplies
6.1 Sample Collection. Same as Method 6, section 6.1, with the exception of the following:
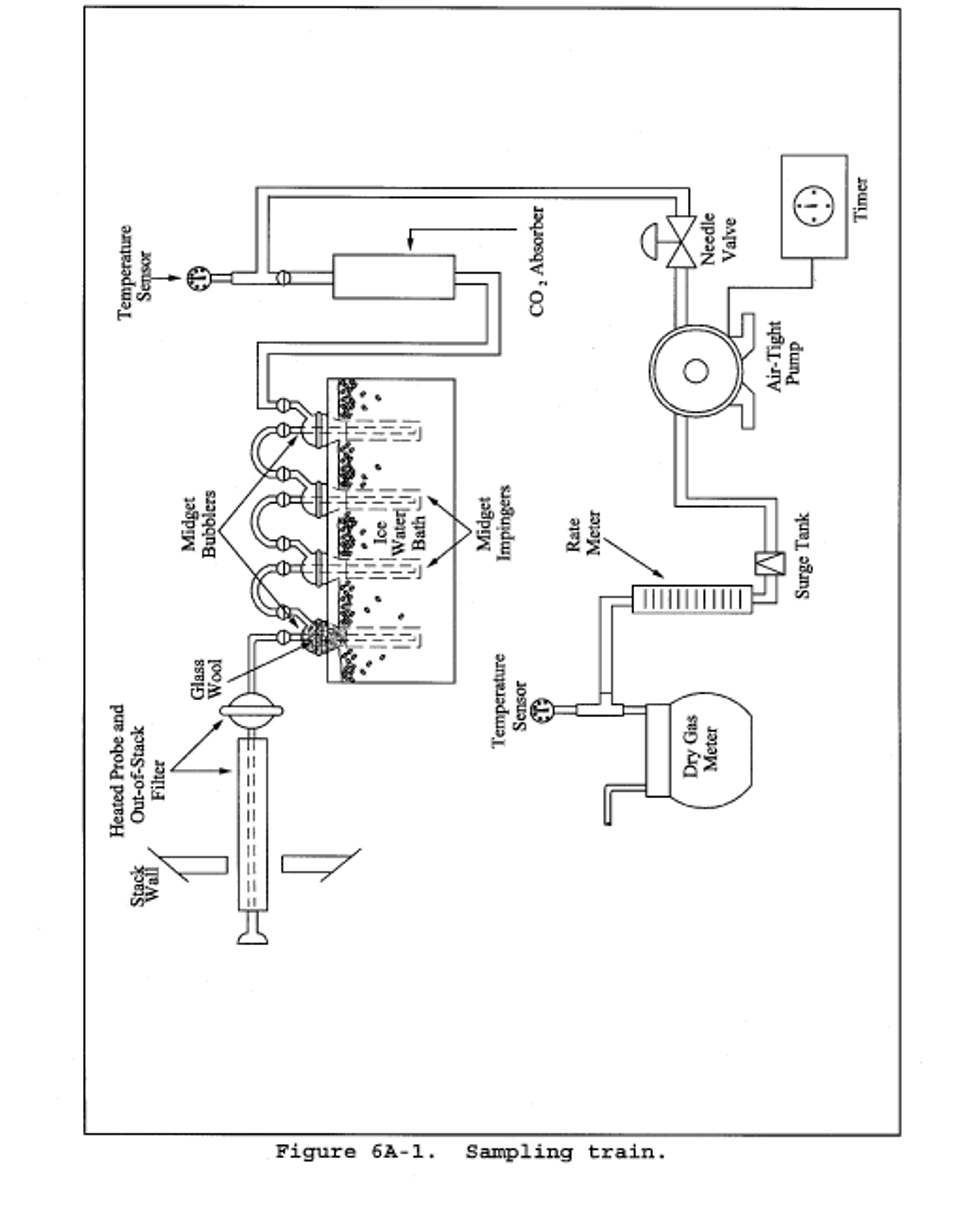
6.1.1 Sampling Train. A schematic of the sampling train used in this method is shown in Figure 6A-1.
6.1.1.1 Impingers and Bubblers. Two 30 = ml midget impingers with a 1 = mm restricted tip and two 30 = ml midget bubblers with unrestricted tips. Other types of impingers and bubblers (e.g., Mae West for SO2 collection and rigid cylinders containing Drierite for moisture absorbers), may be used with proper attention to reagent volumes and levels, subject to the approval of the Administrator.
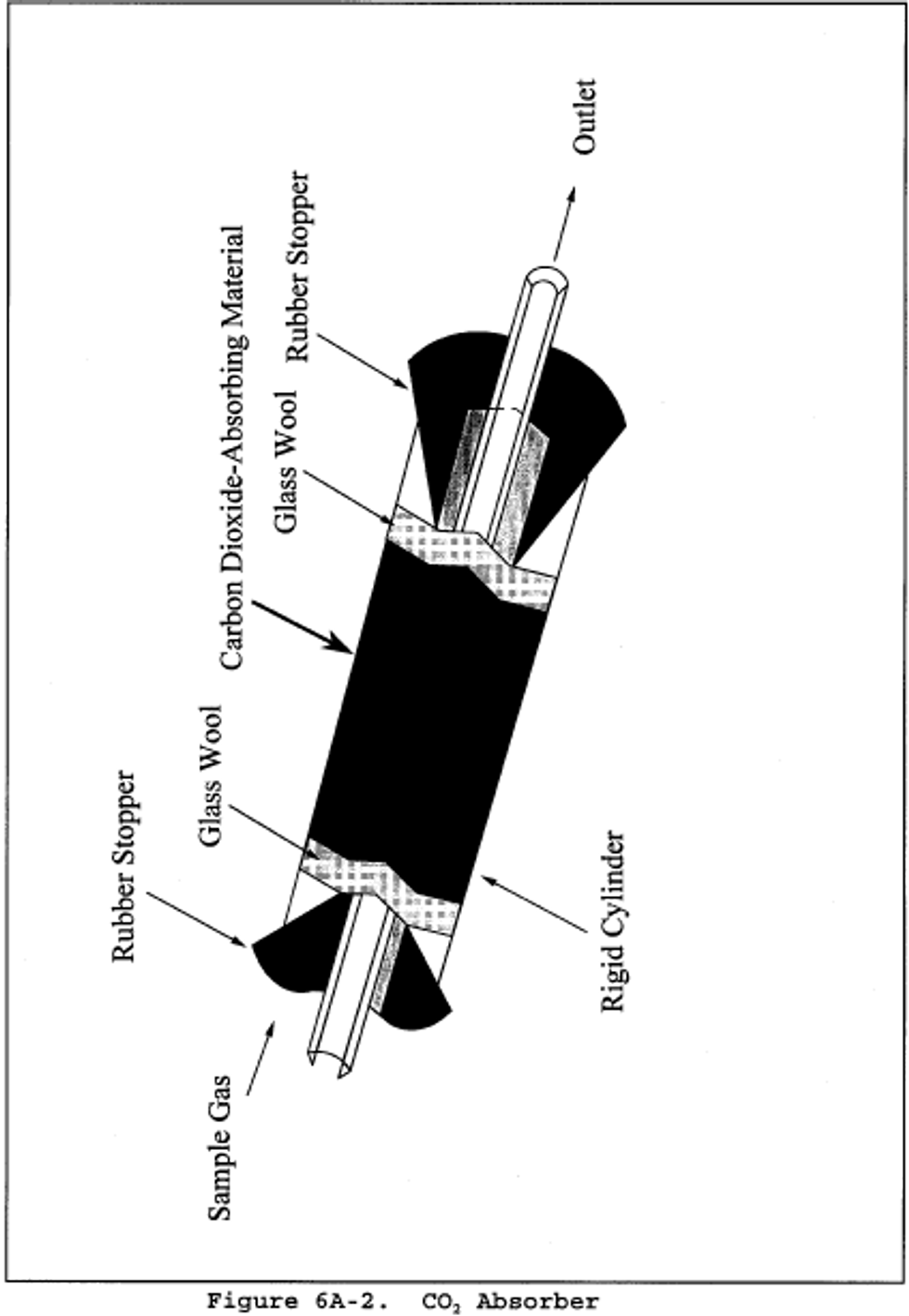
6.1.1.2 CO2 Absorber. A sealable rigid cylinder or bottle with an inside diameter between 30 and 90 mm , a length between 125 and 250 mm, and appropriate connections at both ends. The filter may be a separate heated unit or may be within the heated portion of the probe. If the filter is within the sampling probe, the filter should not be within 15 cm of the probe inlet or any unheated section of the probe, such as the connection to the first bubbler. The probe and filter should be heated to at least 20°C (68°F) above the source temperature, but not greater than 120°C (248°F). The filter temperature (i.e., the sample gas temperature) should be monitored to assure the desired temperature is maintained. A heated Teflon connector may be used to connect the filter holder or probe to the first impinger.
Note:
For applications downstream of wet scrubbers, a heated out-of-stack filter (either borosilicate glass wool or glass fiber mat) is necessary.
6.2 Sample Recovery. Same as Method 6, section 6.2.
6.3 Sample Analysis. Same as Method 6, section 6.3, with the addition of a balance to measure within 0.05 g.
7.0 Reagents and Standards
Note:
Unless otherwise indicated, all reagents must conform to the specifications established by the Committee on Analytical Reagents of the American Chemical Society. Where such specifications are not available, use the best available grade.
7.1 Sample Collection. Same as Method 6, section 7.1, with the addition of the following:
7.1.1 Drierite. Anhydrous calcium sulfate (CaSO4) desiccant, 8 mesh, indicating type is recommended.
Note:
Do not use silica gel or similar desiccant in this application.
7.1.2 CO2 Absorbing Material. Ascarite II. Sodium hydroxide-coated silica, 8- to 20-mesh.
7.2 Sample Recovery and Analysis. Same as Method 6, sections 7.2 and 7.3, respectively.
8.0 Sample Collection, Preservation, Transport, and Storage
8.1 Preparation of Sampling Train.
8.1.1 Measure 15 ml of 80 percent isopropanol into the first midget bubbler and 15 ml of 3 percent hydrogen peroxide into each of the two midget impingers (the second and third vessels in the train) as described in Method 6, section 8.1. Insert the glass wool into the top of the isopropanol bubbler as shown in Figure 6A-1. Place about 25 g of Drierite into the second midget bubbler (the fourth vessel in the train). Clean the outside of the bubblers and impingers and allow the vessels to reach room temperature. Weigh the four vessels simultaneously to the nearest 0.1 g, and record this initial weight (mwi).
8.1.2 With one end of the CO2 absorber sealed, place glass wool into the cylinder to a depth of about 1 cm (0.5 in.). Place about 150 g of CO2 absorbing material in the cylinder on top of the glass wool, and fill the remaining space in the cylinder with glass wool. Assemble the cylinder as shown in figure 6A-2. With the cylinder in a horizontal position, rotate it around the horizontal axis. The CO2 absorbing material should remain in position during the rotation, and no open spaces or channels should be formed. If necessary, pack more glass wool into the cylinder to make the CO2 absorbing material stable. Clean the outside of the cylinder of loose dirt and moisture and allow the cylinder to reach room temperature. Weigh the cylinder to the nearest 0.1 g, and record this initial weight (mai).
8.1.3 Assemble the train as shown in figure 6A-1. Adjust the probe heater to a temperature sufficient to prevent condensation (see note in section 6.1). Place crushed ice and water around the impingers and bubblers. Mount the CO2 absorber outside the water bath in a vertical flow position with the sample gas inlet at the bottom. Flexible tubing (e.g., Tygon) may be used to connect the last SO2 absorbing impinger to the moisture absorber and to connect the moisture absorber to the CO2 absorber. A second, smaller CO2 absorber containing Ascarite II may be added in-line downstream of the primary CO2 absorber as a breakthrough indicator. Ascarite II turns white when CO2 is absorbed.
8.2 Sampling Train Leak-Check Procedure and Sample Collection. Same as Method 6, sections 8.2 and 8.3, respectively.
8.3 Sample Recovery.
8.3.1 Moisture Measurement. Disconnect the isopropanol bubbler, the SO2 impingers, and the moisture absorber from the sample train. Allow about 10 minutes for them to reach room temperature, clean the outside of loose dirt and moisture, and weigh them simultaneously in the same manner as in section 8.1. Record this final weight (mwf).
8.3.2 Peroxide Solution. Discard the contents of the isopropanol bubbler and pour the contents of the midget impingers into a leak-free polyethylene bottle for shipping. Rinse the two midget impingers and connecting tubes with water, and add the washing to the same storage container.
8.3.3 CO2 Absorber. Allow the CO2 absorber to warm to room temperature (about 10 minutes), clean the outside of loose dirt and moisture, and weigh to the nearest 0.1 g in the same manner as in section 8.1. Record this final weight (maf). Discard used Ascarite II material.
9.0 Quality Control
Same as Method 6, section 9.0.
10.0 Calibration and Standardization
Same as Method 6, section 10.0.
11.0 Analytical Procedure
11.1 Sample Analysis. The sample analysis procedure for SO2 is the same as that specified in Method 6, section 11.0.
12.0 Data Analysis and Calculations
Same as Method 6, section 12.0, with the addition of the following:
12.1 Nomenclature.
Cw = Concentration of moisture, percent.
CCO2 = Concentration of CO2, dry basis, percent.
ESO2 = Emission rate of SO2, ng/J (lb/10 6 Btu).
FC = Carbon F-factor from Method 19 for the fuel burned, dscm/J (dscf/10 6 Btu).
mwi = Initial weight of impingers, bubblers, and moisture absorber, g.
mwf = Final weight of impingers, bubblers, and moisture absorber, g.
mai = Initial weight of CO2 absorber, g.
maf = Final weight of CO2 absorber, g.
mSO2 = Mass of SO2 collected, mg.
VCO2(std) = Equivalent volume of CO2 collected at standard conditions, dscm (dscf).
Vw(std) = Equivalent volume of moisture collected at standard conditions, scm (scf).
12.2 CO2 Volume Collected, Corrected to Standard Conditions.
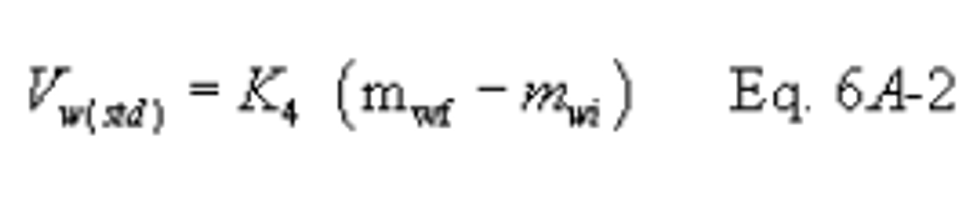
Where:
K3 = Equivalent volume of gaseous CO2 at standard conditions, 5.467 × 10−4 dscm/g (1.930 × 10−2 dscf/g).
12.3 Moisture Volume Collected, Corrected to Standard Conditions.
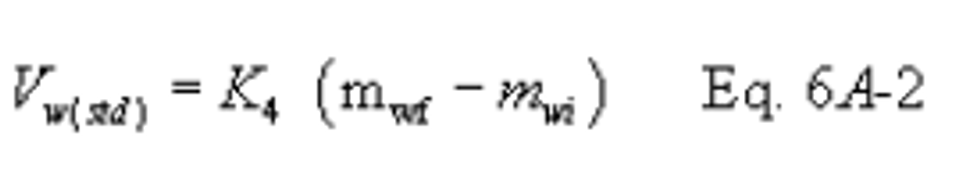
Where:
K4 = Equivalent volume of water vapor at standard conditions, 1.336 × 10−3 scm/g (4.717 × 10−2 scf/g).
12.4 SO2 Concentration.
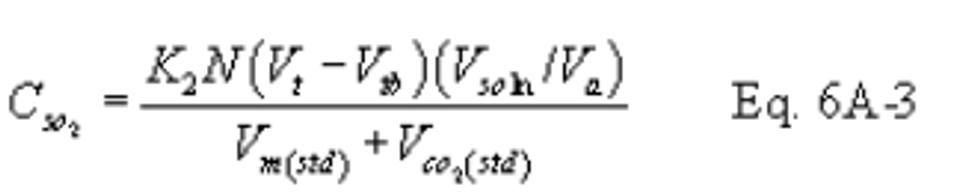
Where:
K2 = 32.03 mg SO2/meq. SO2 (7.061 × 10−5 lb SO2/meq. SO2)
12.5 CO2 Concentration.
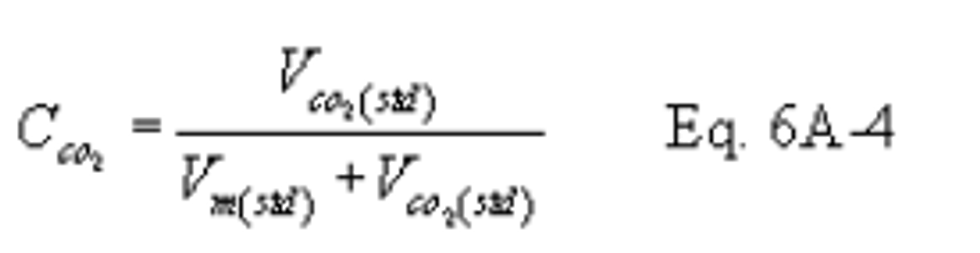
12.6 Moisture Concentration.
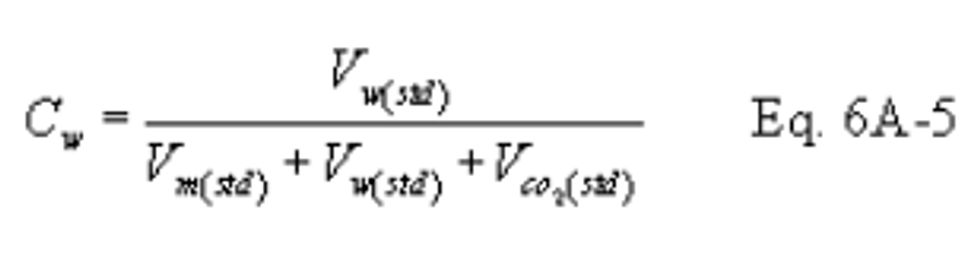
13.0 Method Performance
13.1 Range and Precision. The minimum detectable limit and the upper limit for the measurement of SO2 are the same as for Method 6. For a 20-liter sample, this method has a precision of ±0.5 percent CO2 for concentrations between 2.5 and 25 percent CO2 and ±1.0 percent moisture for moisture concentrations greater than 5 percent.
14.0 Pollution Prevention [Reserved]
15.0 Waste Management [Reserved]
16.0 Alternative Methods
If the only emission measurement desired is in terms of emission rate of SO2 (ng/J or lb/10 6 Btu), an abbreviated procedure may be used. The differences between the above procedure and the abbreviated procedure are described below.
16.1 Sampling Train. The sampling train is the same as that shown in Figure 6A-1 and as described in section 6.1, except that the dry gas meter is not needed.
16.2 Preparation of the Sampling Train. Follow the same procedure as in section 8.1, except do not weigh the isopropanol bubbler, the SO2 absorbing impingers, or the moisture absorber.
16.3 Sampling Train Leak-Check Procedure and Sample Collection. Leak-check and operate the sampling train as described in section 8.2, except that dry gas meter readings, barometric pressure, and dry gas meter temperatures need not be recorded during sampling.
16.4 Sample Recovery. Follow the procedure in section 8.3, except do not weigh the isopropanol bubbler, the SO2 absorbing impingers, or the moisture absorber.
16.5 Sample Analysis. Analysis of the peroxide solution is the same as that described in section 11.1.
16.6 Calculations.
16.6.1 SO2 Collected.
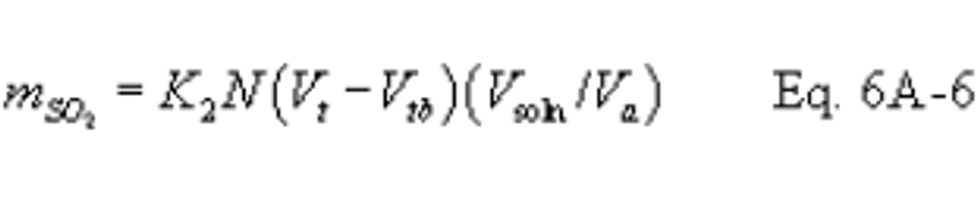
Where:
K2 = 32.03 mg SO2/meq. SO2
K2 = 7.061 × 10−5 lb SO2/meq. SO2
16.6.2 Sulfur Dioxide Emission Rate.
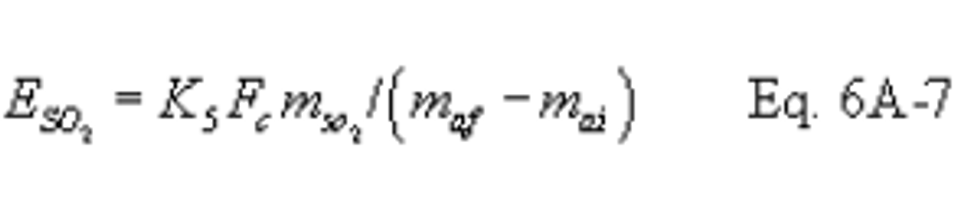
Where:
K5 = 1.829 × 10 9 mg/dscm
K2 = 0.1142 lb/dscf
17.0 References
Same as Method 6, section 17.0, References 1 through 8, with the addition of the following:
1. Stanley, Jon and P.R. Westlin. An Alternate Method for Stack Gas Moisture Determination. Source Evaluation Society Newsletter. 3(4). November 1978.
2. Whittle, Richard N. and P.R. Westlin. Air Pollution Test Report: Development and Evaluation of an Intermittent Integrated SO2/CO2 Emission Sampling Procedure. Environmental Protection Agency, Emission Standard and Engineering Division, Emission Measurement Branch. Research Triangle Park, NC. December 1979. 14 pp.
18.0 Tables, Diagrams, Flowcharts, and Validation Data
Method 6B - Determination of Sulfur Dioxide and Carbon Dioxide Daily Average Emissions From Fossil Fuel Combustion Sources
Note:
This method does not include all of the specifications (e.g., equipment and supplies) and procedures (e.g., sampling and analytical) essential to its performance. Some material is incorporated by reference from other methods in this part. Therefore, to obtain reliable results, persons using this method should have a thorough knowledge of at least the following additional test methods: Method 1, Method 2, Method 3, Method 5, Method 6, and Method 6A.
1.0 Scope and Application
1.1 Analytes.
| Analyte | CAS No. | Sensitivity |
|---|---|---|
| Sulfur dioxide (SO2) | 7449-09-05 | 3.4 mg SO2/m 3 (2.12 × 10−7 lb/ft 3) |
| Carbon dioxide (CO2) | 124-38-9 | N/A |
1.2 Applicability. This method is applicable for the determination of SO2 emissions from combustion sources in terms of concentration (ng/dscm or lb/dscf) and emission rate (ng/J or lb/10 6 Btu), and for the determination of CO2 concentration (percent) on a daily (24 hours) basis.
1.3 Data Quality Objectives. Adherence to the requirements of this method will enhance the quality of the data obtained from air pollutant sampling methods.
2.0 Summary of Method
2.1 A gas sample is extracted from the sampling point in the stack intermittently over a 24-hour or other specified time period. The SO2 fraction is measured by the barium-thorin titration method. Moisture and CO2 fractions are collected in the same sampling train, and are determined gravimetrically.
3.0 Definitions [Reserved]
4.0 Interferences
Same as Method 6, section 4.0.
5.0 Safety
5.1 Disclaimer. This method may involve hazardous materials, operations, and equipment. This test method may not address all of the safety problems associated with its use. It is the responsibility of the user to establish appropriate safety and health practices and determine the applicability of regulatory limitations prior to performing this test method.
5.2 Corrosive Reagents. Same as Method 6, section 5.2.
6.0 Equipment and Supplies
Same as Method 6A, section 6.0, with the following exceptions and additions:
6.1 The isopropanol bubbler is not used. An empty bubbler for the collection of liquid droplets, that does not allow direct contact between the collected liquid and the gas sample, may be included in the sampling train.
6.2 For intermittent operation, include an industrial timer-switch designed to operate in the “on” position at least 2 minutes continuously and “off” the remaining period over a repeating cycle. The cycle of operation is designated in the applicable regulation. At a minimum, the sampling operation should include at least 12, equal, evenly-spaced periods per 24 hours.
6.3 Stainless steel sampling probes, type 316, are not recommended for use with Method 6B because of potential sample contamination due to corrosion. Glass probes or other types of stainless steel, e.g., Hasteloy or Carpenter 20, are recommended for long-term use.
Note:
For applications downstream of wet scrubbers, a heated out-of-stack filter (either borosilicate glass wool or glass fiber mat) is necessary. Probe and filter heating systems capable of maintaining a sample gas temperature of between 20 and 120°C (68 and 248°F) at the filter are also required in these cases. The electric supply for these heating systems should be continuous and separate from the timed operation of the sample pump.
7.0 Reagents and Standards
Same as Method 6A, section 7.0, with the following exceptions:
7.1 Isopropanol is not used for sampling.
7.2 The hydrogen peroxide absorbing solution shall be diluted to no less than 6 percent by volume, instead of 3 percent as specified in Methods 6 and 6A.
7.3 If the Method 6B sampling train is to be operated in a low sample flow condition (less than 100 ml/min or 0.21 ft 3/hr), molecular sieve material may be substituted for Ascarite II as the CO2 absorbing material. The recommended molecular sieve material is Union Carbide 1/16 inch pellets, 5 A°, or equivalent. Molecular sieve material need not be discarded following the sampling run, provided that it is regenerated as per the manufacturer's instruction. Use of molecular sieve material at flow rates higher than 100 ml/min (0.21 ft 3/hr) may cause erroneous CO2 results.
8.0 Sample Collection, Preservation, Transport, and Storage
8.1 Preparation of Sampling Train. Same as Method 6A, section 8.1, with the addition of the following:
8.1.1 The sampling train is assembled as shown in Figure 6A-1 of Method 6A, except that the isopropanol bubbler is not included.
8.1.2 Adjust the timer-switch to operate in the “on” position from 2 to 4 minutes on a 2-hour repeating cycle or other cycle specified in the applicable regulation. Other timer sequences may be used with the restriction that the total sample volume collected is between 25 and 60 liters (0.9 and 2.1 ft 3) for the amounts of sampling reagents prescribed in this method.
8.1.3 Add cold water to the tank until the impingers and bubblers are covered at least two-thirds of their length. The impingers and bubbler tank must be covered and protected from intense heat and direct sunlight. If freezing conditions exist, the impinger solution and the water bath must be protected.
Note:
Sampling may be conducted continuously if a low flow-rate sample pump [20 to 40 ml/min (0.04 to 0.08 ft 3/hr) for the reagent volumes described in this method] is used. If sampling is continuous, the timer-switch is not necessary. In addition, if the sample pump is designed for constant rate sampling, the rate meter may be deleted. The total gas volume collected should be between 25 and 60 liters (0.9 and 2.1 ft 3) for the amounts of sampling reagents prescribed in this method.
8.2 Sampling Train Leak-Check Procedure. Same as Method 6, section 8.2.
8.3 Sample Collection.
8.3.1 The probe and filter (either in-stack, out-of-stack, or both) must be heated to a temperature sufficient to prevent water condensation.
8.3.2 Record the initial dry gas meter reading. To begin sampling, position the tip of the probe at the sampling point, connect the probe to the first impinger (or filter), and start the timer and the sample pump. Adjust the sample flow to a constant rate of approximately 1.0 liter/min (0.035 cfm) as indicated by the rotameter. Observe the operation of the timer, and determine that it is operating as intended (i.e., the timer is in the “on” position for the desired period, and the cycle repeats as required).
8.3.3 One time between 9 a.m. and 11 a.m. during the 24-hour sampling period, record the dry gas meter temperature (Tm) and the barometric pressure (P(bar)).
8.3.4 At the conclusion of the run, turn off the timer and the sample pump, remove the probe from the stack, and record the final gas meter volume reading. Conduct a leak-check as described in section 8.2. If a leak is found, void the test run or use procedures acceptable to the Administrator to adjust the sample volume for leakage. Repeat the steps in sections 8.3.1 to 8.3.4 for successive runs.
8.4 Sample Recovery. The procedures for sample recovery (moisture measurement, peroxide solution, and CO2 absorber) are the same as those in Method 6A, section 8.3.
9.0 Quality Control
Same as Method 6, section 9.0., with the exception of the isopropanol-check.
10.0 Calibration and Standardization
Same as Method 6, section 10.0, with the addition of the following:
10.1 Periodic Calibration Check. After 30 days of operation of the test train, conduct a calibration check according to the same procedures as the post-test calibration check (Method 6, section 10.1.2). If the deviation between initial and periodic calibration factors exceeds 5 percent, use the smaller of the two factors in calculations for the preceding 30 days of data, but use the most recent calibration factor for succeeding test runs.
11.0 Analytical Procedures
11.1 Sample Loss Check and Analysis. Same as Method 6, sections 11.1 and 11.2, respectively.
12.0 Data Analysis and Calculations
Same as Method 6A, section 12.0, except that Pbar and Tm correspond to the values recorded in section 8.3.3 of this method. The values are as follows:
Pbar = Initial barometric pressure for the test period, mm Hg.
Tm = Absolute meter temperature for the test period,°K.
13.0 Method Performance
13.1 Range.
13.1.1 Sulfur Dioxide. Same as Method 6.
13.1.2 Carbon Dioxide. Not determined.
13.2 Repeatability and Reproducibility. EPA-sponsored collaborative studies were undertaken to determine the magnitude of repeatability and reproducibility achievable by qualified testers following the procedures in this method. The results of the studies evolve from 145 field tests including comparisons with Methods 3 and 6. For measurements of emission rates from wet, flue gas desulfurization units in (ng/J), the repeatability (intra-laboratory precision) is 8.0 percent and the reproducibility (inter-laboratory precision) is 11.1 percent.
14.0 Pollution Prevention [Reserved]
15.0 Waste Management [Reserved]
16.0 Alternative Methods
Same as Method 6A, section 16.0, except that the timer is needed and is operated as outlined in this method.
17.0 References
Same as Method 6A, section 17.0, with the addition of the following:
1. Butler, Frank E., et. al. The Collaborative Test of Method 6B: Twenty-Four-Hour Analysis of SO2 and CO2. JAPCA. Vol. 33, No. 10. October 1983.
18.0 Tables, Diagrams, Flowcharts, and Validation Data [Reserved]
Method 6C - Determination of Sulfur Dioxide Emissions From Stationary Sources (Instrumental Analyzer Procedure)
1.0 Scope and Application
What is Method 6C?
Method 6C is a procedure for measuring sulfur dioxide (SO2) in stationary source emissions using a continuous instrumental analyzer. Quality assurance and quality control requirements are included to assure that you, the tester, collect data of known quality. You must document your adherence to these specific requirements for equipment, supplies, sample collection and analysis, calculations, and data analysis.
This method does not completely describe all equipment, supplies, and sampling and analytical procedures you will need but refers to other methods for some of the details. Therefore, to obtain reliable results, you should also have a thorough knowledge of these additional test methods which are found in appendix A to this part:
(a) Method 1 - Sample and Velocity Traverses for Stationary Sources.
(b) Method 4 - Determination of Moisture Content in Stack Gases.
(c) Method 6 - Determination of Sulfur Dioxide Emissions from Stationary Sources.
(d) Method 7E - Determination of Nitrogen Oxides Emissions from Stationary Sources (Instrumental Analyzer Procedure).
1.1 Analytes. What does this method determine? This method measures the concentration of sulfur dioxide.
| Analyte | CAS No. | Sensitivity |
|---|---|---|
| SO2 | 7446-09-5 | Typically <2% of Calibration Span. |
1.2 Applicability. When is this method required? The use of Method 6C may be required by specific New Source Performance Standards, Clean Air Marketing rules, State Implementation Plans, and permits where SO2 concentrations in stationary source emissions must be measured, either to determine compliance with an applicable emission standard or to conduct performance testing of a continuous emission monitoring system (CEMS). Other regulations may also require the use of Method 6C.
1.3 Data Quality Objectives. How good must my collected data be? Refer to section 1.3 of Method 7E.
2.0 Summary of Method
In this method, you continuously sample the effluent gas and convey the sample to an analyzer that measures the concentration of SO2. You must meet the performance requirements of this method to validate your data.
3.0 Definitions
Refer to section 3.0 of Method 7E for the applicable definitions.
4.0 Interferences
Refer to Section 4.0 of Method 7E.
5.0 Safety
Refer to section 5.0 of Method 7E.
6.0 Equipment and Supplies
Figure 7E-1 of Method 7E is a schematic diagram of an acceptable measurement system.
6.1 What do I need for the measurement system? The essential components of the measurement system are the same as those in sections 6.1 and 6.2 of Method 7E, except that the SO2 analyzer described in section 6.2 of this method must be used instead of the analyzer described in section 6.2 of Method 7E. You must follow the noted specifications in section 6.1 of Method 7E.
6.2 What analyzer must I use? You may use an instrument that uses an ultraviolet, non-dispersive infrared, fluorescence, or other detection principle to continuously measure SO2 in the gas stream and meets the performance specifications in section 13.0. The low-range and dual-range analyzer provisions in sections 6.2.8.1 and 6.2.8.2 of Method 7E apply.
7.0 Reagents and Standards
7.1 Calibration Gas. What calibration gases do I need? Refer to section 7.1 of Method 7E for the calibration gas requirements. Example calibration gas mixtures are listed below.
(a) SO2 in nitrogen (N2).
(b) SO2 in air.
(c) SO2 and CO2 in N2.
(d) SO2 andO2 in N2.
(e) SO2/CO2/O2 gas mixture in N2.
(f) CO2/NOX gas mixture in N2.
(g) CO2/SO2/NOX gas mixture in N2.
7.2 Interference Check. What additional reagents do I need for the interference check? The test gases for the interference check are listed in Table 7E-3 of Method 7E. For the alternative interference check, you must use the reagents described in section 7.0 of Method 6.
8.0 Sample Collection, Preservation, Storage, and Transport
8.1 Sampling Site and Sampling Points. You must follow the procedures of section 8.1 of Method 7E.
8.2 Initial Measurement System Performance Tests. You must follow the procedures in section 8.2 of Method 7E. If a dilution-type measurement system is used, the special considerations in section 8.3 of Method 7E also apply.
8.3 Interference Check. You must follow the procedures of section 8.2.7 of Method 7E to conduct an interference check, substituting SO2 for NOX as the method pollutant. For dilution-type measurement systems, you must use the alternative interference check procedure in section 16 and a co-located, unmodified Method 6 sampling train.
8.4 Sample Collection. You must follow the procedures of section 8.4 of Method 7E.
8.5 Post-Run System Bias Check and Drift Assessment. You must follow the procedures of section 8.5 of Method 7E.
9.0 Quality Control
Follow quality control procedures in section 9.0 of Method 7E.
10.0 Calibration and Standardization
Follow the procedures for calibration and standardization in section 10.0 of Method 7E.
11.0 Analytical Procedures
Because sample collection and analysis are performed together (see section 8), additional discussion of the analytical procedure is not necessary.
12.0 Calculations and Data Analysis
You must follow the applicable procedures for calculations and data analysis in section 12.0 of Method 7E as applicable, substituting SO2 for NOX as appropriate.
13.0 Method Performance
13.1 The specifications for the applicable performance checks are the same as in section 13.0 of Method 7E.
13.2 Alternative Interference Check. The results are acceptable if the difference between the Method 6C result and the modified Method 6 result is less than 7.0 percent of the Method 6 result for each of the three test runs. For the purposes of comparison, the Method 6 and 6C results must be expressed in the same units of measure.
14.0 Pollution Prevention [Reserved]
15.0 Waste Management [Reserved]
16.0 Alternative Procedures
16.1 Alternative Interference Check. You may perform an alternative interference check consisting of at least three comparison runs between Method 6C and Method 6. This check validates the Method 6C results at each particular source category (type of facility) where the check is performed. When testing under conditions of low concentrations (<15 ppm), this alternative interference check is not allowed.
Note:
The procedure described below applies to non-dilution sampling systems only. If this alternative interference check is used for a dilution sampling system, use a standard Method 6 sampling train and extract the sample directly from the exhaust stream at points collocated with the Method 6C sample probe.
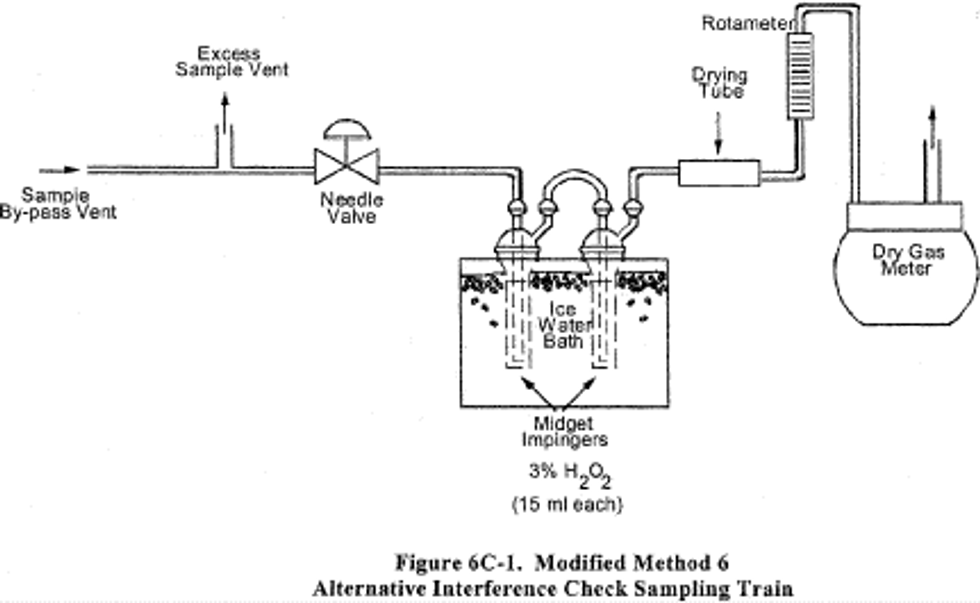
a. Build the modified Method 6 sampling train (flow control valve, two midget impingers containing 3 percent hydrogen peroxide, and dry gas meter) shown in Figure 6C-1. Connect the sampling train to the sample bypass discharge vent. Record the dry gas meter reading before you begin sampling. Simultaneously collect modified Method 6 and Method 6C samples. Open the flow control valve in the modified Method 6 train as you begin to sample with Method 6C. Adjust the Method 6 sampling rate to 1 liter per minute (.10 percent). The sampling time per run must be the same as for Method 6 plus twice the average measurement system response time. If your modified Method 6 train does not include a pump, you risk biasing the results high if you over-pressurize the midget impingers and cause a leak. You can reduce this risk by cautiously increasing the flow rate as sampling begins.
b. After completing a run, record the final dry gas meter reading, meter temperature, and barometric pressure. Recover and analyze the contents of the midget impingers using the procedures in Method 6. Determine the average gas concentration reported by Method 6C for the run.
17.0 References
1. “EPA Traceability Protocol for Assay and Certification of Gaseous Calibration Standards” September 1997 as amended, EPA-600/R-97/121
18.0 Tables, Diagrams, Flowcharts, and Validation Data
Method 7 - Determination of Nitrogen Oxide Emissions From Stationary Sources
Note:
This method does not include all of the specifications (e.g., equipment and supplies) and procedures (e.g., sampling and analytical) essential to its performance. Some material is incorporated by reference from other methods in this part. Therefore, to obtain reliable results, persons using this method should have a thorough knowledge of at least the following additional test methods: Method 1 and Method 5.
1.0 Scope and Application
1.1 Analytes.
| Analyte | CAS No. | Sensitivity |
|---|---|---|
| Nitrogen oxides (NOX), as NO2, including: | ||
| Nitric oxide (NO) | 10102-43-9 | |
| Nitrogen dioxide (NO2) | 10102-44-0 | 2-400 mg/dscm |
1.2 Applicability. This method is applicable for the measurement of nitrogen oxides (NOX) emitted from stationary sources.
1.3 Data Quality Objectives. Adherence to the requirements of this method will enhance the quality of the data obtained from air pollutant sample methods.
2.0 Summary of Method
A grab sample is collected in an evacuated flask containing a dilute sulfuric acid-hydrogen peroxide absorbing solution, and the nitrogen oxides, except nitrous oxide, are measured colorimetrically using the phenoldisulfonic acid (PDS) procedure.
3.0 Definitions [Reserved]
4.0 Interferences
Biased results have been observed when sampling under conditions of high sulfur dioxide concentrations. At or above 2100 ppm SO2, use five times the H2O2 concentration of the Method 7 absorbing solution. Laboratory tests have shown that high concentrations of SO2 (about 2100 ppm) cause low results in Method 7 and 7A. Increasing the H2O2 concentration to five times the original concentration eliminates this bias. However, when no SO2 is present, increasing the concentration by five times results in a low bias.
5.0 Safety
5.1 Disclaimer. This method may involve hazardous materials, operations, and equipment. This test method may not address all of the safety problems associated with its use. It is the responsibility of the user to establish appropriate safety and health practices and to determine the applicability of regulatory limitations prior to performing this test method.
5.2 Corrosive Reagents. The following reagents are hazardous. Personal protective equipment and safe procedures are useful in preventing chemical splashes. If contact occurs, immediately flush with copious amounts of water for at least 15 minutes. Remove clothing under shower and decontaminate. Treat residual chemical burns as thermal burns.
5.2.1 Hydrogen Peroxide (H2O2). Irritating to eyes, skin, nose, and lungs.
5.2.2 Phenoldisulfonic Acid. Irritating to eyes and skin.
5.2.3 Sodium Hydroxide (NaOH). Causes severe damage to eyes and skin. Inhalation causes irritation to nose, throat, and lungs. Reacts exothermically with limited amounts of water.
5.2.4 Sulfuric Acid (H2SO4). Rapidly destructive to body tissue. Will cause third degree burns. Eye damage may result in blindness. Inhalation may be fatal from spasm of the larynx, usually within 30 minutes. May cause lung tissue damage with edema. 1 mg/m 3 for 8 hours will cause lung damage or, in higher concentrations, death. Provide ventilation to limit inhalation. Reacts violently with metals and organics.
5.2.5 Phenol. Poisonous and caustic. Do not handle with bare hands as it is absorbed through the skin.
6.0 Equipment and Supplies
6.1 Sample Collection. A schematic of the sampling train used in performing this method is shown in Figure 7-1. Other grab sampling systems or equipment, capable of measuring sample volume to within 2.0 percent and collecting a sufficient sample volume to allow analytical reproducibility to within 5 percent, will be considered acceptable alternatives, subject to the approval of the Administrator. The following items are required for sample collection:
6.1.1 Probe. Borosilicate glass tubing, sufficiently heated to prevent water condensation and equipped with an in-stack or heated out-of-stack filter to remove particulate matter (a plug of glass wool is satisfactory for this purpose). Stainless steel or Teflon tubing may also be used for the probe. Heating is not necessary if the probe remains dry during the purging period.
6.1.2 Collection Flask. Two-liter borosilicate, round bottom flask, with short neck and 24/40 standard taper opening, protected against implosion or breakage.
6.1.3 Flask Valve. T-bore stopcock connected to a 24/40 standard taper joint.
6.1.4 Temperature Gauge. Dial-type thermometer, or other temperature gauge, capable of measuring 1°C (2°F) intervals from −5 to 50°C (23 to 122°F).
6.1.5 Vacuum Line. Tubing capable of withstanding a vacuum of 75 mm (3 in.) Hg absolute pressure, with “T” connection and T-bore stopcock.
6.1.6 Vacuum Gauge. U-tube manometer, 1 meter (39 in.), with 1 mm (0.04 in.) divisions, or other gauge capable of measuring pressure to within 2.5 mm (0.10 in.) Hg.
6.1.7 Pump. Capable of evacuating the collection flask to a pressure equal to or less than 75 mm (3 in.) Hg absolute.
6.1.8 Squeeze Bulb. One-way.
6.1.9 Volumetric Pipette. 25-ml.
6.1.10 Stopcock and Ground Joint Grease. A high-vacuum, high-temperature chlorofluorocarbon grease is required. Halocarbon 25-5S has been found to be effective.
6.1.11 Barometer. Mercury, aneroid, or other barometer capable of measuring atmospheric pressure to within 2.5 mm (0.1 in.) Hg. See note in Method 5, section 6.1.2.
6.2 Sample Recovery. The following items are required for sample recovery:
6.2.1 Graduated Cylinder. 50-ml with 1 ml divisions.
6.2.2 Storage Containers. Leak-free polyethylene bottles.
6.2.3 Wash Bottle. Polyethylene or glass.
6.2.4 Glass Stirring Rod.
6.2.5 Test Paper for Indicating pH. To cover the pH range of 7 to 14.
6.3 Analysis. The following items are required for analysis:
6.3.1 Volumetric Pipettes. Two 1-ml, two 2-ml, one 3-ml, one 4-ml, two 10-ml, and one 25-ml for each sample and standard.
6.3.2 Porcelain Evaporating Dishes. 175- to 250-ml capacity with lip for pouring, one for each sample and each standard. The Coors No. 45006 (shallowform, 195-ml) has been found to be satisfactory. Alternatively, polymethyl pentene beakers (Nalge No. 1203, 150-ml), or glass beakers (150-ml) may be used. When glass beakers are used, etching of the beakers may cause solid matter to be present in the analytical step; the solids should be removed by filtration.
6.3.3 Steam Bath. Low-temperature ovens or thermostatically controlled hot plates kept below 70°C (160°F) are acceptable alternatives.
6.3.4 Dropping Pipette or Dropper. Three required.
6.3.5 Polyethylene Policeman. One for each sample and each standard.
6.3.6 Graduated Cylinder. 100-ml with 1-ml divisions.
6.3.7 Volumetric Flasks. 50-ml (one for each sample and each standard), 100-ml (one for each sample and each standard, and one for the working standard KNO3 solution), and 1000-ml (one).
6.3.8 Spectrophotometer. To measure at 410 nm.
6.3.9 Graduated Pipette. 10-ml with 0.1-ml divisions.
6.3.10 Test Paper for Indicating pH. To cover the pH range of 7 to 14.
6.3.11 Analytical Balance. To measure to within 0.1 mg.
7.0 Reagents and Standards
Unless otherwise indicated, it is intended that all reagents conform to the specifications established by the Committee on Analytical Reagents of the American Chemical Society, where such specifications are available; otherwise, use the best available grade.
7.1 Sample Collection. The following reagents are required for sampling:
7.1.1 Water. Deionized distilled to conform to ASTM D 1193-77 or 91 Type 3 (incorporated by reference - see §60.17). The KMnO4 test for oxidizable organic matter may be omitted when high concentrations of organic matter are not expected to be present.
7.1.2 Absorbing Solution. Cautiously add 2.8 ml concentrated H2SO4 to a 1-liter flask partially filled with water. Mix well, and add 6 ml of 3 percent hydrogen peroxide, freshly prepared from 30 percent hydrogen peroxide solution. Dilute to 1 liter of water and mix well. The absorbing solution should be used within 1 week of its preparation. Do not expose to extreme heat or direct sunlight.
7.2 Sample Recovery. The following reagents are required for sample recovery:
7.2.1 Water. Same as in 7.1.1.
7.2.2 Sodium Hydroxide, 1 N. Dissolve 40 g NaOH in water, and dilute to 1 liter.
7.3 Analysis. The following reagents and standards are required for analysis:
7.3.1 Water. Same as in 7.1.1.
7.3.2 Fuming Sulfuric Acid. 15 to 18 percent by weight free sulfur trioxide. HANDLE WITH CAUTION.
7.3.3 Phenol. White solid.
7.3.4 Sulfuric Acid. Concentrated, 95 percent minimum assay.
7.3.5 Potassium Nitrate (KNO3). Dried at 105 to 110°C (221 to 230°F) for a minimum of 2 hours just prior to preparation of standard solution.
7.3.6 Standard KNO3 Solution. Dissolve exactly 2.198 g of dried KNO3 in water, and dilute to 1 liter with water in a 1000-ml volumetric flask.
7.3.7 Working Standard KNO3 Solution. Dilute 10 ml of the standard solution to 100 ml with water. One ml of the working standard solution is equivalent to 100 µg nitrogen dioxide (NO2).
7.3.8 Phenoldisulfonic Acid Solution. Dissolve 25 g of pure white phenol solid in 150 ml concentrated sulfuric acid on a steam bath. Cool, add 75 ml fuming sulfuric acid (15 to 18 percent by weight free sulfur trioxide - HANDLE WITH CAUTION), and heat at 100°C (212°F) for 2 hours. Store in a dark, stoppered bottle.
7.3.9 Concentrated Ammonium Hydroxide.
8.0 Sample Collection, Preservation, Storage and Transport
8.1 Sample Collection.
8.1.1 Flask Volume. The volume of the collection flask and flask valve combination must be known prior to sampling. Assemble the flask and flask valve, and fill with water to the stopcock. Measure the volume of water to ±10 ml. Record this volume on the flask.
8.1.2 Pipette 25 ml of absorbing solution into a sample flask, retaining a sufficient quantity for use in preparing the calibration standards. Insert the flask valve stopper into the flask with the valve in the “purge” position. Assemble the sampling train as shown in Figure 7-1, and place the probe at the sampling point. Make sure that all fittings are tight and leak-free, and that all ground glass joints have been greased properly with a high-vacuum, high temperature chlorofluorocarbon-based stopcock grease. Turn the flask valve and the pump valve to their “evacuate” positions. Evacuate the flask to 75 mm (3 in.) Hg absolute pressure, or less. Evacuation to a pressure approaching the vapor pressure of water at the existing temperature is desirable. Turn the pump valve to its “vent” position, and turn off the pump. Check for leakage by observing the manometer for any pressure fluctuation. (Any variation greater than 10 mm (0.4 in.) Hg over a period of 1 minute is not acceptable, and the flask is not to be used until the leakage problem is corrected. Pressure in the flask is not to exceed 75 mm (3 in.) Hg absolute at the time sampling is commenced.) Record the volume of the flask and valve (Vf), the flask temperature (Ti), and the barometric pressure. Turn the flask valve counterclockwise to its “purge” position, and do the same with the pump valve. Purge the probe and the vacuum tube using the squeeze bulb. If condensation occurs in the probe and the flask valve area, heat the probe, and purge until the condensation disappears. Next, turn the pump valve to its “vent” position. Turn the flask valve clockwise to its “evacuate” position, and record the difference in the mercury levels in the manometer. The absolute internal pressure in the flask (Pi) is equal to the barometric pressure less the manometer reading. Immediately turn the flask valve to the “sample” position, and permit the gas to enter the flask until pressures in the flask and sample line (i.e., duct, stack) are equal. This will usually require about 15 seconds; a longer period indicates a plug in the probe, which must be corrected before sampling is continued. After collecting the sample, turn the flask valve to its “purge” position, and disconnect the flask from the sampling train.
8.1.3 Shake the flask for at least 5 minutes.
8.1.4 If the gas being sampled contains insufficient oxygen for the conversion of NO to NO2 (e.g., an applicable subpart of the standards may require taking a sample of a calibration gas mixture of NO in N2), then introduce oxygen into the flask to permit this conversion. Oxygen may be introduced into the flask by one of three methods: (1) Before evacuating the sampling flask, flush with pure cylinder oxygen, then evacuate flask to 75 mm (3 in.) Hg absolute pressure or less; or (2) inject oxygen into the flask after sampling; or (3) terminate sampling with a minimum of 50 mm (2 in.) Hg vacuum remaining in the flask, record this final pressure, and then vent the flask to the atmosphere until the flask pressure is almost equal to atmospheric pressure.
8.2 Sample Recovery. Let the flask sit for a minimum of 16 hours, and then shake the contents for 2 minutes.
8.2.1 Connect the flask to a mercury filled U-tube manometer. Open the valve from the flask to the manometer, and record the flask temperature (Tf), the barometric pressure, and the difference between the mercury levels in the manometer. The absolute internal pressure in the flask (Pf) is the barometric pressure less the manometer reading. Transfer the contents of the flask to a leak-free polyethylene bottle. Rinse the flask twice with 5 ml portions of water, and add the rinse water to the bottle. Adjust the pH to between 9 and 12 by adding 1 N NaOH, dropwise (about 25 to 35 drops). Check the pH by dipping a stirring rod into the solution and then touching the rod to the pH test paper. Remove as little material as possible during this step. Mark the height of the liquid level so that the container can be checked for leakage after transport. Label the container to identify clearly its contents. Seal the container for shipping.
9.0 Quality Control
| Section | Quality control measure | Effect |
|---|---|---|
| 10.1 | Spectrophotometer calibration | Ensure linearity of spectrophotometer response to standards. |
10.0 Calibration and Standardization
10.1 Spectrophotometer.
10.1.1 Optimum Wavelength Determination.
10.1.1.1 Calibrate the wavelength scale of the spectrophotometer every 6 months. The calibration may be accomplished by using an energy source with an intense line emission such as a mercury lamp, or by using a series of glass filters spanning the measuring range of the spectrophotometer. Calibration materials are available commercially and from the National Institute of Standards and Technology. Specific details on the use of such materials should be supplied by the vendor; general information about calibration techniques can be obtained from general reference books on analytical chemistry. The wavelength scale of the spectrophotometer must read correctly within 5 nm at all calibration points; otherwise, repair and recalibrate the spectrophotometer. Once the wavelength scale of the spectrophotometer is in proper calibration, use 410 nm as the optimum wavelength for the measurement of the absorbance of the standards and samples.
10.1.1.2 Alternatively, a scanning procedure may be employed to determine the proper measuring wavelength. If the instrument is a double-beam spectrophotometer, scan the spectrum between 400 and 415 nm using a 200 µg NO2 standard solution in the sample cell and a blank solution in the reference cell. If a peak does not occur, the spectrophotometer is probably malfunctioning and should be repaired. When a peak is obtained within the 400 to 415 nm range, the wavelength at which this peak occurs shall be the optimum wavelength for the measurement of absorbance of both the standards and the samples. For a single-beam spectrophotometer, follow the scanning procedure described above, except scan separately the blank and standard solutions. The optimum wavelength shall be the wavelength at which the maximum difference in absorbance between the standard and the blank occurs.
10.1.2 Determination of Spectrophotometer Calibration Factor Kc. Add 0 ml, 2.0 ml, 4.0 ml, 6.0 ml, and 8.0 ml of the KNO3 working standard solution (1 ml = 100 µg NO2) to a series of five 50-ml volumetric flasks. To each flask, add 25 ml of absorbing solution and 10 ml water. Add 1 N NaOH to each flask until the pH is between 9 and 12 (about 25 to 35 drops). Dilute to the mark with water. Mix thoroughly, and pipette a 25-ml aliquot of each solution into a separate porcelain evaporating dish. Beginning with the evaporation step, follow the analysis procedure of section 11.2 until the solution has been transferred to the 100-ml volumetric flask and diluted to the mark. Measure the absorbance of each solution at the optimum wavelength as determined in section 10.1.1. This calibration procedure must be repeated on each day that samples are analyzed. Calculate the spectrophotometer calibration factor as shown in section 12.2.
10.1.3 Spectrophotometer Calibration Quality Control. Multiply the absorbance value obtained for each standard by the K c factor (reciprocal of the least squares slope) to determine the distance each calibration point lies from the theoretical calibration line. The difference between the calculated concentration values and the actual concentrations ( i.e., 100, 200, 300, and 400 μg NO 2 ) shall be less than 7 percent for all standards.
10.2 Barometer. Calibrate against a mercury barometer or NIST-traceable barometer prior to the field test.
10.3 Temperature Gauge. Calibrate dial thermometers against mercury-in-glass thermometers. An alternative mercury-free thermometer may be used if the thermometer is, at a minimum, equivalent in terms of performance or suitably effective for the specific temperature measurement application.
10.4 Vacuum Gauge. Calibrate mechanical gauges, if used, against a mercury manometer such as that specified in section 6.1.6.
10.5 Analytical Balance. Calibrate against standard weights.
11.0 Analytical Procedures
11.1 Sample Loss Check. Note the level of the liquid in the container, and confirm whether any sample was lost during shipment. Note this on the analytical data sheet. If a noticeable amount of leakage has occurred, either void the sample or use methods, subject to the approval of the Administrator, to correct the final results.
11.2 Sample Preparation. Immediately prior to analysis, transfer the contents of the shipping container to a 50 ml volumetric flask, and rinse the container twice with 5 ml portions of water. Add the rinse water to the flask, and dilute to mark with water; mix thoroughly. Pipette a 25-ml aliquot into the porcelain evaporating dish. Return any unused portion of the sample to the polyethylene storage bottle. Evaporate the 25-ml aliquot to dryness on a steam bath, and allow to cool. Add 2 ml phenoldisulfonic acid solution to the dried residue, and triturate thoroughly with a polyethylene policeman. Make sure the solution contacts all the residue. Add 1 ml water and 4 drops of concentrated sulfuric acid. Heat the solution on a steam bath for 3 minutes with occasional stirring. Allow the solution to cool, add 20 ml water, mix well by stirring, and add concentrated ammonium hydroxide, dropwise, with constant stirring, until the pH is 10 (as determined by pH paper). If the sample contains solids, these must be removed by filtration (centrifugation is an acceptable alternative, subject to the approval of the Administrator) as follows: Filter through Whatman No. 41 filter paper into a 100-ml volumetric flask. Rinse the evaporating dish with three 5-ml portions of water. Filter these three rinses. Wash the filter with at least three 15-ml portions of water. Add the filter washings to the contents of the volumetric flask, and dilute to the mark with water. If solids are absent, the solution can be transferred directly to the 100-ml volumetric flask and diluted to the mark with water.
11.3 Sample Analysis. Mix the contents of the flask thoroughly, and measure the absorbance at the optimum wavelength used for the standards (section 10.1.1), using the blank solution as a zero reference. Dilute the sample and the blank with equal volumes of water if the absorbance exceeds A4, the absorbance of the 400-µg NO2 standard (see section 10.1.3).
12.0 Data Analysis and Calculations
Carry out the calculations, retaining at least one extra significant figure beyond that of the acquired data. Round off figures after final calculations.
12.1 12.1 Nomenclature
A = Absorbance of sample.
A1 = Absorbance of the 100-µg NO2 standard.
A2 = Absorbance of the 200-µg NO2 standard.
A3 = Absorbance of the 300-µg NO2 standard.
A4 = Absorbance of the 400-µg NO2 standard.
C = Concentration of NOX as NO2, dry basis, corrected to standard conditions, mg/dsm 3 (lb/dscf).
F = Dilution factor (i.e., 25/5, 25/10, etc., required only if sample dilution was needed to reduce the absorbance into the range of the calibration).
Kc = Spectrophotometer calibration factor.
M = Mass of NOX as NO2 in gas sample, µg.
Pf = Final absolute pressure of flask, mm Hg (in. Hg).
Pi = Initial absolute pressure of flask, mm Hg (in. Hg).
Pstd = Standard absolute pressure, 760 mm Hg (29.92 in. Hg).
Tf = Final absolute temperature of flask,°K (°R).
Ti = Initial absolute temperature of flask,°K (°R).
Tstd = Standard absolute temperature, 293°K (528°R).
Vsc = Sample volume at standard conditions (dry basis), ml.
Vf = Volume of flask and valve, ml.
Va = Volume of absorbing solution, 25 ml.
12.2 Spectrophotometer Calibration Factor.
12.3 Sample Volume, Dry Basis, Corrected to Standard Conditions.
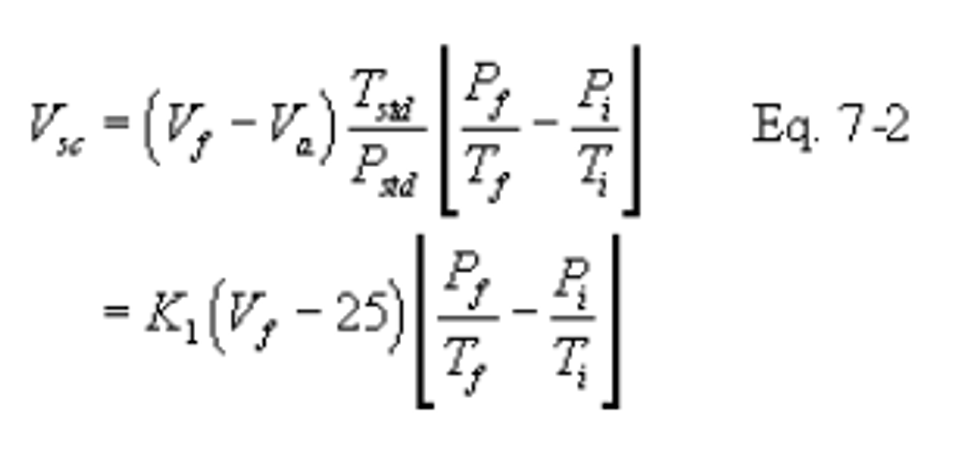
Where:
K1 = 0.3858°K/mm Hg for metric units,
K1 = 17.65°R/in. Hg for English units.
12.4 Total µg NO2 per sample.
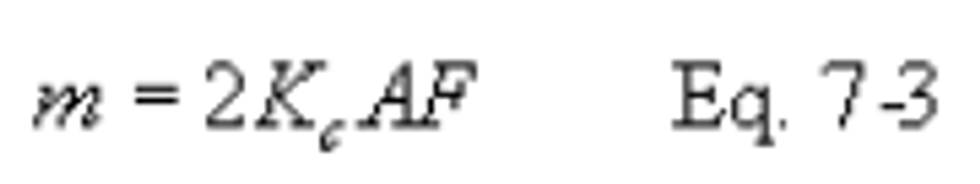
Where:
2 = 50/25, the aliquot factor.
Note:
If other than a 25-ml aliquot is used for analysis, the factor 2 must be replaced by a corresponding factor.
12.5 Sample Concentration, Dry Basis, Corrected to Standard Conditions.
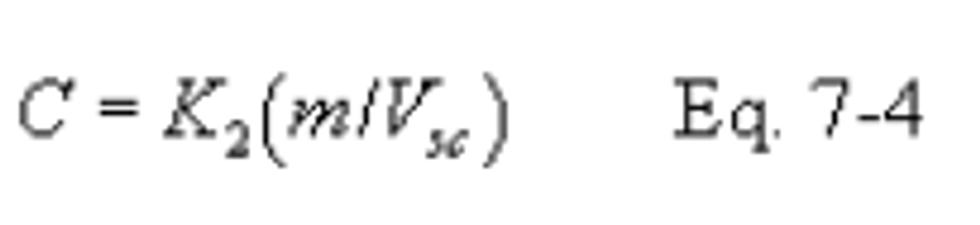
Where:
K2 = 10 3 (mg/m 3)/(µg/ml) for metric units,
K2 = 6.242 × 10−5 (lb/scf)/(µg/ml) for English units.
13.0 Method Performance
13.1 Range. The analytical range of the method has been determined to be 2 to 400 milligrams NOX (as NO2) per dry standard cubic meter, without having to dilute the sample.
14.0 Pollution Prevention [Reserved]
15.0 Waste Management [Reserved]
16.0 References
1. Standard Methods of Chemical Analysis. 6th ed. New York, D. Van Nostrand Co., Inc. 1962. Vol. 1, pp. 329-330.
2. Standard Method of Test for Oxides of Nitrogen in Gaseous Combustion Products (Phenoldisulfonic Acid Procedure). In: 1968 Book of ASTM Standards, Part 26. Philadelphia, PA. 1968. ASTM Designation D 1608-60, pp. 725-729.
3. Jacob, M.B. The Chemical Analysis of Air Pollutants. New York. Interscience Publishers, Inc. 1960. Vol. 10, pp. 351-356.
4. Beatty, R.L., L.B. Berger, and H.H. Schrenk. Determination of Oxides of Nitrogen by the Phenoldisulfonic Acid Method. Bureau of Mines, U.S. Dept. of Interior. R.I. 3687. February 1943.
5. Hamil, H.F. and D.E. Camann. Collaborative Study of Method for the Determination of Nitrogen Oxide Emissions from Stationary Sources (Fossil Fuel-Fired Steam Generators). Southwest Research Institute Report for Environmental Protection Agency. Research Triangle Park, NC. October 5, 1973.
6. Hamil, H.F. and R.E. Thomas. Collaborative Study of Method for the Determination of Nitrogen Oxide Emissions from Stationary Sources (Nitric Acid Plants). Southwest Research Institute Report for Environmental Protection Agency. Research Triangle Park, NC. May 8, 1974.
7. Stack Sampling Safety Manual (Draft). U.S. Environmental Protection Agency, Office of Air Quality Planning and Standards, Research Triangle Park, NC. September 1978.
17.0 Tables, Diagrams, Flowcharts, and Validation Data
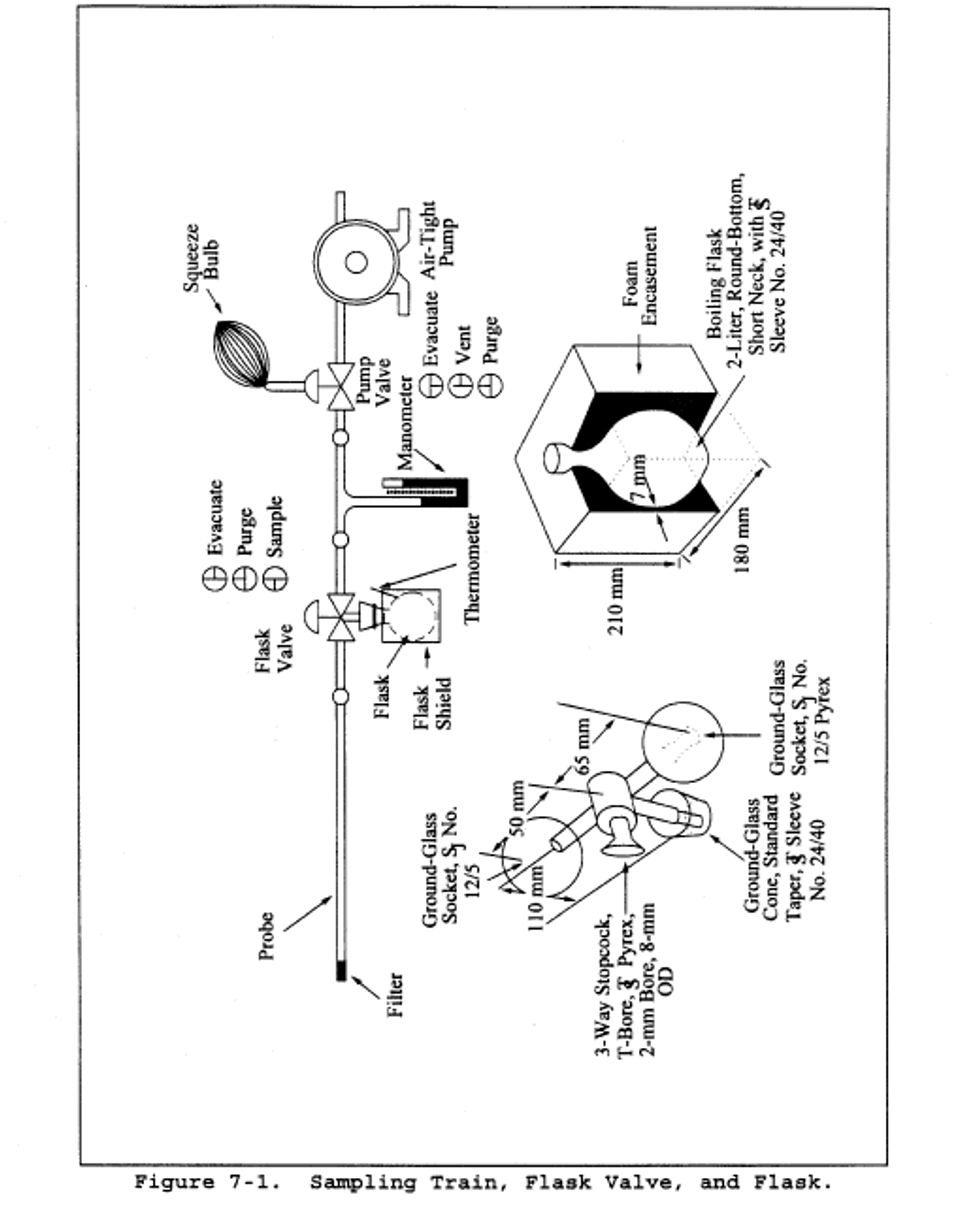
Method 7A - Determination of Nitrogen Oxide Emissions From Stationary Sources (Ion Chromatographic Method)
Note:
This method does not include all of the specifications (e.g., equipment and supplies) and procedures (e.g., sampling and analytical) essential to its performance. Some material is incorporated by reference from other methods in this part. Therefore, to obtain reliable results, persons using this method should have a thorough knowledge of at least the following additional test methods: Method 1, Method 3, Method 5, and Method 7.
1.0 Scope and Application
1.1 Analytes.
| Analyte | CAS No. | Sensitivity |
|---|---|---|
| Nitrogen oxides (NOX), as NO2, including: | ||
| Nitric oxide (NO) | 10102-43-9 | |
| Nitrogen dioxide (NO2) | 10102-44-0 | 65-655 ppmv |
1.2 Applicability. This method is applicable for the determination of NOX emissions from stationary sources.
1.3 Data Quality Objectives. Adherence to the requirements of this method will enhance the quality of the data obtained from air pollutant sampling methods.
2.0 Summary of Method
A grab sample is collected in an evacuated flask containing a dilute sulfuric acid-hydrogen peroxide absorbing solution. The nitrogen oxides, excluding nitrous oxide (N2O), are oxidized to nitrate and measured by ion chromatography.
3.0 Definitions [Reserved]
4.0 Interferences
Biased results have been observed when sampling under conditions of high sulfur dioxide concentrations. At or above 2100 ppm SO2, use five times the H2O2 concentration of the Method 7 absorbing solution. Laboratory tests have shown that high concentrations of SO2 (about 2100 ppm) cause low results in Method 7 and 7A. Increasing the H2O2 concentration to five times the original concentration eliminates this bias. However, when no SO2 is present, increasing the concentration by five times results in a low bias.
5.0 Safety
5.1 This method may involve hazardous materials, operations, and equipment. This test method may not address all of the safety problems associated with its use. It is the responsibility of the user of this test method to establish appropriate safety and health practices and to determine the applicability of regulatory limitations prior to performing this test method.
5.2 Corrosive reagents. The following reagents are hazardous. Personal protective equipment and safe procedures are useful in preventing chemical splashes. If contact occurs, immediately flush with copious amounts of water at least 15 minutes. Remove clothing under shower and decontaminate. Treat residual chemical burns as thermal burns.
5.2.1 Hydrogen Peroxide (H2O2). Irritating to eyes, skin, nose, and lungs.
5.2.2 Sulfuric Acid (H2SO4). Rapidly destructive to body tissue. Will cause third degree burns. Eye damage may result in blindness. Inhalation may be fatal from spasm of the larynx, usually within 30 minutes. May cause lung tissue damage with edema. 3 mg/m 3 will cause lung damage in uninitiated. 1 mg/m 3 for 8 hours will cause lung damage or, in higher concentrations, death. Provide ventilation to limit inhalation. Reacts violently with metals and organics.
6.0 Equipment and Supplies
6.1 Sample Collection. Same as in Method 7, section 6.1.
6.2 Sample Recovery. Same as in Method 7, section 6.2, except the stirring rod and pH paper are not needed.
6.3 Analysis. For the analysis, the following equipment and supplies are required. Alternative instrumentation and procedures will be allowed provided the calibration precision requirement in section 10.1.2 can be met.
6.3.1 Volumetric Pipets. Class A;1-, 2-, 4-, 5-ml (two for the set of standards and one per sample), 6-, 10-, and graduated 5-ml sizes.
6.3.2 Volumetric Flasks. 50-ml (two per sample and one per standard), 200-ml, and 1-liter sizes.
6.3.3 Analytical Balance. To measure to within 0.1 mg.
6.3.4 Ion Chromatograph. The ion chromatograph should have at least the following components:
6.3.4.1 Columns. An anion separation or other column capable of resolving the nitrate ion from sulfate and other species present and a standard anion suppressor column (optional). Suppressor columns are produced as proprietary items; however, one can be produced in the laboratory using the resin available from BioRad Company, 32nd and Griffin Streets, Richmond, California. Peak resolution can be optimized by varying the eluent strength or column flow rate, or by experimenting with alternative columns that may offer more efficient separation. When using guard columns with the stronger reagent to protect the separation column, the analyst should allow rest periods between injection intervals to purge possible sulfate buildup in the guard column.
6.3.4.2 Pump. Capable of maintaining a steady flow as required by the system.
6.3.4.3 Flow Gauges. Capable of measuring the specified system flow rate.
6.3.4.4 Conductivity Detector.
6.3.4.5 Recorder. Compatible with the output voltage range of the detector.
7.0 Reagents and Standards
Unless otherwise indicated, it is intended that all reagents conform to the specifications established by the Committee on Analytical Reagents of the American Chemical Society, where such specifications are available; otherwise, use the best available grade.
7.1 Sample Collection. Same as Method 7, section 7.1.
7.2 Sample Recovery. Same as Method 7, section 7.1.1.
7.3 Analysis. The following reagents and standards are required for analysis:
7.3.1 Water. Same as Method 7, section 7.1.1.
7.3.2 Stock Standard Solution, 1 mg NO2/ml. Dry an adequate amount of sodium nitrate (NaNO3) at 105 to 110°C (221 to 230°F) for a minimum of 2 hours just before preparing the standard solution. Then dissolve exactly 1.847 g of dried NaNO3 in water, and dilute to l liter in a volumetric flask. Mix well. This solution is stable for 1 month and should not be used beyond this time.
7.3.3 Working Standard Solution, 25 µg/ml. Dilute 5 ml of the standard solution to 200 ml with water in a volumetric flask, and mix well.
7.3.4 Eluent Solution. Weigh 1.018 g of sodium carbonate (Na2CO3) and 1.008 g of sodium bicarbonate (NaHCO3), and dissolve in 4 liters of water. This solution is 0.0024 M Na2CO3/0.003 M NaHCO3. Other eluents appropriate to the column type and capable of resolving nitrate ion from sulfate and other species present may be used.
8.0 Sample Collection, Preservation, Storage, and Transport
8.1 Sampling. Same as in Method 7, section 8.1.
8.2 Sample Recovery. Same as in Method 7, section 8.2, except delete the steps on adjusting and checking the pH of the sample. Do not store the samples more than 4 days between collection and analysis.
9.0 Quality Control
| Section | Quality control measure | Effect |
|---|---|---|
| 10.1 | Ion chromatographn calibration | Ensure linearity of ion chromatograph response to standards. |
10.0 Calibration and Standardizations
10.1 Ion Chromatograph.
10.1.1 Determination of Ion Chromatograph Calibration Factor S. Prepare a series of five standards by adding 1.0, 2.0, 4.0, 6.0, and 10.0 ml of working standard solution (25 µg/ml) to a series of five 50-ml volumetric flasks. (The standard masses will equal 25, 50, 100, 150, and 250 µg.) Dilute each flask to the mark with water, and mix well. Analyze with the samples as described in section 11.2, and subtract the blank from each value. Prepare or calculate a linear regression plot of the standard masses in µg (x-axis) versus their peak height responses in millimeters (y-axis). (Take peak height measurements with symmetrical peaks; in all other cases, calculate peak areas.) From this curve, or equation, determine the slope, and calculate its reciprocal to denote as the calibration factor, S.
10.1.2 Ion Chromatograph Calibration Quality Control. If any point on the calibration curve deviates from the line by more than 7 percent of the concentration at that point, remake and reanalyze that standard. This deviation can be determined by multiplying S times the peak height response for each standard. The resultant concentrations must not differ by more than 7 percent from each known standard mass (i.e., 25, 50, 100, 150, and 250 µg).
10.2 Conductivity Detector. Calibrate according to manufacturer's specifications prior to initial use.
10.3 Barometer. Calibrate against a mercury barometer.
10.4 Temperature Gauge. Calibrate dial thermometers against mercury-in-glass thermometers. An alternative mercury-free thermometer may be used if the thermometer is, at a minimum, equivalent in terms of performance or suitably effective for the specific temperature measurement application.
10.5 Vacuum Gauge. Calibrate mechanical gauges, if used, against a mercury manometer such as that specified in section 6.1.6 of Method 7.
10.6 Analytical Balance. Calibrate against standard weights.
11.0 Analytical Procedures
11.1 Sample Preparation.
11.1.1 Note on the analytical data sheet, the level of the liquid in the container, and whether any sample was lost during shipment. If a noticeable amount of leakage has occurred, either void the sample or use methods, subject to the approval of the Administrator, to correct the final results. Immediately before analysis, transfer the contents of the shipping container to a 50-ml volumetric flask, and rinse the container twice with 5 ml portions of water. Add the rinse water to the flask, and dilute to the mark with water. Mix thoroughly.
11.1.2 Pipet a 5-ml aliquot of the sample into a 50-ml volumetric flask, and dilute to the mark with water. Mix thoroughly. For each set of determinations, prepare a reagent blank by diluting 5 ml of absorbing solution to 50 ml with water. (Alternatively, eluent solution may be used instead of water in all sample, standard, and blank dilutions.)
11.2 Analysis.
11.2.1 Prepare a standard calibration curve according to section 10.1.1. Analyze the set of standards followed by the set of samples using the same injection volume for both standards and samples. Repeat this analysis sequence followed by a final analysis of the standard set. Average the results. The two sample values must agree within 5 percent of their mean for the analysis to be valid. Perform this duplicate analysis sequence on the same day. Dilute any sample and the blank with equal volumes of water if the concentration exceeds that of the highest standard.
11.2.2 Document each sample chromatogram by listing the following analytical parameters: injection point, injection volume, nitrate and sulfate retention times, flow rate, detector sensitivity setting, and recorder chart speed.
12.0 Data Analysis and Calculations
Carry out the calculations, retaining at least one extra significant figure beyond that of the acquired data. Round off figures after final calculations.
12.1 Sample Volume. Calculate the sample volume Vsc (in ml), on a dry basis, corrected to standard conditions, using Equation 7-2 of Method 7.
12.2 Sample Concentration of NOX as NO2.
12.2.1 Calculate the sample concentration C (in mg/dscm) as follows:
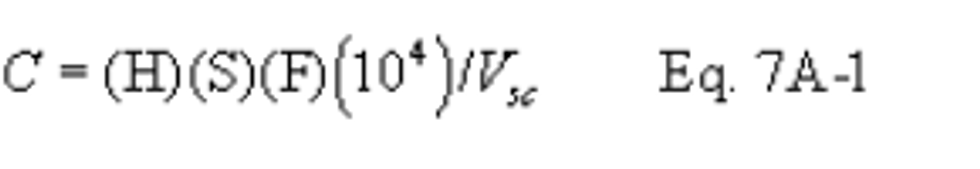
Where:
H = Sample peak height, mm.
S = Calibration factor, µg/mm.
F = Dilution factor (required only if sample dilution was needed to reduce the concentration into the range of calibration), dimensionless.
10 4 = 1:10 dilution times conversion factor of: (mg/10 3 µg)(10 6 ml/m 3).
12.2.2 If desired, the concentration of NO2 may be calculated as ppm NO2 at standard conditions as follows:
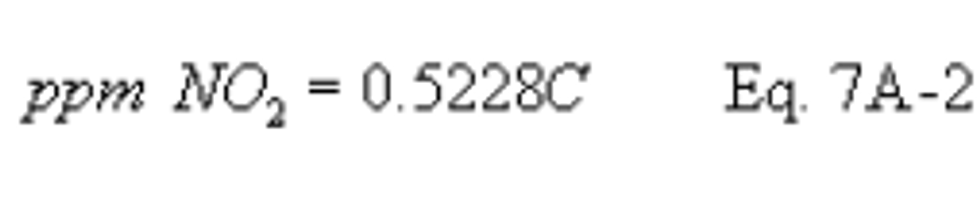
Where:
0.5228 = ml/mg NO2.
13.0 Method Performance
13.1 Range. The analytical range of the method is from 125 to 1250 mg NOX/m 3 as NO2 (65 to 655 ppmv), and higher concentrations may be analyzed by diluting the sample. The lower detection limit is approximately 19 mg/m 3 (10 ppmv), but may vary among instruments.
14.0 Pollution Prevention [Reserved]
15.0 Waste Management [Reserved]
16.0 References
1. Mulik, J.D., and E. Sawicki. Ion Chromatographic Analysis of Environmental Pollutants. Ann Arbor, Ann Arbor Science Publishers, Inc. Vol. 2, 1979.
2. Sawicki, E., J.D. Mulik, and E. Wittgenstein. Ion Chromatographic Analysis of Environmental Pollutants. Ann Arbor, Ann Arbor Science Publishers, Inc. Vol. 1. 1978.
3. Siemer, D.D. Separation of Chloride and Bromide from Complex Matrices Prior to Ion Chromatographic Determination. Anal. Chem. 52(12):1874-1877. October 1980.
4. Small, H., T.S. Stevens, and W.C. Bauman. Novel Ion Exchange Chromatographic Method Using Conductimetric Determination. Anal. Chem. 47(11):1801. 1975.
5. Yu, K.K., and P.R. Westlin. Evaluation of Reference Method 7 Flask Reaction Time. Source Evaluation Society Newsletter. 4(4). November 1979. 10 pp.
6. Stack Sampling Safety Manual (Draft). U.S. Environmental Protection Agency, Office of Air Quality Planning and Standard, Research Triangle Park, NC. September 1978.
17.0 Tables, Diagrams, Flowcharts, and Validation Data [Reserved]
Method 7B - Determination of Nitrogen Oxide Emissions From Stationary Sources (Ultraviolet Spectrophotometric Method)
Note:
This method does not include all of the specifications (e.g., equipment and supplies) and procedures (e.g., sampling and analytical) essential to its performance. Some material is incorporated by reference from other methods in this part. Therefore, to obtain reliable results, persons using this method should have a thorough knowledge of at least the following additional test methods: Method 1, Method 5, and Method 7.
1.0 Scope and Application
1.1 Analytes.
| Analyte | CAS No. | Sensitivity |
|---|---|---|
| Nitrogen oxides (NOX), as NO2, including: | ||
| Nitric oxide (NO) | 10102-43-9 | |
| Nitrogen dioxide (NO2) | 10102-44-0 | 30-786 ppmv |
1.2 Applicability. This method is applicable for the determination of NOX emissions from nitric acid plants.
1.3 Data Quality Objectives. Adherence to the requirements of this method will enhance the quality of the data obtained from air pollutant sampling methods.
2.0 Summary of Method
2.1 A grab sample is collected in an evacuated flask containing a dilute sulfuric acid-hydrogen peroxide absorbing solution; the NOX, excluding nitrous oxide (N2O), are measured by ultraviolet spectrophotometry.
3.0 Definition [Reserved]
4.0 Interferences [Reserved]
5.0 Safety
5.1 This method may involve hazardous materials, operations, and equipment. This test method may not address all of the safety problems associated with its use. It is the responsibility of the user of this test method to establish appropriate safety and health practices and to determine the applicability of regulatory limitations prior to performing this test method.
5.2 Corrosive reagents. The following reagents are hazardous. Personal protective equipment and safe procedures are useful in preventing chemical splashes. If contact occurs, immediately flush with copious amounts of water at least 15 minutes. Remove clothing under shower and decontaminate. Treat residual chemical burn as thermal burn.
5.2.1 Hydrogen Peroxide (H2O2). Irritating to eyes, skin, nose, and lungs.
5.2.2 Sodium Hydroxide (NaOH). Causes severe damage to eyes and skin. Inhalation causes irritation to nose, throat, and lungs. Reacts exothermically with limited amounts of water.
5.2.3 Sulfuric Acid (H2SO4). Rapidly destructive to body tissue. Will cause third degree burns. Eye damage may result in blindness. Inhalation may be fatal from spasm of the larynx, usually within 30 minutes. May cause lung tissue damage with edema. 3 mg/m 3 will cause lung damage in uninitiated. 1 mg/m 3 for 8 hours will cause lung damage or, in higher concentrations, death. Provide ventilation to limit inhalation. Reacts violently with metals and organics.
6.0 Equipment and Supplies
6.1 Sample Collection. Same as Method 7, section 6.1.
6.2 Sample Recovery. The following items are required for sample recovery:
6.2.1 Wash Bottle. Polyethylene or glass.
6.2.2 Volumetric Flasks. 100-ml (one for each sample).
6.3 Analysis. The following items are required for analysis:
6.3.1 Volumetric Pipettes. 5-, 10-, 15-, and 20-ml to make standards and sample dilutions.
6.3.2 Volumetric Flasks. 1000- and 100-ml for preparing standards and dilution of samples.
6.3.3 Spectrophotometer. To measure ultraviolet absorbance at 210 nm.
6.3.4 Analytical Balance. To measure to within 0.1 mg.
7.0 Reagents and Standards
Note:
Unless otherwise indicated, all reagents are to conform to the specifications established by the Committee on Analytical Reagents of the American Chemical Society, where such specifications are available. Otherwise, use the best available grade.
7.1 Sample Collection. Same as Method 7, section 7.1. It is important that the amount of hydrogen peroxide in the absorbing solution not be increased. Higher concentrations of peroxide may interfere with sample analysis.
7.2 Sample Recovery. Same as Method 7, section 7.2.
7.3 Analysis. Same as Method 7, sections 7.3.1, 7.3.3, and 7.3.4, with the addition of the following:
7.3.1 Working Standard KNO3 Solution. Dilute 10 ml of the standard solution to 1000 ml with water. One milliliter of the working standard is equivalent to 10 µg NO2.
8.0 Sample Collection, Preservation, Storage, and Transport
8.1 Sample Collection. Same as Method 7, section 8.1.
8.2 Sample Recovery.
8.2.1 Let the flask sit for a minimum of 16 hours, and then shake the contents for 2 minutes.
8.2.2 Connect the flask to a mercury filled U-tube manometer. Open the valve from the flask to the manometer, and record the flask temperature (Tf), the barometric pressure, and the difference between the mercury levels in the manometer. The absolute internal pressure in the flask (Pf) is the barometric pressure less the manometer reading.
8.2.3 Transfer the contents of the flask to a leak-free wash bottle. Rinse the flask three times with 10-ml portions of water, and add to the bottle. Mark the height of the liquid level so that the container can be checked for leakage after transport. Label the container to identify clearly its contents. Seal the container for shipping.
9.0 Quality Control
| Section | Quality control measure | Effect |
|---|---|---|
| 10.1 | Spectrophotometer calibration | Ensures linearity of spectrophotometer response to standards. |
10.0 Calibration and Standardizations
Same as Method 7, sections 10.2 through 10.5, with the addition of the following:
10.1 Determination of Spectrophotometer Standard Curve. Add 0 ml, 5 ml, 10 ml, 15 ml, and 20 ml of the KNO3 working standard solution (1 ml = 10 µg NO2) to a series of five 100-ml volumetric flasks. To each flask, add 5 ml of absorbing solution. Dilute to the mark with water. The resulting solutions contain 0.0, 50, 100, 150, and 200 µg NO2, respectively. Measure the absorbance by ultraviolet spectrophotometry at 210 nm, using the blank as a zero reference. Prepare a standard curve plotting absorbance vs. µg NO2.
Note:
If other than a 20-ml aliquot of sample is used for analysis, then the amount of absorbing solution in the blank and standards must be adjusted such that the same amount of absorbing solution is in the blank and standards as is in the aliquot of sample used.
10.1.1 Calculate the spectrophotometer calibration factor as follows:
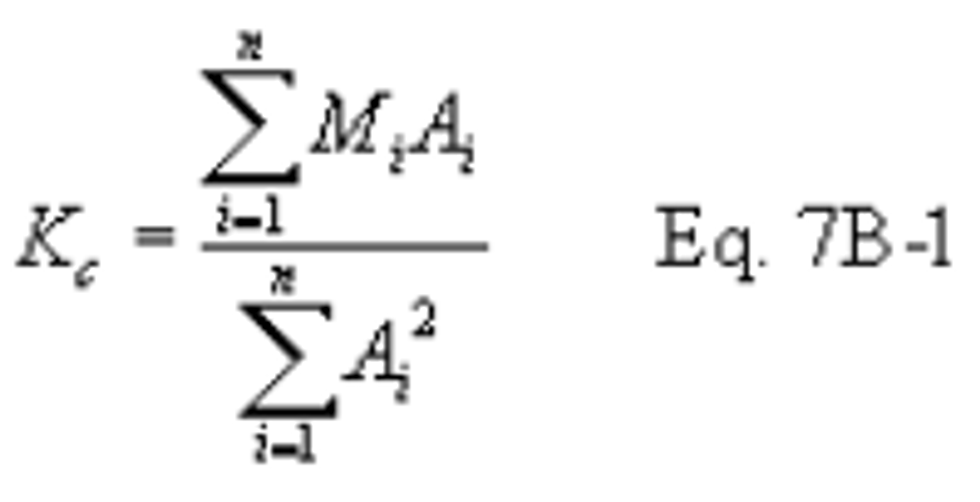
Where:
Mi = Mass of NO2 in standard i, µg.
Ai = Absorbance of NO2 standard i.
n = Total number of calibration standards.
10.1.2 For the set of calibration standards specified here, Equation 7B-1 simplifies to the following:
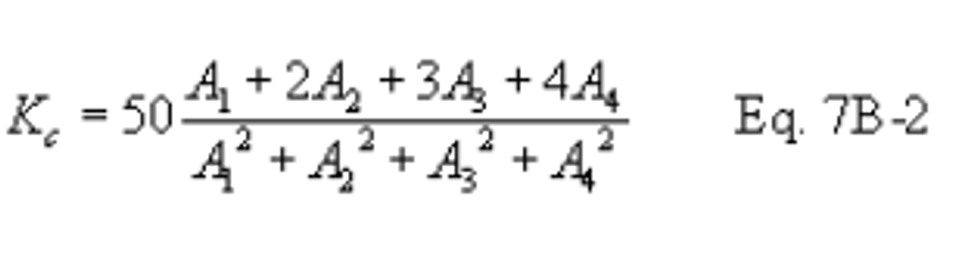
10.2 Spectrophotometer Calibration Quality Control. Multiply the absorbance value obtained for each standard by the Kc factor (reciprocal of the least squares slope) to determine the distance each calibration point lies from the theoretical calibration line. The difference between the calculated concentration values and the actual concentrations (i.e., 50, 100, 150, and 200 µg NO2) should be less than 7 percent for all standards.
11.0 Analytical Procedures
11.1 Sample Loss Check. Note the level of the liquid in the container, and confirm whether any sample was lost during shipment. Note this on the analytical data sheet. If a noticeable amount of leakage has occurred, either void the sample or use methods, subject to the approval of the Administrator, to correct the final results.
11.2 Sample Preparation. Immediately prior to analysis, transfer the contents of the shipping container to a 100-ml volumetric flask, and rinse the container twice with 5-ml portions of water. Add the rinse water to the flask, and dilute to mark with water.
11.3 Sample Analysis. Mix the contents of the flask thoroughly and pipette a 20 ml-aliquot of sample into a 100-ml volumetric flask. Dilute to the mark with water. Using the blank as zero reference, read the absorbance of the sample at 210 nm.
11.4 Audit Sample Analysis. Same as Method 7, section 11.4, except that a set of audit samples must be analyzed with each set of compliance samples or once per analysis day, or once per week when averaging continuous samples.
12.0 Data Analysis and Calculations
Same as Method 7, section 12.0, except replace section 12.3 with the following:
12.1 Total µg NO2 Per Sample.
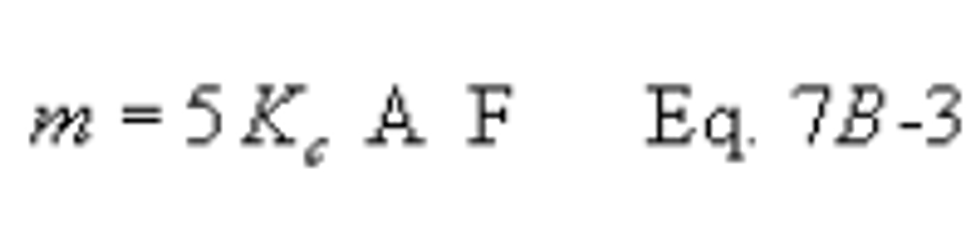
Where:
5 = 100/20, the aliquot factor.
Note:
If other than a 20-ml aliquot is used for analysis, the factor 5 must be replaced by a corresponding factor.
13.0 Method Performance
13.1 Range. The analytical range of the method as outlined has been determined to be 57 to 1500 milligrams NOX (as NO2) per dry standard cubic meter, or 30 to 786 parts per million by volume (ppmv) NOX.
14.0 Pollution Prevention [Reserved]
15.0 Waste Management [Reserved]
16.0 References
1. National Institute for Occupational Safety and Health. Recommendations for Occupational Exposure to Nitric Acid. In: Occupational Safety and Health Reporter. Washington, D.C. Bureau of National Affairs, Inc. 1976. p. 149.
2. Rennie, P.J., A.M. Sumner, and F.B. Basketter. Determination of Nitrate in Raw, Potable, and Waste Waters by Ultraviolet Spectrophotometry. Analyst. 104:837. September 1979.
17.0 Tables, Diagrams, Flowcharts, and Validation Data [Reserved]
Method 7C - Determination of Nitrogen Oxide Emissions From Stationary Sources (Alkaline Permanganate/Colorimetric Method)
Note:
This method does not include all of the specifications (e.g., equipment and supplies) and procedures (e.g., sampling and analytical) essential to its performance. Some material is incorporated by reference from other methods in this part. Therefore, to obtain reliable results, persons using this method should have a thorough knowledge of at least the following additional test methods: Method 1, Method 3, Method 6 and Method 7.
1.0 Scope and Application
1.1 Analytes.
| Analyte | CAS no. | Sensitivity |
|---|---|---|
| Nitrogen oxides (NOX), as NO2, including: | ||
| Nitric oxide (NO) | 10102-43-9 | |
| Nitrogen dioxide (NO2) | 10102-44-07 | ppmv |
1.2 Applicability. This method applies to the measurement of NOX emissions from fossil-fuel fired steam generators, electric utility plants, nitric acid plants, or other sources as specified in the regulations.
1.3 Data Quality Objectives. Adherence to the requirements of this method will enhance the quality of the data obtained from air pollutant sampling methods.
2.0 Summary of Method
An integrated gas sample is extracted from the stack and passed through impingers containing an alkaline potassium permanganate solution; NOX (NO NO2) emissions are oxidized to NO2 and NO3. Then NO3 −is reduced to NO2 −with cadmium, and the NO2 −is analyzed colorimetrically.
3.0 Definitions [Reserved]
4.0 Interferences
Possible interferents are sulfur dioxides (SO2) and ammonia (NH3).
4.1 High concentrations of SO2 could interfere because SO2 consumes MnO4 (as does NOX) and, therefore, could reduce the NOX collection efficiency. However, when sampling emissions from a coal-fired electric utility plant burning 2.1 percent sulfur coal with no control of SO2 emissions, collection efficiency was not reduced. In fact, calculations show that sampling 3000 ppm SO2 will reduce the MnO4 concentration by only 5 percent if all the SO2 is consumed in the first impinger.
4.2 Ammonia (NH3) is slowly oxidized to NO3 − by the absorbing solution. At 100 ppm NH3 in the gas stream, an interference of 6 ppm NOX (11 mg NO2/m 3) was observed when the sample was analyzed 10 days after collection. Therefore, the method may not be applicable to plants using NH3 injection to control NOX emissions unless means are taken to correct the results. An equation has been developed to allow quantification of the interference and is discussed in Reference 5 of section 16.0.
5.0 Safety
5.1 Disclaimer. This method may involve hazardous materials, operations, and equipment. This test method may not address all of the safety problems associated with its use. It is the responsibility of the user of this test method to establish appropriate safety and health practices and to determine the applicability of regulatory limitations prior to performing this test method.
5.2 Corrosive Reagents. The following reagents are hazardous. Personal protective equipment and safe procedures are useful in preventing chemical splashes. If contact occurs, immediately flush with copious amounts of water for at least 15 minutes. Remove clothing under shower and decontaminate. Treat residual chemical burns as thermal burns.
5.2.1 Hydrochloric Acid (HCl). Highly toxic and corrosive. Causes severe damage to skin. Vapors are highly irritating to eyes, skin, nose, and lungs, causing severe damage. May cause bronchitis, pneumonia, or edema of lungs. Exposure to vapor concentrations of 0.13 to 0.2 percent can be lethal in minutes. Will react with metals, producing hydrogen.
5.2.2 Oxalic Acid (COOH)2. Poisonous. Irritating to eyes, skin, nose, and throat.
5.2.3 Sodium Hydroxide (NaOH). Causes severe damage to eye tissues and to skin. Inhalation causes irritation to nose, throat, and lungs. Reacts exothermically with small amounts of water.
5.2.4 Potassium Permanganate (KMnO4). Caustic, strong oxidizer. Avoid bodily contact with.
6.0 Equipment and Supplies
6.1 Sample Collection and Sample Recovery. A schematic of the Method 7C sampling train is shown in Figure 7C-1, and component parts are discussed below. Alternative apparatus and procedures are allowed provided acceptable accuracy and precision can be demonstrated to the satisfaction of the Administrator.
6.1.1 Probe. Borosilicate glass tubing, sufficiently heated to prevent water condensation and equipped with an in-stack or heated out-of-stack filter to remove particulate matter (a plug of glass wool is satisfactory for this purpose). Stainless steel or Teflon tubing may also be used for the probe.
6.1.2 Impingers. Three restricted-orifice glass impingers, having the specifications given in Figure 7C-2, are required for each sampling train. The impingers must be connected in series with leak-free glass connectors. Stopcock grease may be used, if necessary, to prevent leakage. (The impingers can be fabricated by a glass blower if not available commercially.)
6.1.3 Glass Wool, Stopcock Grease, Drying Tube, Valve, Pump, Barometer, and Vacuum Gauge and Rotameter. Same as in Method 6, sections 6.1.1.3, 6.1.1.4, 6.1.1.6, 6.1.1.7, 6.1.1.8, 6.1.2, and 6.1.3, respectively.
6.1.4 Rate Meter. Rotameter, or equivalent, accurate to within 2 percent at the selected flow rate of between 400 and 500 ml/min (0.014 to 0.018 cfm). For rotameters, a range of 0 to 1 liter/min (0 to 0.035 cfm) is recommended.
6.1.5 Volume Meter. Dry gas meter (DGM) capable of measuring the sample volume under the sampling conditions of 400 to 500 ml/min (0.014 to 0.018 cfm) for 60 minutes within an accuracy of 2 percent.
6.1.6 Filter. To remove NOX from ambient air, prepared by adding 20 g of 5-angstrom molecular sieve to a cylindrical tube (e.g., a polyethylene drying tube).
6.1.7 Polyethylene Bottles. 1-liter, for sample recovery.
6.1.8 Funnel and Stirring Rods. For sample recovery.
6.2 Sample Preparation and Analysis.
6.2.1 Hot Plate. Stirring type with 50- by 10-mm Teflon-coated stirring bars.
6.2.2 Beakers. 400-, 600-, and 1000-ml capacities.
6.2.3 Filtering Flask. 500-ml capacity with side arm.
6.2.4 Buchner Funnel. 75-mm ID, with spout equipped with a 13-mm ID by 90-mm long piece of Teflon tubing to minimize possibility of aspirating sample solution during filtration.
6.2.5 Filter Paper. Whatman GF/C, 7.0-cm diameter.
6.2.6 Stirring Rods.
6.2.7 Volumetric Flasks. 100-, 200- or 250-, 500-, and 1000-ml capacity.
6.2.8 Watch Glasses. To cover 600- and 1000-ml beakers.
6.2.9 Graduated Cylinders. 50- and 250-ml capacities.
6.2.10 Pipettes. Class A.
6.2.11 pH Meter. To measure pH from 0.5 to 12.0.
6.2.12 Burette. 50-ml with a micrometer type stopcock. (The stopcock is Catalog No. 8225-t-05, Ace Glass, Inc., Post Office Box 996, Louisville, Kentucky 50201.) Place a glass wool plug in bottom of burette. Cut off burette at a height of 43 cm (17 in.) from the top of plug, and have a blower attach a glass funnel to top of burette such that the diameter of the burette remains essentially unchanged. Other means of attaching the funnel are acceptable.
6.2.13 Glass Funnel. 75-mm ID at the top.
6.2.14 Spectrophotometer. Capable of measuring absorbance at 540 nm; 1-cm cells are adequate.
6.2.15 Metal Thermometers. Bimetallic thermometers, range 0 to 150°C (32 to 300°F).
6.2.16 Culture Tubes. 20-by 150-mm, Kimax No. 45048.
6.2.17 Parafilm “M.” Obtained from American Can Company, Greenwich, Connecticut 06830.
6.2.18 CO2 Measurement Equipment. Same as in Method 3, section 6.0.
7.0 Reagents and Standards
Unless otherwise indicated, it is intended that all reagents conform to the specifications established by the Committee on Analytical Reagents of the American Chemical Society, where such specifications are available; otherwise, use the best available grade.
7.1 Sample Collection.
7.1.1 Water. Deionized distilled to conform to ASTM Specification D 1193-77 or 91 Type 3 (incorporated by reference - see §60.17).
7.1.2 Potassium Permanganate, 4.0 Percent (w/w), Sodium Hydroxide, 2.0 Percent (w/w) solution (KMnO4/NaOH solution). Dissolve 40.0 g of KMnO4 and 20.0 g of NaOH in 940 ml of water.
7.2 Sample Preparation and Analysis.
7.2.1 Water. Same as in section 7.1.1.
7.2.2 Oxalic Acid Solution. Dissolve 48 g of oxalic acid [(COOH)2·2H2O] in water, and dilute to 500 ml. Do not heat the solution.
7.2.3 Sodium Hydroxide, 0.5 N. Dissolve 20 g of NaOH in water, and dilute to 1 liter.
7.2.4 Sodium Hydroxide, 10 N. Dissolve 40 g of NaOH in water, and dilute to 100 ml.
7.2.5 Ethylenediamine Tetraacetic Acid (EDTA) Solution, 6.5 percent (w/v). Dissolve 6.5 g of EDTA (disodium salt) in water, and dilute to 100 ml. Dissolution is best accomplished by using a magnetic stirrer.
7.2.6 Column Rinse Solution. Add 20 ml of 6.5 percent EDTA solution to 960 ml of water, and adjust the pH to between 11.7 and 12.0 with 0.5 N NaOH.
7.2.7 Hydrochloric Acid (HCl), 2 N. Add 86 ml of concentrated HCl to a 500 ml-volumetric flask containing water, dilute to volume, and mix well. Store in a glass-stoppered bottle.
7.2.8 Sulfanilamide Solution. Add 20 g of sulfanilamide (melting point 165 to 167°C (329 to 333°F)) to 700 ml of water. Add, with mixing, 50 ml concentrated phosphoric acid (85 percent), and dilute to 1000 ml. This solution is stable for at least 1 month, if refrigerated.
7.2.9 N-(1-Naphthyl)-Ethylenediamine Dihydrochloride (NEDA) Solution. Dissolve 0.5 g of NEDA in 500 ml of water. An aqueous solution should have one absorption peak at 320 nm over the range of 260 to 400 nm. NEDA that shows more than one absorption peak over this range is impure and should not be used. This solution is stable for at least 1 month if protected from light and refrigerated.
7.2.10 Cadmium. Obtained from Matheson Coleman and Bell, 2909 Highland Avenue, Norwood, Ohio 45212, as EM Laboratories Catalog No. 2001. Prepare by rinsing in 2 N HCl for 5 minutes until the color is silver-grey. Then rinse the cadmium with water until the rinsings are neutral when tested with pH paper. CAUTION: H2 is liberated during preparation. Prepare in an exhaust hood away from any flame or combustion source.
7.2.11 Sodium Nitrite (NaNO2) Standard Solution, Nominal Concentration, 1000 µg NO2−/ml. Desiccate NaNO2 overnight. Accurately weigh 1.4 to 1.6 g of NaNO2 (assay of 97 percent NaNO2 or greater), dissolve in water, and dilute to 1 liter. Calculate the exact NO2-concentration using Equation 7C-1 in section 12.2. This solution is stable for at least 6 months under laboratory conditions.
7.2.12 Potassium Nitrate (KNO3) Standard Solution. Dry KNO3 at 110°C (230°F) for 2 hours, and cool in a desiccator. Accurately weigh 9 to 10 g of KNO3 to within 0.1 mg, dissolve in water, and dilute to 1 liter. Calculate the exact NO3 − concentration using Equation 7C-2 in section 12.3. This solution is stable for 2 months without preservative under laboratory conditions.
7.2.13 Spiking Solution. Pipette 7 ml of the KNO3 standard into a 100-ml volumetric flask, and dilute to volume.
7.2.14 Blank Solution. Dissolve 2.4 g of KMnO4 and 1.2 g of NaOH in 96 ml of water. Alternatively, dilute 60 ml of KMnO4/NaOH solution to 100 ml.
8.0 Sample Collection, Preservation, Storage, and Transport
8.1 Preparation of Sampling Train. Add 200 ml of KMnO4/NaOH solution (Section 7.1.2) to each of three impingers, and assemble the train as shown in Figure 7C-1. Adjust the probe heater to a temperature sufficient to prevent water condensation.
8.2 Leak-Checks. Same as in Method 6, section 8.2.
8.3 Sample Collection.
8.3.1 Record the initial DGM reading and barometric pressure. Determine the sampling point or points according to the appropriate regulations (e.g., §60.46(b)(5) of 40 CFR Part 60). Position the tip of the probe at the sampling point, connect the probe to the first impinger, and start the pump. Adjust the sample flow to a value between 400 and 500 ml/min (0.014 and 0.018 cfm). CAUTION: DO NOT EXCEED THESE FLOW RATES. Once adjusted, maintain a constant flow rate during the entire sampling run. Sample for 60 minutes. For relative accuracy (RA) testing of continuous emission monitors, the minimum sampling time is 1 hour, sampling 20 minutes at each traverse point.
Note:
When the SO2 concentration is greater than 1200 ppm, the sampling time may have to be reduced to 30 minutes to eliminate plugging of the impinger orifice with MnO2. For RA tests with SO2 greater than 1200 ppm, sample for 30 minutes (10 minutes at each point).
8.3.2 Record the DGM temperature, and check the flow rate at least every 5 minutes. At the conclusion of each run, turn off the pump, remove the probe from the stack, and record the final readings. Divide the sample volume by the sampling time to determine the average flow rate. Conduct the mandatory post-test leak-check. If a leak is found, void the test run, or use procedures acceptable to the Administrator to adjust the sample volume for the leakage.
8.4 CO2 Measurement. During sampling, measure the CO2 content of the stack gas near the sampling point using Method 3. The single-point grab sampling procedure is adequate, provided the measurements are made at least three times (near the start, midway, and before the end of a run), and the average CO2 concentration is computed. The Orsat or Fyrite analyzer may be used for this analysis.
8.5 Sample Recovery. Disconnect the impingers. Pour the contents of the impingers into a 1-liter polyethylene bottle using a funnel and a stirring rod (or other means) to prevent spillage. Complete the quantitative transfer by rinsing the impingers and connecting tubes with water until the rinsings are clear to light pink, and add the rinsings to the bottle. Mix the sample, and mark the solution level. Seal and identify the sample container.
9.0 Quality Control
| Section | Quality control measure | Effect |
|---|---|---|
| 8.2, 10.1-10.3 | Sampling equipment leak-check and calibration | Ensure accurate measurement of sample volume. |
| 10.4 | Spectrophotometer calibration | Ensure linearity of spectrophotometer response to standards |
| 11.3 | Spiked sample analysis. | Ensure reduction efficiency of column. |
10.0 Calibration and Standardizations
10.1 Volume Metering System. Same as Method 6, section 10.1. For detailed instructions on carrying out these calibrations, it is suggested that section 3.5.2 of Reference 4 of section 16.0 be consulted.
10.2 Temperature Sensors and Barometer. Same as in Method 6, sections 10.2 and 10.4, respectively.
10.3 Check of Rate Meter Calibration Accuracy (Optional). Disconnect the probe from the first impinger, and connect the filter. Start the pump, and adjust the rate meter to read between 400 and 500 ml/min (0.014 and 0.018 cfm). After the flow rate has stabilized, start measuring the volume sampled, as recorded by the dry gas meter and the sampling time. Collect enough volume to measure accurately the flow rate. Then calculate the flow rate. This average flow rate must be less than 500 ml/min (0.018 cfm) for the sample to be valid; therefore, it is recommended that the flow rate be checked as above prior to each test.
10.4 Spectrophotometer.
10.4.1 Dilute 5.0 ml of the NaNO2 standard solution to 200 ml with water. This solution nominally contains 25 µg NO2 −/ml. Use this solution to prepare calibration standards to cover the range of 0.25 to 3.00 µg NO2 −/ml. Prepare a minimum of three standards each for the linear and slightly nonlinear (described below) range of the curve. Use pipettes for all additions.
10.4.2 Measure the absorbance of the standards and a water blank as instructed in section 11.5. Plot the net absorbance vs. µg NO2 −/ml. Draw a smooth curve through the points. The curve should be linear up to an absorbance of approximately 1.2 with a slope of approximately 0.53 absorbance units/µg NO2 −/ml. The curve should pass through the origin. The curve is slightly nonlinear from an absorbance of 1.2 to 1.6.
11.0 Analytical Procedures
11.1 Sample Stability. Collected samples are stable for at least four weeks; thus, analysis must occur within 4 weeks of collection.
11.2 Sample Preparation.
11.2.1 Prepare a cadmium reduction column as follows: Fill the burette with water. Add freshly prepared cadmium slowly, with tapping, until no further settling occurs. The height of the cadmium column should be 39 cm (15 in). When not in use, store the column under rinse solution.
Note:
The column should not contain any bands of cadmium fines. This may occur if regenerated cadmium is used and will greatly reduce the column lifetime.
11.2.2 Note the level of liquid in the sample container, and determine whether any sample was lost during shipment. If a noticeable amount of leakage has occurred, the volume lost can be determined from the difference between initial and final solution levels, and this value can then be used to correct the analytical result. Quantitatively transfer the contents to a 1-liter volumetric flask, and dilute to volume.
11.2.3 Take a 100-ml aliquot of the sample and blank (unexposed KMnO4/NaOH) solutions, and transfer to 400-ml beakers containing magnetic stirring bars. Using a pH meter, add concentrated H2SO4 with stirring until a pH of 0.7 is obtained. Allow the solutions to stand for 15 minutes. Cover the beakers with watch glasses, and bring the temperature of the solutions to 50°C (122°F). Keep the temperature below 60°C (140°F). Dissolve 4.8 g of oxalic acid in a minimum volume of water, approximately 50 ml, at room temperature. Do not heat the solution. Add this solution slowly, in increments, until the KMnO4 solution becomes colorless. If the color is not completely removed, prepare some more of the above oxalic acid solution, and add until a colorless solution is obtained. Add an excess of oxalic acid by dissolving 1.6 g of oxalic acid in 50 ml of water, and add 6 ml of this solution to the colorless solution. If suspended matter is present, add concentrated H2SO4 until a clear solution is obtained.
11.2.4 Allow the samples to cool to near room temperature, being sure that the samples are still clear. Adjust the pH to between 11.7 and 12.0 with 10 N NaOH. Quantitatively transfer the mixture to a Buchner funnel containing GF/C filter paper, and filter the precipitate. Filter the mixture into a 500-ml filtering flask. Wash the solid material four times with water. When filtration is complete, wash the Teflon tubing, quantitatively transfer the filtrate to a 500-ml volumetric flask, and dilute to volume. The samples are now ready for cadmium reduction. Pipette a 50-ml aliquot of the sample into a 150-ml beaker, and add a magnetic stirring bar. Pipette in 1.0 ml of 6.5 percent EDTA solution, and mix.
11.3 Determine the correct stopcock setting to establish a flow rate of 7 to 9 ml/min of column rinse solution through the cadmium reduction column. Use a 50-ml graduated cylinder to collect and measure the solution volume. After the last of the rinse solution has passed from the funnel into the burette, but before air entrapment can occur, start adding the sample, and collect it in a 250-ml graduated cylinder. Complete the quantitative transfer of the sample to the column as the sample passes through the column. After the last of the sample has passed from the funnel into the burette, start adding 60 ml of column rinse solution, and collect the rinse solution until the solution just disappears from the funnel. Quantitatively transfer the sample to a 200-ml volumetric flask (a 250-ml flask may be required), and dilute to volume. The samples are now ready for NO2-analysis.
Note:
Two spiked samples should be run with every group of samples passed through the column. To do this, prepare two additional 50-ml aliquots of the sample suspected to have the highest NO2-concentration, and add 1 ml of the spiking solution to these aliquots. If the spike recovery or column efficiency (see section 12.2) is below 95 percent, prepare a new column, and repeat the cadmium reduction.
11.5 Sample Analysis. Pipette 10 ml of sample into a culture tube. Pipette in 10 ml of sulfanilamide solution and 1.4 ml of NEDA solution. Cover the culture tube with parafilm, and mix the solution. Prepare a blank in the same manner using the sample from treatment of the unexposed KMnO4/NaOH solution. Also, prepare a calibration standard to check the slope of the calibration curve. After a 10-minute color development interval, measure the absorbance at 540 nm against water. Read µg NO2 −/ml from the calibration curve. If the absorbance is greater than that of the highest calibration standard, use less than 10 ml of sample, and repeat the analysis. Determine the NO2 −concentration using the calibration curve obtained in section 10.4.
Note:
Some test tubes give a high blank NO2 − value but culture tubes do not.
11.6 Audit Sample Analysis. Same as in Method 7, section 11.4.
12.0 Data Analysis and Calculations
Carry out calculations, retaining at least one extra significant figure beyond that of the acquired data. Round off figures after final calculation.
12.1 Nomenclature.
B = Analysis of blank, µg NO2 −/ml.
C = Concentration of NOX as NO2, dry basis, mg/dsm 3.
E = Column efficiency, dimensionless
K2 = 10−3 mg/µg.
m = Mass of NOX, as NO2, in sample, µg.
Pbar = Barometric pressure, mm Hg (in. Hg).
Pstd = Standard absolute pressure, 760 mm Hg (29.92 in. Hg).
s = Concentration of spiking solution, µg NO3/ml.
S = Analysis of sample, µg NO2 −/ml.
Tm = Average dry gas meter absolute temperature,°K.
Tstd = Standard absolute temperature, 293°K (528°R).
Vm(std) = Dry gas volume measured by the dry gas meter, corrected to standard conditions, dscm (dscf).
Vm = Dry gas volume as measured by the dry gas meter, scm (scf).
x = Analysis of spiked sample, µg NO2 −/ml.
X = Correction factor for CO2 collection = 100/(100 − %CO2(V/V)).
y = Analysis of unspiked sample, µg NO2 −/ml.
Y = Dry gas meter calibration factor.
1.0 ppm NO = 1.247 mg NO/m 3 at STP.
1.0 ppm NO2 = 1.912 mg NO2/m 3 at STP.
1 ft 3 = 2.832 × 10−2 m 3.
12.2 NO2 Concentration. Calculate the NO2 concentration of the solution (see section 7.2.11) using the following equation:
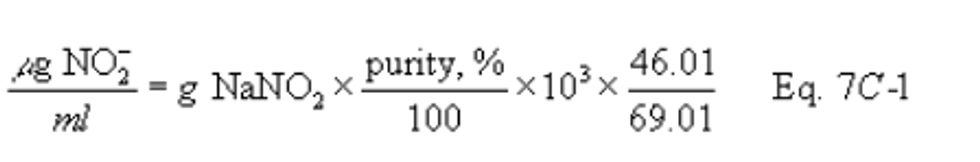
12.3 NO3 Concentration. Calculate the NO3 concentration of the KNO3 solution (see section 7.2.12) using the following equation:
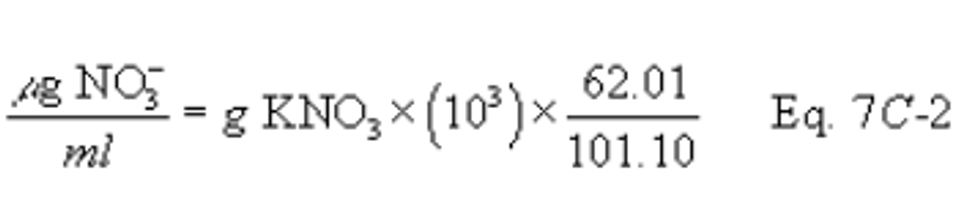
12.4 Sample Volume, Dry Basis, Corrected to Standard Conditions.
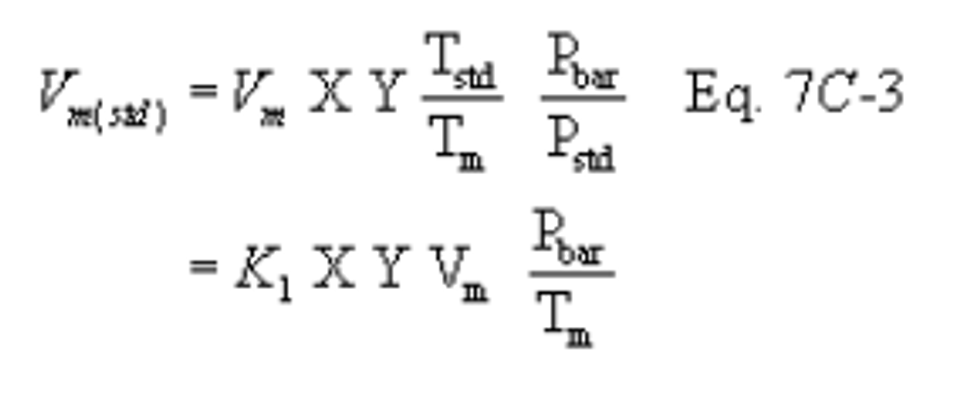
Where:
K1 = 0.3855°K/mm Hg for metric units.
K1 = 17.65°R/in. Hg for English units.
12.5 Efficiency of Cadmium Reduction Column. Calculate this value as follows:
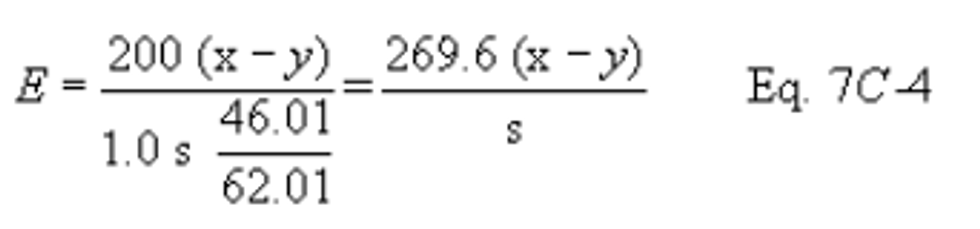
Where:
200 = Final volume of sample and blank after passing through the column, ml.
1.0 = Volume of spiking solution added, ml.
46.01=µg NO2 −/µmole.
62.01=µg NO3 −/µmole.
12.6 Total µg NO2.
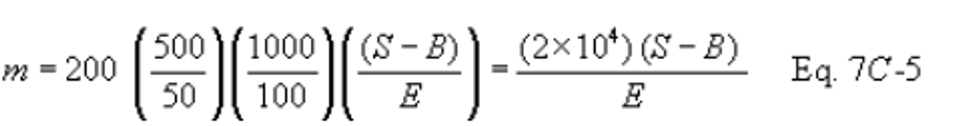
Where:
500 = Total volume of prepared sample, ml.
50 = Aliquot of prepared sample processed through cadmium column, ml.
100 = Aliquot of KMnO4/NaOH solution, ml.
1000 = Total volume of KMnO4/NaOH solution, ml.
12.7 Sample Concentration.
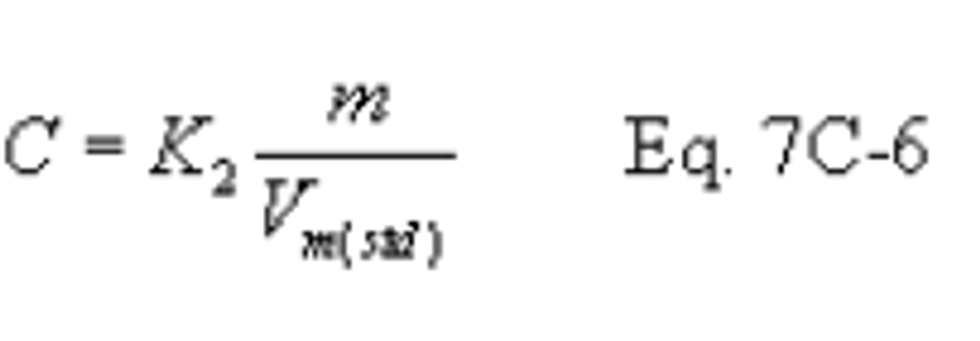
13.0 Method Performance
13.1 Precision. The intra-laboratory relative standard deviation for a single measurement is 2.8 and 2.9 percent at 201 and 268 ppm NOX, respectively.
13.2 Bias. The method does not exhibit any bias relative to Method 7.
13.3 Range. The lower detectable limit is 13 mg NOX/m 3, as NO2 (7 ppm NOX) when sampling at 500 ml/min for 1 hour. No upper limit has been established; however, when using the recommended sampling conditions, the method has been found to collect NOX emissions quantitatively up to 1782 mg NOX/m 3, as NO2 (932 ppm NOX).
14.0 Pollution Prevention [Reserved]
15.0 Waste Management [Reserved]
16.0 References
1. Margeson, J.H., W.J. Mitchell, J.C. Suggs, and M.R. Midgett. Integrated Sampling and Analysis Methods for Determining NOX Emissions at Electric Utility Plants. U.S. Environmental Protection Agency, Research Triangle Park, NC. Journal of the Air Pollution Control Association. 32:1210-1215. 1982.
2. Memorandum and attachment from J.H. Margeson, Source Branch, Quality Assurance Division, Environmental Monitoring Systems Laboratory, to The Record, EPA. March 30, 1983. NH3 Interference in Methods 7C and 7D.
3. Margeson, J.H., J.C. Suggs, and M.R. Midgett. Reduction of Nitrate to Nitrite with Cadmium. Anal. Chem. 52:1955-57. 1980.
4. Quality Assurance Handbook for Air Pollution Measurement Systems. Volume III - Stationary Source Specific Methods. U.S. Environmental Protection Agency. Research Triangle Park, NC. Publication No. EPA-600/4-77-027b. August 1977.
5. Margeson, J.H., et al. An Integrated Method for Determining NOX Emissions at Nitric Acid Plants. Analytical Chemistry. 47 (11):1801. 1975.
17.0 Tables, Diagrams, Flowcharts, and Validation Data
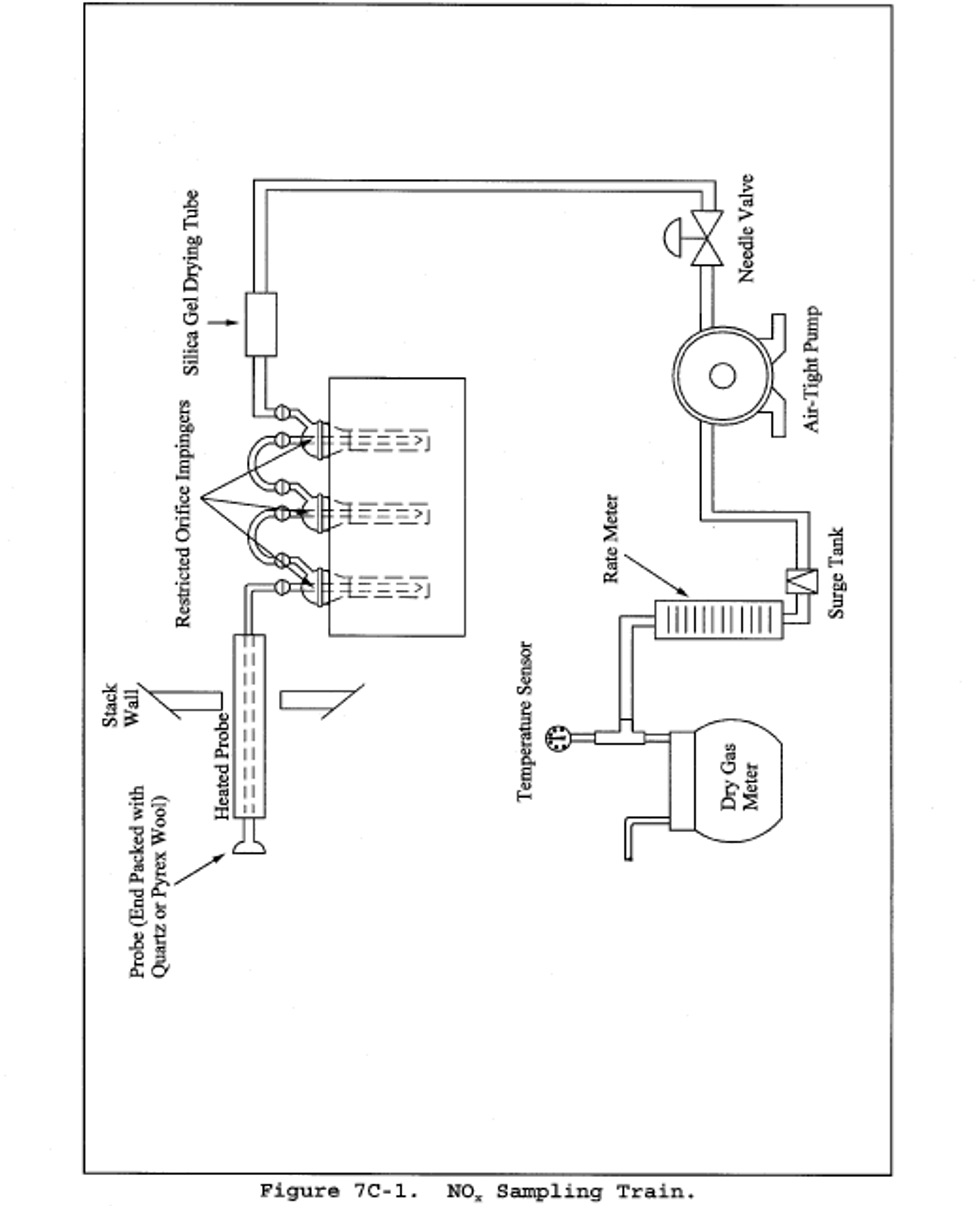
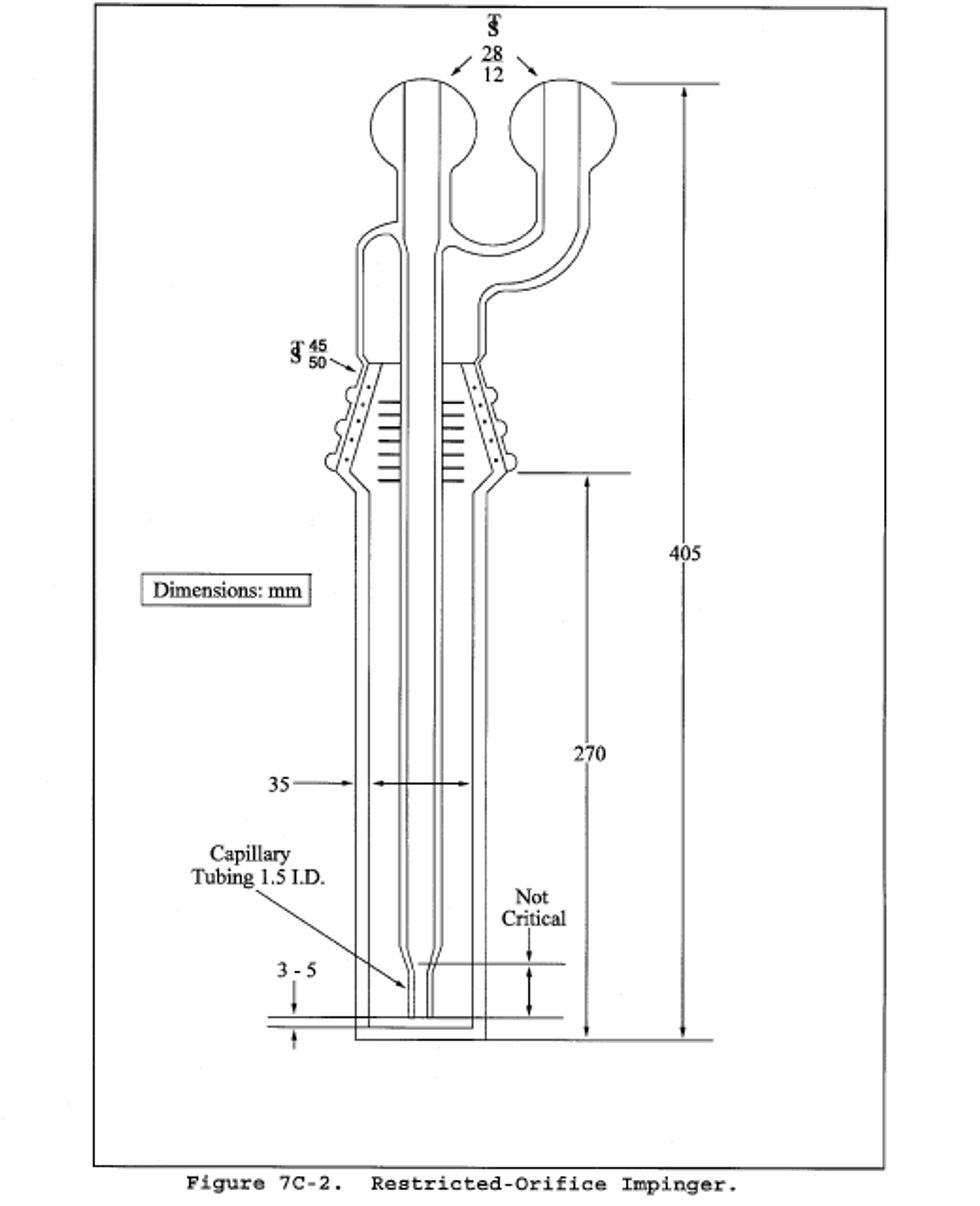
Method 7D - Determination of Nitrogen Oxide Emissions From Stationary Sources (Alkaline-Permanganate/Ion Chromatographic Method)
Note:
This method is not inclusive with respect to specifications (e.g., equipment and supplies) and procedures (e.g., sampling and analytical) essential to its performance. Some material is incorporated by reference from other methods in this part. Therefore, to obtain reliable results, persons using this method should have a thorough knowledge of at least the following additional test methods: Method 1, Method 3, Method 6, Method 7, and Method 7C.
1.0 Scope and Application
1.1 Analytes.
| Analyte | CAS No. | Sensitivity |
|---|---|---|
| Nitrogen oxides (NOX), as NO2, including: | ||
| Nitric oxide (NO) | 10102-43-9 | |
| Nitrogen dioxide (NO2) | 10102-44-0 | 7 ppmv |
1.2 Applicability. This method applies to the measurement of NOX emissions from fossil-fuel fired steam generators, electric utility plants, nitric acid plants, or other sources as specified in the regulations.
1.3 Data Quality Objectives. Adherence to the requirements of this method will enhance the quality of the data obtained from air pollutant sampling methods.
2.0 Summary of Method
An integrated gas sample is extracted from the stack and passed through impingers containing an alkaline-potassium permanganate solution; NOX (NO NO2) emissions are oxidized to NO3 −. Then NO3 − is analyzed by ion chromatography.
3.0 Definitions [Reserved]
4.0 Interferences
Same as in Method 7C, section 4.0.
5.0 Safety
5.1 Disclaimer. This method may involve hazardous materials, operations, and equipment. This test method may not address all of the safety problems associated with its use. It is the responsibility of the user of this test method to establish appropriate safety and health practices and to determine the applicability of regulatory limitations prior to performing this test method.
5.2 Corrosive reagents. The following reagents are hazardous. Personal protective equipment and safe procedures are useful in preventing chemical splashes. If contact occurs, immediately flush with copious amounts of water for at least 15 minutes. Remove clothing under shower and decontaminate. Treat residual chemical burns as thermal burns.
5.2.1 Hydrogen Peroxide (H2O2). Irritating to eyes, skin, nose, and lungs. 30% H2O2 is a strong oxidizing agent; avoid contact with skin, eyes, and combustible material. Wear gloves when handling.
5.2.2 Sodium Hydroxide (NaOH). Causes severe damage to eye tissues and to skin. Inhalation causes irritation to nose, throat, and lungs. Reacts exothermically with limited amounts of water.
5.2.3 Potassium Permanganate (KMnO4). Caustic, strong oxidizer. Avoid bodily contact with.
6.0 Equipment and Supplies
6.1 Sample Collection and Sample Recovery. Same as Method 7C, section 6.1. A schematic of the sampling train used in performing this method is shown in Figure 7C-1 of Method 7C.
6.2 Sample Preparation and Analysis.
6.2.1 Magnetic Stirrer. With 25- by 10-mm Teflon-coated stirring bars.
6.2.2 Filtering Flask. 500-ml capacity with sidearm.
6.2.3 Buchner Funnel. 75-mm ID, with spout equipped with a 13-mm ID by 90-mm long piece of Teflon tubing to minimize possibility of aspirating sample solution during filtration.
6.2.4 Filter Paper. Whatman GF/C, 7.0-cm diameter.
6.2.5 Stirring Rods.
6.2.6 Volumetric Flask. 250-ml.
6.2.7 Pipettes. Class A.
6.2.8 Erlenmeyer Flasks. 250-ml.
6.2.9 Ion Chromatograph. Equipped with an anion separator column to separate NO3 −, H3 suppressor, and necessary auxiliary equipment. Nonsuppressed and other forms of ion chromatography may also be used provided that adequate resolution of NO3 − is obtained. The system must also be able to resolve and detect NO2 −.
7.0 Reagents and Standards
Note:
Unless otherwise indicated, it is intended that all reagents conform to the specifications established by the Committee on Analytical Reagents of the American Chemical Society, where such specifications are available; otherwise, use the best available grade.
7.1 Sample Collection.
7.1.1 Water. Deionized distilled to conform to ASTM specification D 1193-77 or 91 Type 3 (incorporated by reference - see §60.17).
7.1.2 Potassium Permanganate, 4.0 Percent (w/w), Sodium Hydroxide, 2.0 Percent (w/w). Dissolve 40.0 g of KMnO4 and 20.0 g of NaOH in 940 ml of water.
7.2 Sample Preparation and Analysis.
7.2.1 Water. Same as in section 7.1.1.
7.2.2 Hydrogen Peroxide (H2O2), 5 Percent. Dilute 30 percent H2O2 1:5 (v/v) with water.
7.2.3 Blank Solution. Dissolve 2.4 g of KMnO4 and 1.2 g of NaOH in 96 ml of water. Alternatively, dilute 60 ml of KMnO4/NaOH solution to 100 ml.
7.2.4 KNO3 Standard Solution. Dry KNO3 at 110°C for 2 hours, and cool in a desiccator. Accurately weigh 9 to 10 g of KNO3 to within 0.1 mg, dissolve in water, and dilute to 1 liter. Calculate the exact NO3 − concentration using Equation 7D-1 in section 12.2. This solution is stable for 2 months without preservative under laboratory conditions.
7.2.5 Eluent, 0.003 M NaHCO3/0.0024 M Na2CO3. Dissolve 1.008 g NaHCO3 and 1.018 g Na2CO3 in water, and dilute to 4 liters. Other eluents capable of resolving nitrate ion from sulfate and other species present may be used.
8.0 Sample Collection, Preservation, Transport, and Storage.
8.1 Sampling. Same as in Method 7C, section 8.1.
8.2 Sample Recovery. Same as in Method 7C, section 8.2.
8.3 Sample Preparation for Analysis.
Note:
Samples must be analyzed within 28 days of collection.
8.3.1 Note the level of liquid in the sample container, and determine whether any sample was lost during shipment. If a noticeable amount of leakage has occurred, the volume lost can be determined from the difference between initial and final solution levels, and this value can then be used to correct the analytical result. Quantitatively transfer the contents to a 1-liter volumetric flask, and dilute to volume.
8.3.2 Sample preparation can be started 36 hours after collection. This time is necessary to ensure that all NO2 − is converted to NO3 − in the collection solution. Take a 50-ml aliquot of the sample and blank, and transfer to 250-ml Erlenmeyer flasks. Add a magnetic stirring bar. Adjust the stirring rate to as fast a rate as possible without loss of solution. Add 5 percent H2O2 in increments of approximately 5 ml using a 5-ml pipette. When the KMnO4 color appears to have been removed, allow the precipitate to settle, and examine the supernatant liquid. If the liquid is clear, the H2O2 addition is complete. If the KMnO4 color persists, add more H2O2, with stirring, until the supernatant liquid is clear.
Note:
The faster the stirring rate, the less volume of H2O2 that will be required to remove the KMnO4.) Quantitatively transfer the mixture to a Buchner funnel containing GF/C filter paper, and filter the precipitate. The spout of the Buchner funnel should be equipped with a 13-mm ID by 90-mm long piece of Teflon tubing. This modification minimizes the possibility of aspirating sample solution during filtration. Filter the mixture into a 500-ml filtering flask. Wash the solid material four times with water. When filtration is complete, wash the Teflon tubing, quantitatively transfer the filtrate to a 250-ml volumetric flask, and dilute to volume. The sample and blank are now ready for NO3 −analysis.
9.0 Quality Control
| Section | Quality control measure | Effect |
|---|---|---|
| 8.2, 10.1-10.3 | Sampling equipment leak-check and calibration | Ensure accurate measurement of sample volume. |
| 10.4 | Spectrophotometer calibration | Ensure linearity of spectrophotometer response to standards. |
| 11.3 | Spiked sample analysis | Ensure reduction efficiency of column. |
10.0 Calibration and Standardizations
10.1 Dry Gas Meter (DGM) System.
10.1.1 Initial Calibration. Same as in Method 6, section 10.1.1. For detailed instructions on carrying out this calibration, it is suggested that section 3.5.2 of Citation 4 in section 16.0 of Method 7C be consulted.
10.1.2 Post-Test Calibration Check. Same as in Method 6, section 10.1.2.
10.2 Thermometers for DGM and Barometer. Same as in Method 6, sections 10.2 and 10.4, respectively.
10.3 Ion Chromatograph.
10.3.1 Dilute a given volume (1.0 ml or greater) of the KNO3 standard solution to a convenient volume with water, and use this solution to prepare calibration standards. Prepare at least four standards to cover the range of the samples being analyzed. Use pipettes for all additions. Run standards as instructed in section 11.2. Determine peak height or area, and plot the individual values versus concentration in µg NO3 −/ml.
10.3.2 Do not force the curve through zero. Draw a smooth curve through the points. The curve should be linear. With the linear curve, use linear regression to determine the calibration equation.
11.0 Analytical Procedures
11.1 The following chromatographic conditions are recommended: 0.003 M NaHCO3/0.0024 Na2CO3 eluent solution (Section 7.2.5), full scale range, 3 µMHO; sample loop, 0.5 ml; flow rate, 2.5 ml/min. These conditions should give a NO3 − retention time of approximately 15 minutes (Figure 7D-1).
11.2 Establish a stable baseline. Inject a sample of water, and determine whether any NO3 − appears in the chromatogram. If NO3 − is present, repeat the water load/injection procedure approximately five times; then re-inject a water sample and observe the chromatogram. When no NO3 − is present, the instrument is ready for use. Inject calibration standards. Then inject samples and a blank. Repeat the injection of the calibration standards (to compensate for any drift in response of the instrument). Measure the NO3 − peak height or peak area, and determine the sample concentration from the calibration curve.
12.0 Data Analysis and Calculations
Carry out calculations, retaining at least one extra significant figure beyond that of the acquired data. Round off figures after final calculation.
12.1 Nomenclature. Same as in Method 7C, section 12.1.
12.2 NO3 − concentration. Calculate the NO3 − concentration in the KNO3 standard solution (see section 7.2.4) using the following equation:
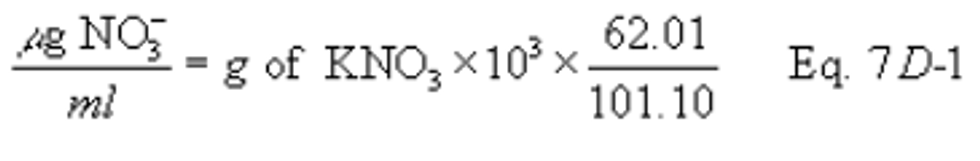
12.3 Sample Volume, Dry Basis, Corrected to Standard Conditions. Same as in Method 7C, section 12.4.
12.4 Total µg NO2 Per Sample.
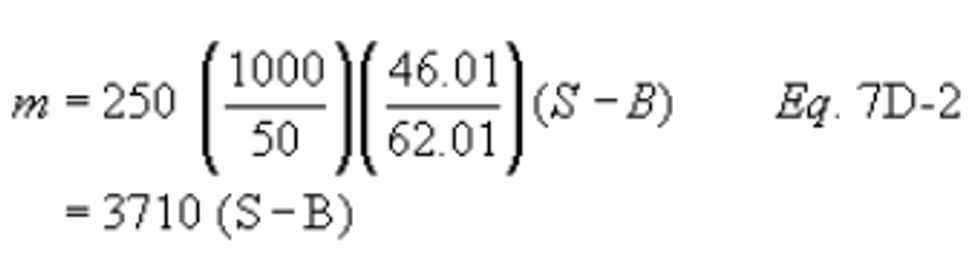
Where:
250 = Volume of prepared sample, ml.
1000 = Total volume of KMnO4 solution, ml.
50 = Aliquot of KMnO4/NaOH solution, ml.
46.01 = Molecular weight of NO3 −.
62.01 = Molecular weight of NO3 −.
12.5 Sample Concentration. Same as in Method 7C, section 12.7.
13.0 Method Performance
13.1 Precision. The intra-laboratory relative standard deviation for a single measurement is approximately 6 percent at 200 to 270 ppm NOX.
13.2 Bias. The method does not exhibit any bias relative to Method 7.
13.3 Range. The lower detectable limit is similar to that of Method 7C. No upper limit has been established; however, when using the recommended sampling conditions, the method has been found to collect NOX emissions quantitatively up to 1782 mg NOX/m 3, as NO2 (932 ppm NOX).
14.0 Pollution Prevention [Reserved]
15.0 Waste Management [Reserved]
16.0 References
Same as Method 7C, section 16.0, References 1, 2, 4, and 5.
17.0 Tables, Diagrams, Flowcharts, and Validation Data
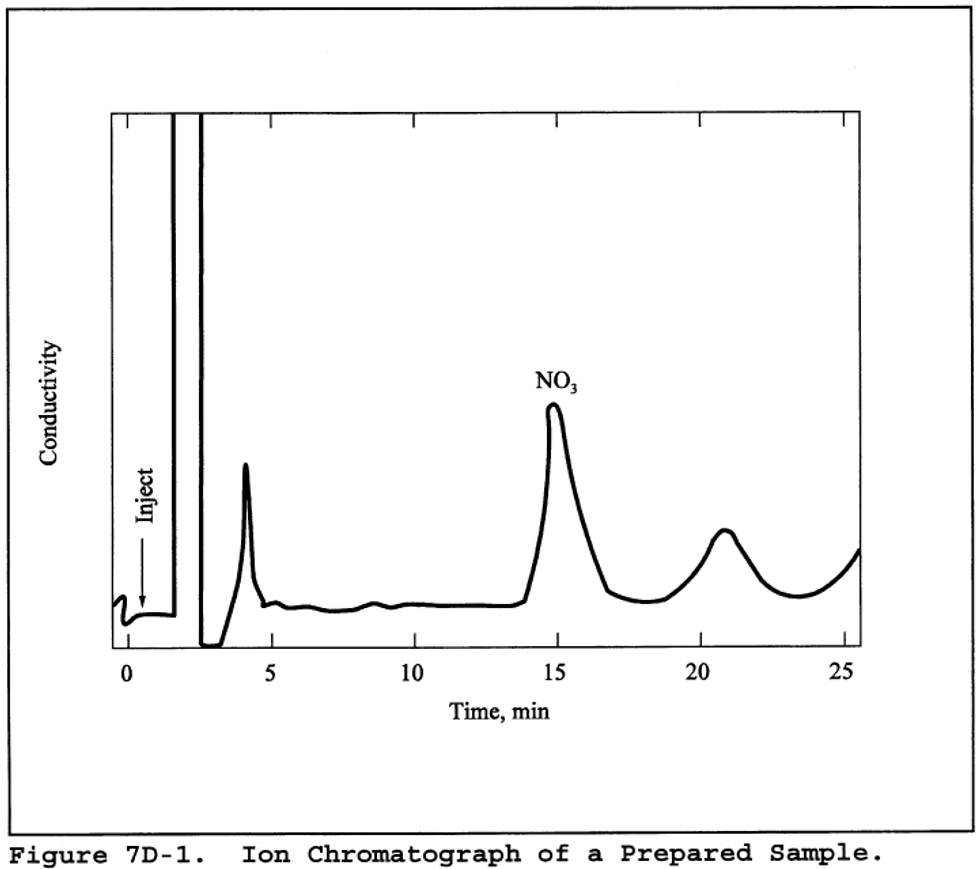
Method 7E - Determination of Nitrogen Oxides Emissions From Stationary Sources (Instrumental Analyzer Procedure)
1.0 Scope and Application
What is Method 7E?
Method 7E is a procedure for measuring nitrogen oxides (NOX) in stationary source emissions using a continuous instrumental analyzer. Quality assurance and quality control requirements are included to assure that you, the tester, collect data of known quality. You must document your adherence to these specific requirements for equipment, supplies, sample collection and analysis, calculations, and data analysis. This method does not completely describe all equipment, supplies, and sampling and analytical procedures you will need but refers to other methods for some of the details. Therefore, to obtain reliable results, you should also have a thorough knowledge of these additional test methods which are found in appendix A to this part:
(a) Method 1 - Sample and Velocity Traverses for Stationary Sources.
(b) Method 4 - Determination of Moisture Content in Stack Gases.
1.1 Analytes. What does this method determine? This method measures the concentration of nitrogen oxides as NO2.
| Analyte | CAS No. | Sensitivity |
|---|---|---|
| Nitric oxide (NO) | 10102-43-9 | Typically <2% of |
| Nitrogen dioxide (NO2) | 10102-44-0 | Calibration Span. |
1.2 Applicability. When is this method required? The use of Method 7E may be required by specific New Source Performance Standards, Clean Air Marketing rules, State Implementation Plans, and permits where measurement of NOX concentrations in stationary source emissions is required, either to determine compliance with an applicable emissions standard or to conduct performance testing of a continuous monitoring system (CEMS). Other regulations may also require the use of Method 7E.
1.3 Data Quality Objectives (DQO). How good must my collected data be? Method 7E is designed to provide high-quality data for determining compliance with Federal and State emission standards and for relative accuracy testing of CEMS. In these and other applications, the principal objective is to ensure the accuracy of the data at the actual emission levels encountered. To meet this objective, the use of EPA traceability protocol calibration gases and measurement system performance tests are required.
1.4 Data Quality Assessment for Low Emitters. Is performance relief granted when testing low-emission units? Yes. For low-emitting sources, there are alternative performance specifications for analyzer calibration error, system bias, drift, and response time. Also, the alternative dynamic spiking procedure in section 16 may provide performance relief for certain low-emitting units.
2.0 Summary of Method
In this method, a sample of the effluent gas is continuously sampled and conveyed to the analyzer for measuring the concentration of NOX. You may measure NO and NO2 separately or simultaneously together but, for the purposes of this method, NOX is the sum of NO and NO2. You must meet the performance requirements of this method to validate your data.
3.0 Definitions
3.1 Analyzer Calibration Error, for non-dilution systems, means the difference between the manufacturer certified concentration of a calibration gas and the measured concentration of the same gas when it is introduced into the analyzer in direct calibration mode.
3.2 Calibration Curve means the relationship between an analyzer's response to the injection of a series of calibration gases and the actual concentrations of those gases.
3.3 Calibration Gas means the gas mixture containing NOX at a known concentration and produced and certified in accordance with “EPA Traceability Protocol for Assay and Certification of Gaseous Calibration Standards,” September 1997, as amended August 25, 1999, EPA-600/R-97/121 or more recent updates. The tests for analyzer calibration error, drift, and system bias require the use of calibration gas prepared according to this protocol. If a zero gas is used for the low-level gas, it must meet the requirements under the definition for “zero air material” in 40 CFR 72.2 in place of being prepared by the traceability protocol.
3.3.1 Low-Level Gas means a calibration gas with a concentration that is less than 20 percent of the calibration span and may be a zero gas.
3.3.2 Mid-Level Gas means a calibration gas with a concentration that is 40 to 60 percent of the calibration span.
3.3.3 High-Level Gas means a calibration gas with a concentration that is equal to the calibration span.
3.4 Calibration Span means the upper limit of the analyzer's calibration that is set by the choice of high-level calibration gas. No valid run average concentration may exceed the calibration span. To the extent practicable, the measured emissions should be between 20 to 100 percent of the selected calibration span. This may not be practicable in some cases of low-concentration measurements or testing for compliance with an emission limit when emissions are substantially less than the limit. In such cases, calibration spans that are practicable to achieving the data quality objectives without being excessively high should be chosen.
3.5 Centroidal Area means the central area of the stack or duct that is no greater than 1 percent of the stack or duct cross section. This area has the same geometric shape as the stack or duct.
3.6 Converter Efficiency Gas means a calibration gas with a known NO or NO2 concentration and of Traceability Protocol quality.
3.7 Data Recorder means the equipment that permanently records the concentrations reported by the analyzer.
3.8 Direct Calibration Mode means introducing the calibration gases directly into the analyzer (or into the assembled measurement system at a point downstream of all sample conditioning equipment) according to manufacturer's recommended calibration procedure. This mode of calibration applies to non-dilution-type measurement systems.
3.9 Drift means the difference between the pre- and post-run system bias (or system calibration error) checks at a specific calibration gas concentration level (i.e. low-, mid- or high-).
3.10 Gas Analyzer means the equipment that senses the gas being measured and generates an output proportional to its concentration.
3.11 Interference Check means the test to detect analyzer responses to compounds other than the compound of interest, usually a gas present in the measured gas stream, that is not adequately accounted for in the calibration procedure and may cause measurement bias.
3.12 Low-Concentration Analyzer means any analyzer that operates with a calibration span of 20 ppm NOX or lower. Each analyzer model used routinely to measure low NOX concentrations must pass a manufacturer's stability test (MST). An MST subjects the analyzer to a range of line voltages and temperatures that reflect potential field conditions to demonstrate its stability following procedures similar to those provided in 40 CFR 53.23. Ambient-level analyzers are exempt from the MST requirements of section 16.3. A copy of this information must be included in each test report. Table 7E-5 lists the criteria to be met.
3.13 Measurement System means all of the equipment used to determine the NOX concentration. The measurement system comprises six major subsystems: Sample acquisition, sample transport, sample conditioning, calibration gas manifold, gas analyzer, and data recorder.
3.14 Response Time means the time it takes the measurement system to respond to a change in gas concentration occurring at the sampling point when the system is operating normally at its target sample flow rate or dilution ratio.
3.15 Run means a series of gas samples taken successively from the stack or duct. A test normally consists of a specific number of runs.
3.16 System Bias means the difference between a calibration gas measured in direct calibration mode and in system calibration mode. System bias is determined before and after each run at the low- and mid- or high-concentration levels. For dilution-type systems, pre- and post-run system calibration error is measured rather than system bias.
3.17 System Calibration Error applies to dilution-type systems and means the difference between the measured concentration of low-, mid-, or high-level calibration gas and the certified concentration for each gas when introduced in system calibration mode. For dilution-type systems, a 3-point system calibration error test is conducted in lieu of the analyzer calibration error test, and 2-point system calibration error tests are conducted in lieu of system bias tests.
3.18 System Calibration Mode means introducing the calibration gases into the measurement system at the probe, upstream of the filter and all sample conditioning components.
3.19 Test refers to the series of runs required by the applicable regulation.
4.0 Interferences
Note that interferences may vary among instruments and that instrument-specific interferences must be evaluated through the interference test.
5.0 Safety
What safety measures should I consider when using this method? This method may require you to work with hazardous materials and in hazardous conditions. We encourage you to establish safety procedures before using the method. Among other precautions, you should become familiar with the safety recommendations in the gas analyzer user's manual. Occupational Safety and Health Administration (OSHA) regulations concerning cylinder and noxious gases may apply. Nitric oxide and NO2 are toxic and dangerous gases. Nitric oxide is immediately converted to NO2 upon reaction with air. Nitrogen dioxide is a highly poisonous and insidious gas. Inflammation of the lungs from exposure may cause only slight pain or pass unnoticed, but the resulting edema several days later may cause death. A concentration of 100 ppm is dangerous for even a short exposure, and 200 ppm may be fatal. Calibration gases must be handled with utmost care and with adequate ventilation. Emission-level exposure to these gases should be avoided.
6.0 Equipment and Supplies
The performance criteria in this method will be met or exceeded if you are properly using equipment designed for this application.
6.1 What do I need for the measurement system? You may use any equipment and supplies meeting the following specifications:
(1) Sampling system components that are not evaluated in the system bias or system calibration error test must be glass, Teflon, or stainless steel. Other materials are potentially acceptable, subject to approval by the Administrator.
(2) The interference, calibration error, and system bias criteria must be met.
(3) Sample flow rate must be maintained within 10 percent of the flow rate at which the system response time was measured.
(4) All system components (excluding sample conditioning components, if used) must maintain the sample temperature above the moisture dew point. Ensure minimal contact between any condensate and the sample gas. Section 6.2 provides example equipment specifications for a NOX measurement system. Figure 7E-1 is a diagram of an example dry-basis measurement system that is likely to meet the method requirements and is provided as guidance. For wet-basis systems, you may use alternative equipment and supplies as needed (some of which are described in Section 6.2), provided that the measurement system meets the applicable performance specifications of this method.
6.2 Measurement System Components
6.2.1 Sample Probe. Glass, stainless steel, or other approved material, of sufficient length to traverse the sample points.
6.2.2 Particulate Filter. An in-stack or out-of-stack filter. The filter must be made of material that is non-reactive to the gas being sampled. The filter media for out-of-stack filters must be included in the system bias test. The particulate filter requirement may be waived in applications where no significant particulate matter is expected (e.g., for emission testing of a combustion turbine firing natural gas).
6.2.3 Sample Line. The sample line from the probe to the conditioning system/sample pump should be made of Teflon or other material that does not absorb or otherwise alter the sample gas. For a dry-basis measurement system (as shown in Figure 7E-1), the temperature of the sample line must be maintained at a sufficiently high level to prevent condensation before the sample conditioning components. For wet-basis measurement systems, the temperature of the sample line must be maintained at a sufficiently high level to prevent condensation before the analyzer.
6.2.4 Conditioning Equipment. For dry basis measurements, a condenser, dryer or other suitable device is required to remove moisture continuously from the sample gas. Any equipment needed to heat the probe or sample line to avoid condensation prior to the sample conditioning component is also required.
For wet basis systems, you must keep the sample above its dew point either by: (1) Heating the sample line and all sample transport components up to the inlet of the analyzer (and, for hot-wet extractive systems, also heating the analyzer) or (2) by diluting the sample prior to analysis using a dilution probe system. The components required to do either of the above are considered to be conditioning equipment.
6.2.5 Sampling Pump. For systems similar to the one shown in Figure 7E-1, a leak-free pump is needed to pull the sample gas through the system at a flow rate sufficient to minimize the response time of the measurement system. The pump may be constructed of any material that is non-reactive to the gas being sampled. For dilution-type measurement systems, an ejector pump (eductor) is used to create a vacuum that draws the sample through a critical orifice at a constant rate.
6.2.6 Calibration Gas Manifold. Prepare a system to allow the introduction of calibration gases either directly to the gas analyzer in direct calibration mode or into the measurement system, at the probe, in system calibration mode, or both, depending upon the type of system used. In system calibration mode, the system should be able to flood the sampling probe and vent excess gas. Alternatively, calibration gases may be introduced at the calibration valve following the probe. Maintain a constant pressure in the gas manifold. For in-stack dilution-type systems, a gas dilution subsystem is required to transport large volumes of purified air to the sample probe and a probe controller is needed to maintain the proper dilution ratio.
6.2.7 Sample Gas Manifold. For the type of system shown in Figure 7E-1, the sample gas manifold diverts a portion of the sample to the analyzer, delivering the remainder to the by-pass discharge vent. The manifold should also be able to introduce calibration gases directly to the analyzer (except for dilution-type systems). The manifold must be made of material that is non-reactive to the gas sampled or the calibration gas and be configured to safely discharge the bypass gas.
6.2.8 NO X Analyzer. An instrument that continuously measures NOX in the gas stream and meets the applicable specifications in section 13.0. An analyzer that operates on the principle of chemiluminescence with an NO2 to NO converter is one example of an analyzer that has been used successfully in the past. Analyzers operating on other principles may also be used provided the performance criteria in section 13.0 are met.
6.2.8.1 Dual Range Analyzers. For certain applications, a wide range of gas concentrations may be encountered, necessitating the use of two measurement ranges. Dual-range analyzers are readily available for these applications. These analyzers are often equipped with automated range-switching capability, so that when readings exceed the full-scale of the low measurement range, they are recorded on the high range. As an alternative to using a dual-range analyzer, you may use two segments of a single, large measurement scale to serve as the low and high ranges. In all cases, when two ranges are used, you must quality-assure both ranges using the proper sets of calibration gases. You must also meet the interference, calibration error, system bias, and drift checks. However, we caution that when you use two segments of a large measurement scale for dual range purposes, it may be difficult to meet the performance specifications on the low range due to signal-to-noise ratio considerations.
6.2.8.2 Low Concentration Analyzer. When an analyzer is routinely calibrated with a calibration span of 20 ppmv or less, the manufacturer's stability test (MST) is required. See Table 7E-5 for test parameters.
6.2.9 Data Recording. A strip chart recorder, computerized data acquisition system, digital recorder, or data logger for recording measurement data may be used.
7.0 Reagents and Standards
7.1 Calibration Gas. What calibration gases do I need? Your calibration gas must be NO in N2 and certified (or recertified) within an uncertainty of 2.0 percent in accordance with “EPA Traceability Protocol for Assay and Certification of Gaseous Calibration Standards” September 1997, as amended August 25, 1999, EPA-600/R-97/121. Blended gases meeting the Traceability Protocol are allowed if the additional gas components are shown not to interfere with the analysis. If a zero gas is used for the low-level gas, it must meet the requirements under the definition for “zero air material” in 40 CFR 72.2. The calibration gas must not be used after its expiration date. Except for applications under part 75 of this chapter, it is acceptable to prepare calibration gas mixtures from EPA Traceability Protocol gases in accordance with Method 205 in appendix M to part 51 of this chapter. For part 75 applications, the use of Method 205 is subject to the approval of the Administrator. The goal and recommendation for selecting calibration gases is to bracket the sample concentrations. The following calibration gas concentrations are required:
7.1.1 High-Level Gas. This concentration is chosen to set the calibration span as defined in Section 3.4.
7.1.2 Mid-Level Gas. 40 to 60 percent of the calibration span.
7.1.3 Low-Level Gas. Less than 20 percent of the calibration span.
7.1.4 Converter Efficiency Gas. What reagents do I need for the converter efficiency test? The converter efficiency gas is a manufacturer-certified gas with a concentration sufficient to show NO2 conversion at the concentrations encountered in the source. A test gas concentration in the 40 to 60 ppm range is suggested, but other concentrations may be more appropriate to specific sources. For the test described in section 8.2.4.1, NO2 is required. For the alternative converter efficiency tests in section 16.2, NO is required.
7.2 Interference Check. What reagents do I need for the interference check? Use the appropriate test gases listed in Table 7E-3 or others not listed that can potentially interfere (as indicated by the test facility type, instrument manufacturer, etc.) to conduct the interference check. These gases should be manufacturer certified but do not have to be prepared by the EPA traceability protocol.
8.0 Sample Collection, Preservation, Storage, and Transport
Emission Test Procedure
Since you are allowed to choose different options to comply with some of the performance criteria, it is your responsibility to identify the specific options you have chosen, to document that the performance criteria for that option have been met, and to identify any deviations from the method.
8.1 What sampling site and sampling points do I select?
8.1.1 Unless otherwise specified in an applicable regulation or by the Administrator, when this method is used to determine compliance with an emission standard, conduct a stratification test as described in section 8.1.2 to determine the sampling traverse points to be used. For performance testing of continuous emission monitoring systems, follow the sampling site selection and traverse point layout procedures described in the appropriate performance specification or applicable regulation (e.g., Performance Specification 2 in appendix B to this part).
8.1.2 Determination of Stratification. Perform a stratification test at each test site to determine the appropriate number of sample traverse points. If testing for multiple pollutants or diluents at the same site, a stratification test using only one pollutant or diluent satisfies this requirement. A stratification test is not required for small stacks that are less than 4 inches in diameter. To test for stratification, use a probe of appropriate length to measure the NOX (or pollutant of interest) concentration at 12 traverse points located according to Table 1-1 or Table 1-2 of Method 1. Alternatively, you may measure at three points on a line passing through the centroidal area. Space the three points at 16.7, 50.0, and 83.3 percent of the measurement line. Sample for a minimum of twice the system response time (see section 8.2.6) at each traverse point. Calculate the individual point and mean NOX concentrations. If the concentration at each traverse point differs from the mean concentration for all traverse points by no more than: ±5.0 percent of the mean concentration; or ±0.5 ppm (whichever is less restrictive), the gas stream is considered unstratified, and you may collect samples from a single point that most closely matches the mean. If the 5.0 percent or 0.5 ppm criterion is not met, but the concentration at each traverse point differs from the mean concentration for all traverse points by not more than: ±10.0 percent of the mean concentration; or ±1.0 ppm (whichever is less restrictive), the gas stream is considered to be minimally stratified and you may take samples from three points. Space the three points at 16.7, 50.0, and 83.3 percent of the measurement line. Alternatively, if a 12-point stratification test was performed and the emissions were shown to be minimally stratified (all points within ± 10.0 percent of their mean or within ±1.0 ppm), and if the stack diameter (or equivalent diameter, for a rectangular stack or duct) is greater than 2.4 meters (7.8 ft), then you may use 3-point sampling and locate the three points along the measurement line exhibiting the highest average concentration during the stratification test at 0.4, 1.2 and 2.0 meters from the stack or duct wall. If the gas stream is found to be stratified because the 10.0 percent or 1.0 ppm criterion for a 3-point test is not met, locate 12 traverse points for the test in accordance with Table 1-1 or Table 1-2 of Method 1.
8.2 Initial Measurement System Performance Tests. What initial performance criteria must my system meet before I begin collecting samples? Before measuring emissions, perform the following procedures:
(a) Calibration gas verification,
(b) Measurement system preparation,
(c) Calibration error test,
(d) NO2 to NO conversion efficiency test, if applicable,
(e) System bias check,
(f) System response time test, and
(g) Interference check
8.2.1 Calibration Gas Verification. How must I verify the concentrations of my calibration gases? Obtain a certificate from the gas manufacturer documenting the quality of the gas. Confirm that the manufacturer certification is complete and current. Ensure that your calibration gas certifications have not expired. This documentation should be available on-site for inspection. To the extent practicable, select a high-level gas concentration that will result in the measured emissions being between 20 and 100 percent of the calibration span.
8.2.2 Measurement System Preparation. How do I prepare my measurement system? Assemble, prepare, and precondition the measurement system according to your standard operating procedure. Adjust the system to achieve the correct sampling rate or dilution ratio (as applicable).
8.2.3 Calibration Error Test. How do I confirm my analyzer calibration is correct? After you have assembled, prepared and calibrated your sampling system and analyzer, you must conduct a 3-point analyzer calibration error test (or a 3-point system calibration error test for dilution systems) before the first run and again after any failed system bias test (or 2-point system calibration error test for dilution systems) or failed drift test. Introduce the low-, mid-, and high-level calibration gases sequentially. For non-dilution-type measurement systems, introduce the gases in direct calibration mode. For dilution-type measurement systems, introduce the gases in system calibration mode.
(1) For non-dilution systems, you may adjust the system to maintain the correct flow rate at the analyzer during the test, but you may not make adjustments for any other purpose. For dilution systems, you must operate the measurement system at the appropriate dilution ratio during all system calibration error checks, and may make only the adjustments necessary to maintain the proper ratio.
(2) Record the analyzer's response to each calibration gas on a form similar to Table 7E-1. For each calibration gas, calculate the analyzer calibration error using Equation 7E-1 in section 12.2 or the system calibration error using Equation 7E-3 in section 12.4 (as applicable). The calibration error specification in section 13.1 must be met for the low-, mid-, and high-level gases. If the calibration error specification is not met, take corrective action and repeat the test until an acceptable 3-point calibration is achieved.
8.2.4 NO 2 to NO Conversion Efficiency Test. Before or after each field test, you must conduct an NO2 to NO conversion efficiency test if your system converts NO2 to NO before analyzing for NOX. You may risk testing multiple facilities before performing this test provided you pass this test at the conclusion of the final facility test. A failed final conversion efficiency test in this case will invalidate all tests performed subsequent to the test in which the converter efficiency test was passed. Follow the procedures in section 8.2.4.1, or 8.2.4.2. If desired, the converter efficiency factor derived from this test may be used to correct the test results for converter efficiency if the NO2 fraction in the measured test gas is known. Use Equation 7E-8 in section 12.8 for this correction.
8.2.4.1 Introduce NO2 converter efficiency gas to the analyzer in direct calibration mode and record the NOX concentration displayed by the analyzer. Calculate the converter efficiency using Equation 7E-7 in section 12.7. The specification for converter efficiency in section 13.5 must be met. The user is cautioned that state-of-the-art NO2 calibration gases may have limited shelf lives, and this could affect the ability to pass the 90-percent conversion efficiency requirement.
8.2.4.2 Alternatively, either of the procedures for determining conversion efficiency using NO in section 16.2 may be used.
8.2.5 Initial System Bias and System Calibration Error Checks. Before sampling begins, determine whether the high-level or mid-level calibration gas best approximates the emissions and use it as the upscale gas. Introduce the upscale gas at the probe upstream of all sample conditioning components in system calibration mode. Record the time it takes for the measured concentration to increase to a value that is at least 95 percent or within 0.5 ppm (whichever is less restrictive) of a stable response for both the low-level and upscale gases. Continue to observe the gas concentration reading until it has reached a final, stable value. Record this value on a form similar to Table 7E-2.
(1) Next, introduce the low-level gas in system calibration mode and record the time required for the concentration response to decrease to a value that is within 5.0 percent or 0.5 ppm (whichever is less restrictive) of the certified low-range gas concentration. If the low-level gas is a zero gas, use the procedures described above and observe the change in concentration until the response is 0.5 ppm or 5.0 percent of the upscale gas concentration (whichever is less restrictive).
(2) Continue to observe the low-level gas reading until it has reached a final, stable value and record the result on a form similar to Table 7E-2. Operate the measurement system at the normal sampling rate during all system bias checks. Make only the adjustments necessary to achieve proper calibration gas flow rates at the analyzer.
(3) From these data, calculate the measurement system response time (see section 8.2.6) and then calculate the initial system bias using Equation 7E-2 in section 12.3. For dilution systems, calculate the system calibration error in lieu of system bias using equation 7E-3 in section 12.4. See section 13.2 for acceptable performance criteria for system bias and system calibration error. If the initial system bias (or system calibration error) specification is not met, take corrective action. Then, you must repeat the applicable calibration error test from section 8.2.3 and the initial system bias (or 2-point system calibration error) check until acceptable results are achieved, after which you may begin sampling.
(Note: For dilution-type systems, data from the 3-point system calibration error test described in section 8.2.3 may be used to meet the initial 2-point system calibration error test requirement of this section, if the calibration gases were injected as described in this section, and if response time data were recorded).
8.2.6 Measurement System Response Time. As described in section 8.2.5, you must determine the measurement system response time during the initial system bias (or 2-point system calibration error) check. Observe the times required to achieve 95 percent of a stable response for both the low-level and upscale gases. The longer interval is the response time.
8.2.7 Interference Check. Conduct an interference response test of the gas analyzer prior to its initial use in the field. If you have multiple analyzers of the same make and model, you need only perform this alternative interference check on one analyzer. You may also meet the interference check requirement if the instrument manufacturer performs this or a similar check on an analyzer of the same make and model of the analyzer that you use and provides you with documented results.
(1) You may introduce the appropriate interference test gases (that are potentially encountered during a test; see examples in Table 7E-3) into the analyzer separately or as mixtures. Test the analyzer with the interference gas alone at the highest concentration expected at a test source and again with the interference gas and NOX at a representative NOX test concentration. For analyzers measuring NOX greater than 20 ppm, use a calibration gas with a NOX concentration of 80 to 100 ppm and set this concentration equal to the calibration span. For analyzers measuring less than 20 ppm NOX, select an NO concentration for the calibration span that reflects the emission levels at the sources to be tested, and perform the interference check at that level. Measure the total interference response of the analyzer to these gases in ppmv. Record the responses and determine the interference using Table 7E-4. The specification in section 13.4 must be met.
(2) A copy of this data, including the date completed and signed certification, must be available for inspection at the test site and included with each test report. This interference test is valid for the life of the instrument unless major analytical components (e.g., the detector) are replaced with different model parts. If major components are replaced with different model parts, the interference gas check must be repeated before returning the analyzer to service. If major components are replaced, the interference gas check must be repeated before returning the analyzer to service. The tester must ensure that any specific technology, equipment, or procedures that are intended to remove interference effects are operating properly during testing.
8.3 Dilution-Type Systems - Special Considerations. When a dilution-type measurement system is used, there are three important considerations that must be taken into account to ensure the quality of the emissions data. First, the critical orifice size and dilution ratio must be selected properly so that the sample dew point will be below the sample line and analyzer temperatures. Second, a high-quality, accurate probe controller must be used to maintain the dilution ratio during the test. The probe controller should be capable of monitoring the dilution air pressure, eductor vacuum, and sample flow rates. Third, differences between the molecular weight of calibration gas mixtures and the stack gas molecular weight must be addressed because these can affect the dilution ratio and introduce measurement bias.
8.4 Sample Collection.
(1) Position the probe at the first sampling point. Purge the system for at least two times the response time before recording any data. Then, traverse all required sampling points, sampling at each point for an equal length of time and maintaining the appropriate sample flow rate or dilution ratio (as applicable). You must record at least one valid data point per minute during the test run.
(2) Each time the probe is removed from the stack and replaced, you must recondition the sampling system for at least two times the system response time prior to your next recording. If the average of any run exceeds the calibration span value, that run is invalid.
(3) You may satisfy the multipoint traverse requirement by sampling sequentially using a single-hole probe or a multi-hole probe designed to sample at the prescribed points with a flow within 10 percent of mean flow rate. Notwithstanding, for applications under part 75 of this chapter, the use of multi-hole probes is subject to the approval of the Administrator.
8.5 Post-Run System Bias Check and Drift Assessment.
How do I confirm that each sample I collect is valid? After each run, repeat the system bias check or 2-point system calibration error check (for dilution systems) to validate the run. Do not make adjustments to the measurement system (other than to maintain the target sampling rate or dilution ratio) between the end of the run and the completion of the post-run system bias or system calibration error check. Note that for all post-run system bias or 2-point system calibration error checks, you may inject the low-level gas first and the upscale gas last, or vice-versa. If conducting a relative accuracy test or relative accuracy test audit, consisting of nine runs or more, you may risk sampling for up to three runs before performing the post-run bias or system calibration error check provided you pass this test at the conclusion of the group of three runs. A failed post-run bias or system calibration error check in this case will invalidate all runs subsequent to the last passed check. When conducting a performance or compliance test, you must perform a post-run system bias or system calibration error check after each individual test run.
(1) If you do not pass the post-run system bias (or system calibration error) check, then the run is invalid. You must diagnose and fix the problem and pass another calibration error test (Section 8.2.3) and system bias (or 2-point system calibration error) check (Section 8.2.5) before repeating the run. Record the system bias (or system calibration error) results on a form similar to Table 7E-2.
(2) After each run, calculate the low-level and upscale drift, using Equation 7E-4 in section 12.5. If the post-run low- and upscale bias (or 2-point system calibration error) checks are passed, but the low-or upscale drift exceeds the specification in section 13.3, the run data are valid, but a 3-point calibration error test and a system bias (or 2-point system calibration error) check must be performed and passed before any more test runs are done.
(3) For dilution systems, data from a 3-point system calibration error test may be used to met the pre-run 2-point system calibration error requirement for the first run in a test sequence. Also, the post-run bias (or 2-point calibration error) check data may be used as the pre-run data for the next run in the test sequence at the discretion of the tester.
8.6 Alternative Interference and System Bias Checks (Dynamic Spike Procedure). If I want to use the dynamic spike procedure to validate my data, what procedure should I follow? Except for applications under part 75 of this chapter, you may use the dynamic spiking procedure and requirements provided in section 16.1 during each test as an alternative to the interference check and the pre- and post-run system bias checks. The calibration error test is still required under this option. Use of the dynamic spiking procedure for Part 75 applications is subject to the approval of the Administrator.
8.7 Moisture correction. You must determine the moisture content of the flue gas and correct the measured gas concentrations to a dry basis using Method 4 or other appropriate methods, subject to the approval of the Administrator, when the moisture basis (wet or dry) of the measurements made with this method is different from the moisture basis of either: (1) The applicable emissions limit; or (2) the CEMS being evaluated for relative accuracy. Moisture correction is also required if the applicable limit is in lb/mmBtu and the moisture basis of the Method 7E NOX analyzer is different from the moisture basis of the Method 3A diluent gas (CO2 or O2) analyzer.
9.0 Quality Control
What quality control measures must I take?
The following table is a summary of the mandatory, suggested, and alternative quality assurance and quality control measures and the associated frequency and acceptance criteria. All of the QC data, along with the sample run data, must be documented and included in the test report.
| Status | Process or element | QA/QC specification | Acceptance criteria | Checking frequency |
|---|---|---|---|---|
| S | Identify Data User | Regulatory Agency or other primary end user of data | Before designing test. | |
| S | Analyzer Design | Analyzer resolution or sensitivity | <2.0% of full-scale range | Manufacturer design. |
| M | Interference gas check | Sum of responses ≤2.5% of calibration span Alternatively, sum of responses: | ||
| ≤0.5 ppmv for calibration spans of 5 to 10 ppmv | ||||
| ≤0.2 ppmv for calibration spans <5 ppmv | ||||
| See Table 7E-3 | ||||
| M | Calibration Gases | Traceability protocol (G1, G2) | Valid certificate required Uncertainty ≤2.0% of tag value | |
| M | High-level gas | Equal to the calibration span | Each test. | |
| M | Mid-level gas | 40 to 60% of calibration span | Each test. | |
| M | Low-level gas | <20% of calibration span | Each test. | |
| S | Data Recorder Design | Data resolution | ≤0.5% of full-scale range | Manufacturer design. |
| S | Sample Extraction | Probe material | SS or quartz if stack >500°F | East test. |
| M | Sample Extraction | Probe, filter and sample line temperature | For dry-basis analyzers, keep sample above the dew point by heating, prior to sample conditioning | Each run. |
| For wet-basis analyzers, keep sample above dew point at all times, by heating or dilution | ||||
| S | Sample Extraction | Calibration valve material | SS | Each test. |
| S | Sample Extraction | Sample pump material | Inert to sample constituents | Each test. |
| S | Sample Extraction | Manifolding material | Inert to sample constituents | Each test. |
| S | Moisture Removal | Equipment efficiency | <5% target compound removal | Verified through system bias check. |
| S | Particulate Removal | Filter inertness | Pass system bias check | Each bias check. |
| M | Analyzer & Calibration Gas Performance | Analyzer calibration error (of 3-point system calibration error for dilution systems) | Within ±2.0 percent of the calibration span of the analyzer for the low-, mid-, and high-level calibration gases | Before initial run and after a failed system bias test or drift test. |
| Alternative specification: ≤0.5 ppmv absolute difference | ||||
| M | System Performance | System bias (or pre- and post-run 2-point system calibration error for dilution (Systems) | Within ±5.0% of the analyzer calibration span for low-sacle and upscale calibration gases | Before and after each run. |
| Alternative specification: ≤0.5 ppmv absolute difference | ||||
| M | System Performance | System response time | Determines minimum sampling time per point | During initial sampling system bias test. |
| M | System Performance | Drift | ≤3.0% of calibration span for low-level and mid- or high-level gases | After each test run. |
| Alternative specification: ≤0.5 ppmv absolute difference | ||||
| M | System Performance | NO2-NO conversion efficiency | ≥90% of certified test gas concentration | Before or after each test. |
| M | System Performance | Purge time | ≥2 times system response time | Before starting the first run and when probe is removed from and re-inserted into the stack. |
| M | System Performance | Minimum sample time at each point | Two times the system response time | Each sample point. |
| M | System Performance | Stable sample flow rate (surrogate for maintaining system response time) | Within 10% of flow rate established during system response time check | Each run. |
| M | Sample Point Selection | Stratification test | All points within: | Prior to first run. |
| ±5% of mean for 1-point sampling | ||||
| ±10% of mean for 3-point | ||||
| Alternatively, all points within: | ||||
| ±0.5 ppm of mean for 1-point sampling | ||||
| ±1.0 ppm of mean for 3-point sampling | ||||
| A | Multiple sample points simultaneously | No. of openings in probe | Multi-hole probe with verifiable constant flow through all holes within 10% of mean flow rate (requires Administrative approval for Part 75) | Each run. |
| M | Data Recording | Frequency | ≤1 minute average | During run. |
| S | Data Parameters | Sample concentration range | All 1-minute averages within calibration span | Each run. |
| M | Date Parameters | Average concentration for the run | Run average ≤calibration span | Each run. |
| S = Suggest. M = Mandatory. A = Alternative. Agency. | ||||
10.0 Calibration and Standardization
What measurement system calibrations are required?
(1) The initial 3-point calibration error test as described in section 8.2.3 and the system bias (or system calibration error) checks described in section 8.2.5 are required and must meet the specifications in section 13 before you start the test. Make all necessary adjustments to calibrate the gas analyzer and data recorder. Then, after the test commences, the system bias or system calibration error checks described in section 8.5 are required before and after each run. Your analyzer must be calibrated for all species of NOX that it detects. Analyzers that measure NO and NO2 separately without using a converter must be calibrated with both NO and NO2.
(2) You must include a copy of the manufacturer's certification of the calibration gases used in the testing as part of the test report. This certification must include the 13 documentation requirements in the EPA Traceability Protocol For Assay and Certification of Gaseous Calibration Standards, September 1997, as amended August 25, 1999. When Method 205 is used to produce diluted calibration gases, you must document that the specifications for the gas dilution system are met for the test. You must also include the date of the most recent dilution system calibration against flow standards and the name of the person or manufacturer who carried out the calibration in the test report.
11.0 Analytical Procedures
Because sample collection and analysis are performed together (see section 8), additional discussion of the analytical procedure is not necessary.
12.0 Calculations and Data Analysis
You must follow the procedures for calculations and data analysis listed in this section.
12.1 Nomenclature. The terms used in the equations are defined as follows:
ACE = Analyzer calibration error, percent of calibration span.
BWS = Moisture content of sample gas as measured by Method 4 or other approved method, percent/100.
CAvg = Average unadjusted gas concentration indicated by data recorder for the test run, ppmv.
CD = Pollutant concentration adjusted to dry conditions, ppmv.
CDir = Measured concentration of a calibration gas (low, mid, or high) when introduced in direct calibration mode, ppmv.
CGas = Average effluent gas concentration adjusted for bias, ppmv.
CM = Average of initial and final system calibration bias (or 2-point system calibration error) check responses for the upscale calibration gas, ppmv.
CMA = Actual concentration of the upscale calibration gas, ppmv.
CNative = NOX concentration in the stack gas as calculated in section 12.6, ppmv.
CO = Average of the initial and final system calibration bias (or 2-point system calibration error) check responses from the low-level (or zero) calibration gas, ppmv.
COA = Actual concentration of the low-level calibration gas, ppmv.
CS = Measured concentration of a calibration gas (low, mid, or high) when introduced in system calibration mode, ppmv.
CSS = Concentration of NOX measured in the spiked sample, ppmv.
CSpike = Concentration of NOX in the undiluted spike gas, ppmv.
CCalc = Calculated concentration of NOX in the spike gas diluted in the sample, ppmv.
CV = Manufacturer certified concentration of a calibration gas (low, mid, or high), ppmv.
CW = Pollutant concentration measured under moist sample conditions, wet basis, ppmv.
CS = Calibration span, ppmv.
D = Drift assessment, percent of calibration span.
DF = Dilution system dilution factor or spike gas dilution factor, dimensionless.
EffNO2 = NO2 to NO converter efficiency, percent.
NOXCorr = The NOX concentration corrected for the converter efficiency, ppmv.
NOXFinal = The final NOX concentration observed during the converter efficiency test in section 16.2.2, ppmv.
NOXPeak = The highest NOX concentration observed during the converter efficiency test in section 16.2.2, ppmv.
QSpike = Flow rate of spike gas introduced in system calibration mode, L/min.
QTotal = Total sample flow rate during the spike test, L/min.
R = Spike recovery, percent.
SB = System bias, percent of calibration span.
SBi = Pre-run system bias, percent of calibration span.
SBfinal = Post-run system bias, percent of calibration span.
SCE = System calibration error, percent of calibration span.
SCEi = Pre-run system calibration error, percent of calibration span.
SCEFinal = Post-run system calibration error, percent of calibration span.
12.2 Analyzer Calibration Error. For non-dilution systems, use Equation 7E-1 to calculate the analyzer calibration error for the low-, mid-, and high-level calibration gases.
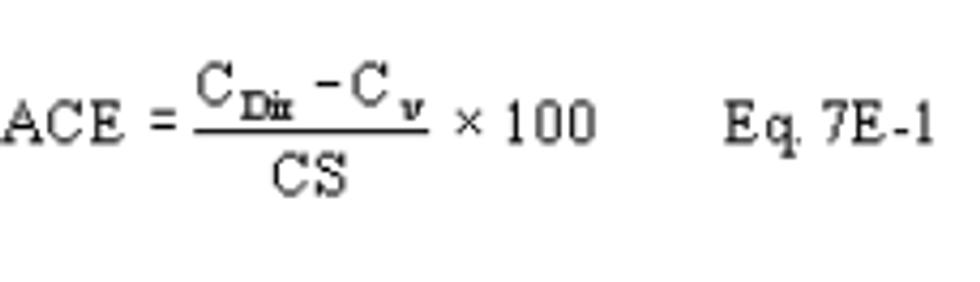
12.3 System Bias. For non-dilution systems, use Equation 7E-2 to calculate the system bias separately for the low-level and upscale calibration gases.

12.4 System Calibration Error. Use Equation 7E-3 to calculate the system calibration error for dilution systems. Equation 7E-3 applies to both the initial 3-point system calibration error test and the subsequent 2-point calibration error checks between test runs. In this equation, the term “Cs” refers to the diluted calibration gas concentration measured by the analyzer.
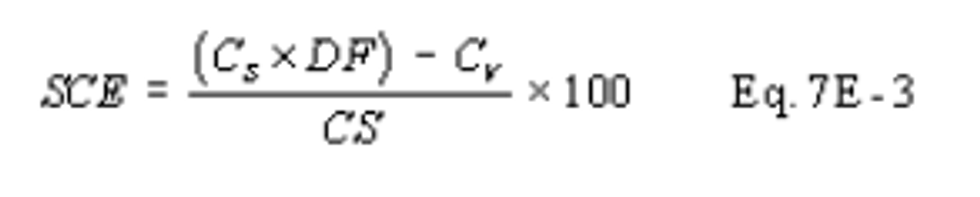
12.5 Drift Assessment. Use Equation 7E-4 to separately calculate the low-level and upscale drift over each test run. For dilution systems, replace “SBfinal” and “SBi” with “SCEfinal” and “SCEi”, respectively, to calculate and evaluate drift.
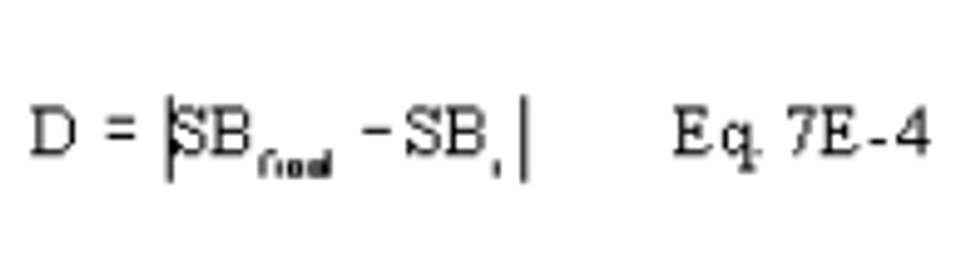
12.6 Effluent Gas Concentration. For each test run, calculate Cavg, the arithmetic average of all valid NOX concentration values (e.g., 1-minute averages). Then adjust the value of Cavg for bias using Equation 7E-5a if you use a non-zero gas as your low-level calibration gas, or Equation 7E-5b if you use a zero gas as your low-level calibration gas.
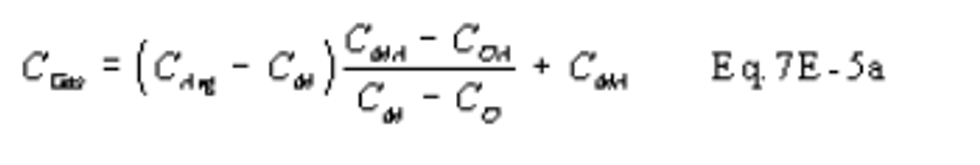
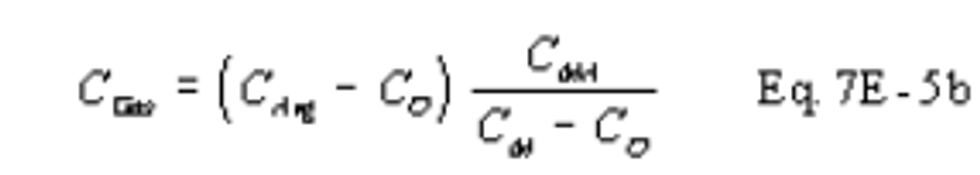
12.7 NO 2 - NO Conversion Efficiency. If the NOX converter efficiency test described in section 8.2.4.1 is performed, calculate the efficiency using Equation 7E-7.
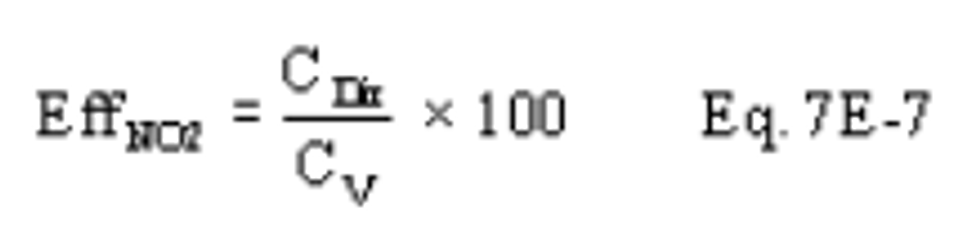
12.8 NO2 - NO Conversion Efficiency Correction. If desired, calculate the total NOX concentration with a correction for converter efficiency using Equation 7E-8.
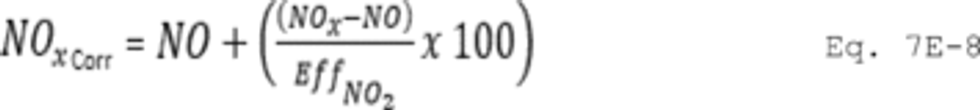
12.9 Alternative NO 2 Converter Efficiency. If the alternative procedure of section 16.2.2 is used, determine the NOX concentration decrease from NOXPeak after the minimum 30-minute test interval using Equation 7E-9. This decrease from NOXPeak must meet the requirement in section 13.5 for the converter to be acceptable.
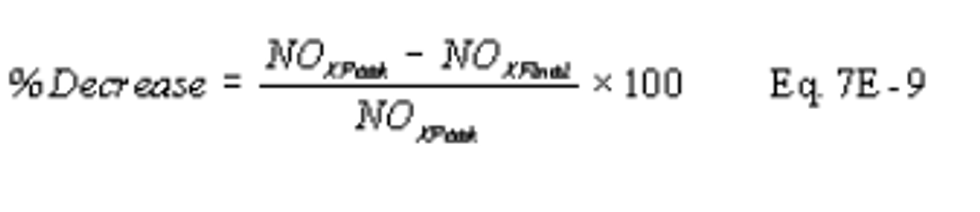
12.10 Moisture Correction. Use Equation 7E-10 if your measurements need to be corrected to a dry basis.
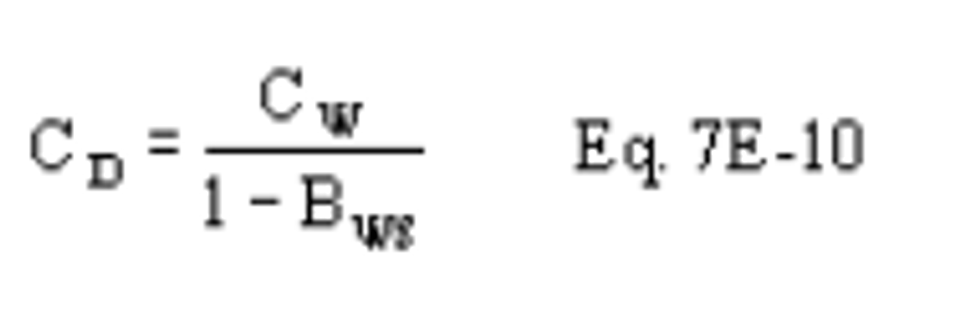
12.11 Calculated Spike Gas Concentration and Spike Recovery for the Example Alternative Dynamic Spiking Procedure in section 16.1.3. Use Equation 7E-11 to determine the calculated spike gas concentration. Use Equation 7E-12 to calculate the spike recovery.
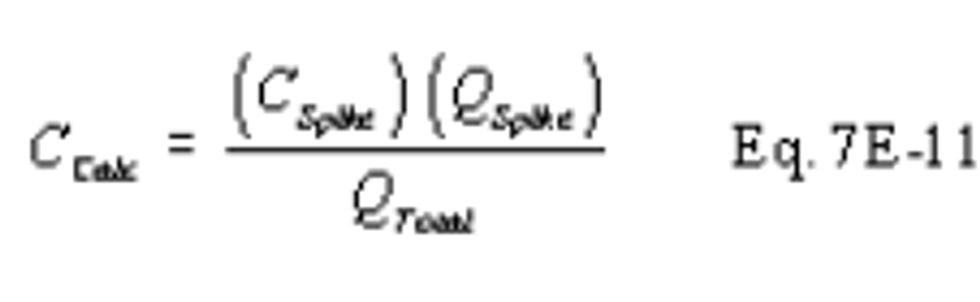
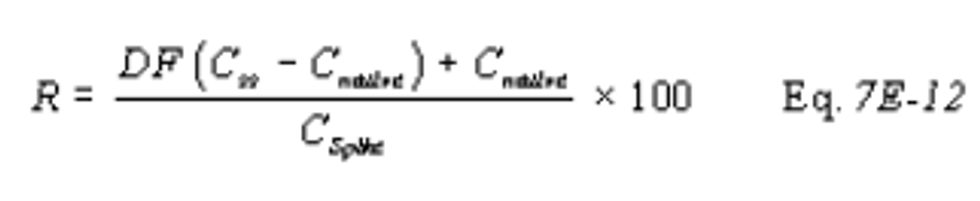
13.0 Method Performance
13.1 Calibration Error. This specification is applicable to both the analyzer calibration error and the 3-point system calibration error tests described in section 8.2.3. At each calibration gas level (low, mid, and high) the calibration error must either be within ±2.0 percent of the calibration span. Alternatively, the results are acceptable if |Cdir − Cv| or |Cs−Cv| (as applicable) is ≤0.5 ppmv.
13.2 System Bias. This specification is applicable to both the system bias and 2-point system calibration error tests described in section 8.2.5 and 8.5. The pre- and post-run system bias (or system calibration error) must be within ±5.0 percent of the calibration span for the low-level and upscale calibration gases. Alternatively, the results are acceptable if | Cs −Cdir | is ≤0.5 ppmv or if | Cs− Cv | is ≤0.5 ppmv (as applicable).
13.3 Drift. For each run, the low-level and upscale drift must be less than or equal to 3.0 percent of the calibration span. The drift is also acceptable if the pre- and post-run bias (or the pre- and post-run system calibration error) responses do not differ by more than 0.5 ppmv at each gas concentration (i.e. | Cs post-run− Cs pre-run | ≤0.5 ppmv).
13.4 Interference Check. The total interference response (i.e., the sum of the interference responses of all tested gaseous components) must not be greater than 2.50 percent of the calibration span for the analyzer tested. In summing the interferences, use the larger of the absolute values obtained for the interferent tested with and without the pollutant present. The results are also acceptable if the sum of the responses does not exceed 0.5 ppmv for a calibration span of 5 to 10 ppmv, or 0.2 ppmv for a calibration span <5 ppmv.
13.5 NO 2 to NO Conversion Efficiency Test (as applicable). The NO2 to NO conversion efficiency, calculated according to Equation 7E-7, must be greater than or equal to 90 percent. The alternative conversion efficiency check, described in section 16.2.2 and calculated according to Equation 7E-9, must not result in a decrease from NOXPeak by more than 2.0 percent.
13.6 Alternative Dynamic Spike Procedure. Recoveries of both pre-test spikes and post-test spikes must be within 100 ±10 percent. If the absolute difference between the calculated spike value and measured spike value is equal to or less than 0.20 ppmv, then the requirements of the ADSC are met.
14.0 Pollution Prevention [Reserved]
15.0 Waste Management [Reserved]
16.0 Alternative Procedures
16.1 Dynamic Spike Procedure. Except for applications under part 75 of this chapter, you may use a dynamic spiking procedure to validate your test data for a specific test matrix in place of the interference check and pre- and post-run system bias checks. For part 75 applications, use of this procedure is subject to the approval of the Administrator. Best results are obtained for this procedure when source emissions are steady and not varying. Fluctuating emissions may render this alternative procedure difficult to pass. To use this alternative, you must meet the following requirements.
16.1.1 Procedure Documentation. You must detail the procedure you followed in the test report, including how the spike was measured, added, verified during the run, and calculated after the test.
16.1.2 Spiking Procedure Requirements. The spikes must be prepared from EPA Traceability Protocol gases. Your procedure must be designed to spike field samples at two target levels both before and after the test. Your target spike levels should bracket the average sample NOX concentrations. The higher target concentration must be less than the calibration span. You must collect at least 5 data points for each target concentration. The spiking procedure must be performed before the first run and repeated after the last run of the test program.
16.1.3 Example Spiking Procedure. Determine the NO concentration needed to generate concentrations that are 50 and 150 percent of the anticipated NOX concentration in the stack at the total sampling flow rate while keeping the spike flow rate at or below 10 percent of this total. Use a mass flow meter (accurate within 2.0 percent) to generate these NO spike gas concentrations at a constant flow rate. Use Equation 7E-11 in section 12.11 to determine the calculated spike concentration in the collected sample.
(1) Prepare the measurement system and conduct the analyzer calibration error test as described in sections 8.2.2 and 8.2.3. Following the sampling procedures in section 8.1, determine the stack NOX concentration and use this concentration as the average stack concentration (Cavg) for the first spike level, or if desired, for both pre-test spike levels. Introduce the first level spike gas into the system in system calibration mode and begin sample collection. Wait for at least two times the system response time before measuring the spiked sample concentration. Then record at least five successive 1-minute averages of the spiked sample gas. Monitor the spike gas flow rate and maintain at the determined addition rate. Average the five 1-minute averages and determine the spike recovery using Equation 7E-12. Repeat this procedure for the other pre-test spike level. The recovery at each level must be within the limits in section 13.6 before proceeding with the test.
(2) Conduct the number of runs required for the test. Then repeat the above procedure for the post-test spike evaluation. The last run of the test may serve as the average stack concentration for the post-test spike test calculations. The results of the post-test spikes must meet the limits in section 13.6.
16.2 Alternative NO 2 to NO Conversion Efficiency Procedures. You may use either of the following procedures to determine converter efficiency in place of the procedure in section 8.2.4.1.
16.2.1 The procedure for determining conversion efficiency using NO in 40 CFR 86.123-78.
16.2.2 Bag Procedure. Perform the analyzer calibration error test to document the calibration (both NO and NOX modes, as applicable). Fill a Tedlar or equivalent bag approximately half full with either ambient air, pure oxygen, or an oxygen standard gas with at least 19.5 percent by volume oxygen content. Fill the remainder of the bag with mid- to high-level NO in N2 (or other appropriate concentration) calibration gas. (Note that the concentration of the NO standard should be sufficiently high enough for the diluted concentration to be easily and accurately measured on the scale used. The size of the bag should be large enough to accommodate the procedure and time required. Verify through the manufacturer that the Tedlar alternative is suitable for NO and make this verifed information available for inspection.)
(1) Immediately attach the bag to the inlet of the NOX analyzer (or external converter if used). In the case of a dilution-system, introduce the gas at a point upstream of the dilution assembly. Measure the NOX concentration for a period of 30 minutes. If the NOX concentration drops more than 2 percent absolute from the peak value observed, then the NO2 converter has failed to meet the criteria of this test. Take corrective action. The highest NOX value observed is considered to be NOXPeak. The final NOX value observed is considered to be NOXfinal.
(2) [Reserved]
16.3 Manufacturer's Stability Test. A manufacturer's stability test is required for all analyzers that routinely measure emissions below 20 ppmv and is optional but recommended for other analyzers. This test evaluates each analyzer model by subjecting it to the tests listed in Table 7E-5 following procedures similar to those in 40 CFR 53.23 for thermal stability and insensitivity to supply voltage variations. If the analyzer will be used under temperature conditions that are outside the test conditions in Table B-4 of Part 53.23, alternative test temperatures that better reflect the analyzer field environment should be used. Alternative procedures or documentation that establish the analyzer's stability over the appropriate line voltages and temperatures are acceptable.
17.0 References
1. “ERA Traceability Protocol for Assay and Certification of Gaseous Calibration Standards” September 1997 as amended, ERA-600/R-97/121.
18.0 Tables, Diagrams, Flowcharts, and Validation Data
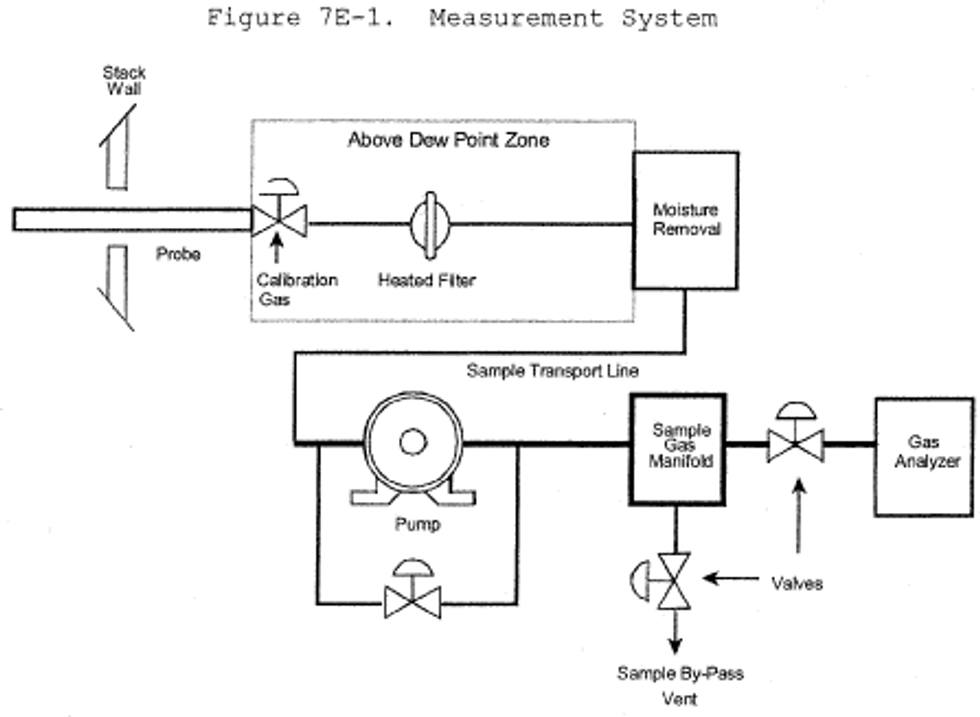
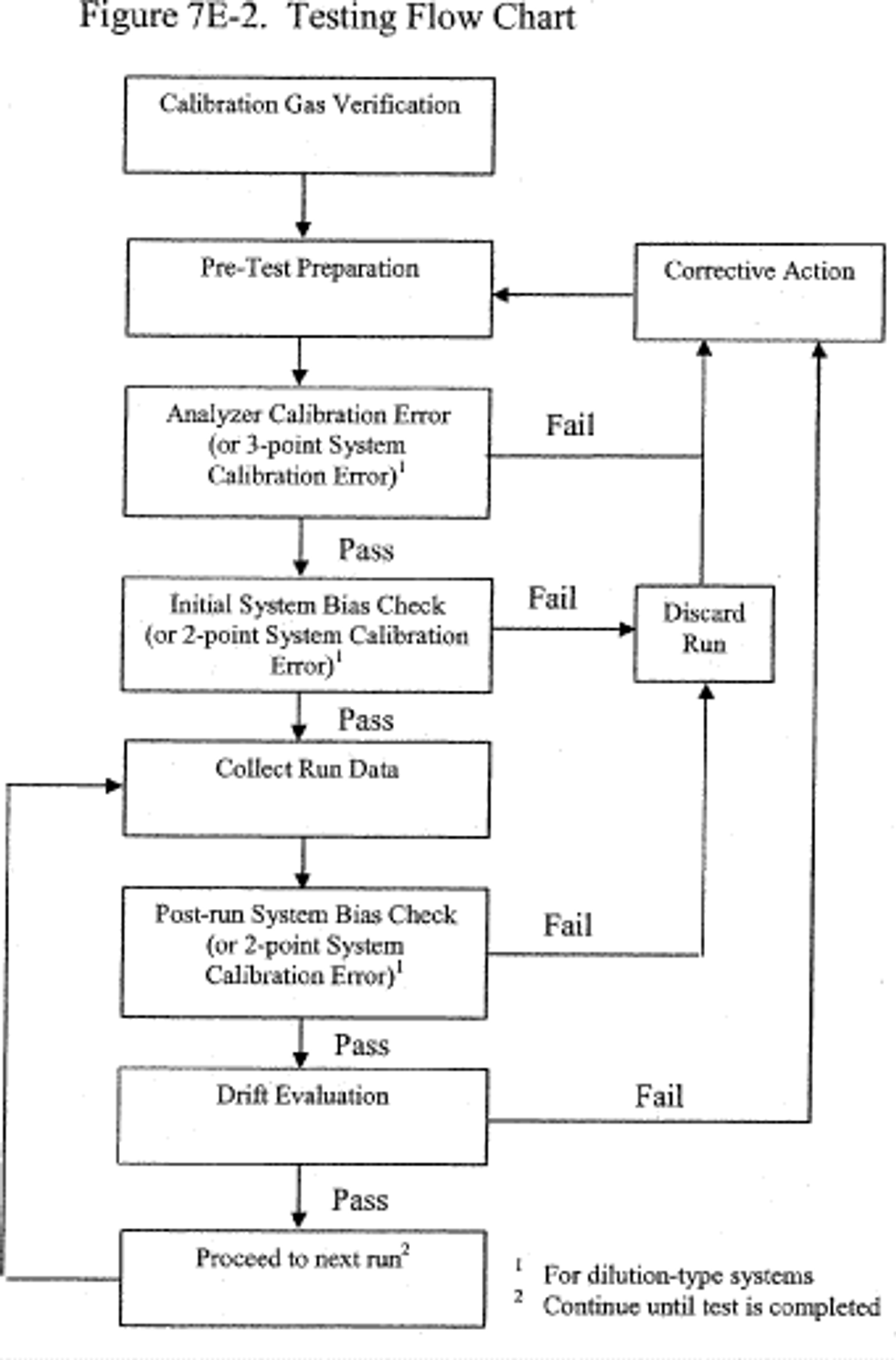

| Potential interferent gas 1 | Concentrations 2 sample conditioning type | |
|---|---|---|
| Hot wet | Dried | |
| CO2 | 5 and 15% | 5 and 15% |
| H2O | 25% | 1% |
| NO | 15 ppmv | 15 ppmv |
| NO2 | 15 ppmv | 15 ppmv |
| N2O | 10 ppmv | 10 ppmv |
| CO | 50 ppmv | 50 ppmv |
| NH3 | 10 ppmv | 10 ppmv |
| CH4 | 50 ppmv | 50 ppmv |
| SO2 | 20 ppmv | 20 ppmv |
| H2 | 50 ppmv | 50 ppmv |
| HCl | 10 ppmv | 10 ppmv |
| 1 Any applicable gas may be eliminated or tested at a reduced level if the manufacturer has provided reliable means for limiting or scrubbing that gas to a specified level. 2 As practicable, gas concentrations should be the highest expected at test sites. | ||
Table 7E-4 - Interference Response
Date of Test:
Analyzer Type:
Model No.:
Serial No:
Calibration Span:
| Test gas type | Concentration (ppm) | Analyzer response |
|---|---|---|
| Sum of Responses | ||
| % of Calibration Span | ||
| Test description | Acceptance criteria (note 1) |
|---|---|
| Thermal Stability | Temperature range when drift does not exceed 3.0% of analyzer range over a 12-hour run when measured with NOX present @ 80% of calibration span. |
| Fault Conditions | Identify conditions which, when they occur, result in performance which is not in compliance with the Manufacturer's Stability Test criteria. These are to be indicated visually or electrically to alert the operator of the problem. |
| Insensitivity to Supply Voltage Variations | ±10.0% (or manufacturers alternative) variation from nominal voltage must produce a drift of ≤2.0% of calibration span for either zero or concentration ≥80% NOX present. |
| Analyzer Calibration Error | For a low-, medium-, and high-calibration gas, the difference between the manufacturer certified value and the analyzer response in direct calibration mode, no more than 2.0% of calibration span. |
| Note 1: If the instrument is to be used as a Low Range analyzer, all tests must be performed at a calibration span of 20 ppm or less. | |
Method 8 - Determination of Sulfuric Acid and Sulfur Dioxide Emissions From Stationary Sources
Note:
This method does not include all of the specifications (e.g., equipment and supplies) and procedures (e.g., sampling and analytical) essential to its performance. Some material is incorporated by reference from other methods in this part. Therefore, to obtain reliable results, persons using this method should have a thorough knowledge of at least the following additional test methods: Method 1, Method 2, Method 3, Method 5, and Method 6.
1.0 Scope and Application
1.1 Analytes.
| Analyte | CAS No. | Sensitivity |
|---|---|---|
| Sulfuric acid, including: Sulfuric acid (H2SO4) mist, Sulfur trioxide (SO3) | 7664-93-9, 7449-11-9 | 0.05 mg/m 3 (0.03 × 10−7 lb/ft 3). |
| Sulfur dioxide (SO2) | 7449-09-5 | 1.2 mg/m 3 (3 × 10−9 lb/ft 3). |
1.2 Applicability. This method is applicable for the determination of H2SO4 (including H2SO4 mist and SO3) and gaseous SO2 emissions from stationary sources.
Note:
Filterable particulate matter may be determined along with H2SO4 and SO2 (subject to the approval of the Administrator) by inserting a heated glass fiber filter between the probe and isopropanol impinger (see section 6.1.1 of Method 6). If this option is chosen, particulate analysis is gravimetric only; sulfuric acid is not determined separately.
1.3 Data Quality Objectives. Adherence to the requirements of this method will enhance the quality of the data obtained from air pollutant sampling methods.
2.0 Summary of Method
A gas sample is extracted isokinetically from the stack. The H2SO4 and the SO2 are separated, and both fractions are measured separately by the barium-thorin titration method.
3.0 Definitions [Reserved]
4.0 Interferences
4.1 Possible interfering agents of this method are fluorides, free ammonia, and dimethyl aniline. If any of these interfering agents is present (this can be determined by knowledge of the process), alternative methods, subject to the approval of the Administrator, are required.
5.0 Safety
5.1 Disclaimer. This method may involve hazardous materials, operations, and equipment. This test method may not address all of the safety problems associated with its use. It is the responsibility of the user of this test method to establish appropriate safety and health practices and determine the applicability of regulatory limitations prior to performing this test method.
5.2 Corrosive reagents. Same as Method 6, section 5.2.
6.0 Equipment and Supplies
6.1 Sample Collection. Same as Method 5, section 6.1, with the following additions and exceptions:
6.1.1 Sampling Train. A schematic of the sampling train used in this method is shown in Figure 8-1; it is similar to the Method 5 sampling train, except that the filter position is different, and the filter holder does not have to be heated. See Method 5, section 6.1.1, for details and guidelines on operation and maintenance.
6.1.1.1 Probe Nozzle. Borosilicate or quartz glass with a sharp, tapered leading edge and coupled to the probe liner using a polytetrafluoroethylene (PTFE) or glass-lined union (e.g., fused silica, Slico, or equivalent). When the stack temperature exceeds 210°C (410°F), a leak-free ground glass fitting or other leak free, non-contaminating fitting must be used to couple the nozzle to the probe liner. It is also acceptable to use a one-piece glass nozzle/liner assembly. The angle of the taper shall be ≤30°, and the taper shall be on the outside to preserve a constant internal diameter. The probe nozzle shall be of the button-hook or elbow design, unless otherwise specified by the Administrator. Other materials of construction may be used, subject to the approval of the Administrator. A range of nozzle sizes suitable for isokinetic sampling should be available. Typical nozzle sizes range from 0.32 to 1.27 cm ( 1/8 to 1/2 in) inside diameter (ID) in increments of 0.16 cm ( 1/16 in). Larger nozzles sizes are also available if higher volume sampling trains are used.
6.1.1.2 Probe Liner. Borosilicate or quartz glass, with a heating system to prevent visible condensation during sampling. Do not use metal probe liners.
6.1.1.3 Filter Holder. Borosilicate glass, with a glass frit filter support and a silicone rubber gasket. Other gasket materials (e.g., Teflon or Viton) may be used, subject to the approval of the Administrator. The holder design shall provide a positive seal against leakage from the outside or around the filter. The filter holder shall be placed between the first and second impingers. Do not heat the filter holder.
6.1.1.4 Impingers. Four, of the Greenburg-Smith design, as shown in Figure 8-1. The first and third impingers must have standard tips. The second and fourth impingers must be modified by replacing the insert with an approximately 13-mm ( 1/2-in.) ID glass tube, having an unconstricted tip located 13 mm ( 1/2 in.) from the bottom of the impinger. Similar collection systems, subject to the approval of the Administrator, may be used.
6.1.1.5 Temperature Sensor. Thermometer, or equivalent, to measure the temperature of the gas leaving the impinger train to within 1°C (2°F).
6.2 Sample Recovery. The following items are required for sample recovery:
6.2.1 Wash Bottles. Two polyethylene or glass bottles, 500-ml.
6.2.2 Graduated Cylinders. Two graduated cylinders (volumetric flasks may be used), 250-ml, 1-liter.
6.2.3 Storage Bottles. Leak-free polyethylene bottles, 1-liter size (two for each sampling run).
6.2.4 Trip Balance. 500-g capacity, to measure to ±0.5 g (necessary only if a moisture content analysis is to be done).
6.3 Analysis. The following items are required for sample analysis:
6.3.1 Pipettes. Volumetric 10-ml, 100-ml.
6.3.2 Burette. 50-ml.
6.3.3 Erlenmeyer Flask. 250-ml (one for each sample, blank, and standard).
6.3.4 Graduated Cylinder. 100-ml.
6.3.5 Dropping Bottle. To add indicator solution, 125-ml size.
7.0 Reagents and Standards
Note:
Unless otherwise indicated, all reagents are to conform to the specifications established by the Committee on Analytical Reagents of the American Chemical Society, where such specifications are available. Otherwise, use the best available grade.
7.1 Sample Collection. The following reagents are required for sample collection:
7.1.1 Filters and Silica Gel. Same as in Method 5, sections 7.1.1 and 7.1.2, respectively.
7.1.2 Water. Same as in Method 6, section 7.1.1.
7.1.3 Isopropanol, 80 Percent by Volume. Mix 800 ml of isopropanol with 200 ml of water.
Note:
Check for peroxide impurities using the procedure outlined in Method 6, section 7.1.2.1.
7.1.4 Hydrogen Peroxide (H 2O 2), 3 Percent by Volume. Dilute 100 ml of 30 percent H2O2) to 1 liter with water. Prepare fresh daily.
7.1.5 Crushed Ice.
7.2 Sample Recovery. The reagents and standards required for sample recovery are:
7.2.1 Water. Same as in section 7.1.2.
7.2.2 Isopropanol, 80 Percent. Same as in section 7.1.3.
7.3 Sample Analysis. Same as Method 6, section 7.3.
8.0 Sample Collection, Preservation, Storage, and Transport
8.1 Pretest Preparation. Same as Method 5, section 8.1, except that filters should be inspected but need not be desiccated, weighed, or identified. If the effluent gas can be considered dry (i.e., moisture-free), the silica gel need not be weighed.
8.2 Preliminary Determinations. Same as Method 5, section 8.2.
8.3 Preparation of Sampling Train. Same as Method 5, section 8.3, with the following exceptions:
8.3.1 Use Figure 8-1 instead of Figure 5-1.
8.3.2 Replace the second sentence of Method 5, section 8.3.1 with: Place 100 ml of 80 percent isopropanol in the first impinger, 100 ml of 3 percent H2O2 in both the second and third impingers; retain a portion of each reagent for use as a blank solution. Place about 200 g of silica gel in the fourth impinger.
8.3.3 Ignore any other statements in section 8.3 of Method 5 that are obviously not applicable to the performance of Method 8.
Note:
If moisture content is to be determined by impinger analysis, weigh each of the first three impingers (plus absorbing solution) to the nearest 0.5 g, and record these weights. Weigh also the silica gel (or silica gel plus container) to the nearest 0.5 g, and record.)
8.4 Metering System Leak-Check Procedure. Same as Method 5, section 8.4.1.
8.5 Pretest Leak-Check Procedure. Follow the basic procedure in Method 5, section 8.4.2, noting that the probe heater shall be adjusted to the minimum temperature required to prevent condensation, and also that verbage such as “* * * plugging the inlet to the filter holder * * * ” found in section 8.4.2.2 of Method 5 shall be replaced by “ * * * plugging the inlet to the first impinger * * * ”. The pretest leak-check is recommended, but is not required.
8.6 Sampling Train Operation. Follow the basic procedures in Method 5, section 8.5, in conjunction with the following special instructions:
8.6.1 Record the data on a sheet similar to that shown in Figure 8-2 (alternatively, Figure 5-2 in Method 5 may be used). The sampling rate shall not exceed 0.030 m 3/min (1.0 cfm) during the run. Periodically during the test, observe the connecting line between the probe and first impinger for signs of condensation. If condensation does occur, adjust the probe heater setting upward to the minimum temperature required to prevent condensation. If component changes become necessary during a run, a leak-check shall be performed immediately before each change, according to the procedure outlined in section 8.4.3 of Method 5 (with appropriate modifications, as mentioned in section 8.5 of this method); record all leak rates. If the leakage rate(s) exceeds the specified rate, the tester shall either void the run or plan to correct the sample volume as outlined in section 12.3 of Method 5. Leak-checks immediately after component changes are recommended, but not required. If these leak-checks are performed, the procedure in section 8.4.2 of Method 5 (with appropriate modifications) shall be used.
8.6.2 After turning off the pump and recording the final readings at the conclusion of each run, remove the probe from the stack. Conduct a post-test (mandatory) leak-check as outlined in section 8.4.4 of Method 5 (with appropriate modifications), and record the leak rate. If the post-test leakage rate exceeds the specified acceptable rate, either correct the sample volume, as outlined in section 12.3 of Method 5, or void the run.
8.6.3 Drain the ice bath and, with the probe disconnected, purge the remaining part of the train by drawing clean ambient air through the system for 15 minutes at the average flow rate used for sampling.
Note:
Clean ambient air can be provided by passing air through a charcoal filter. Alternatively, ambient air (without cleaning) may be used.
8.7 Calculation of Percent Isokinetic. Same as Method 5, section 8.6.
8.8 Sample Recovery. Proper cleanup procedure begins as soon as the probe is removed from the stack at the end of the sampling period. Allow the probe to cool. Treat the samples as follows:
8.8.1 Container No. 1.
8.8.1.1 If a moisture content analysis is to be performed, clean and weigh the first impinger (plus contents) to the nearest 0.5 g, and record this weight.
8.8.1.2 Transfer the contents of the first impinger to a 250-ml graduated cylinder. Rinse the probe, first impinger, all connecting glassware before the filter, and the front half of the filter holder with 80 percent isopropanol. Add the isopropanol rinse solution to the cylinder. Dilute the contents of the cylinder to 225 ml with 80 percent isopropanol, and transfer the cylinder contents to the storage container. Rinse the cylinder with 25 ml of 80 percent isopropanol, and transfer the rinse to the storage container. Add the filter to the solution in the storage container and mix. Seal the container to protect the solution against evaporation. Mark the level of liquid on the container, and identify the sample container.
8.8.2 Container No. 2.
8.8.2.1 If a moisture content analysis is to be performed, clean and weigh the second and third impingers (plus contents) to the nearest 0.5 g, and record the weights. Also, weigh the spent silica gel (or silica gel plus impinger) to the nearest 0.5 g, and record the weight.
8.8.2.2 Transfer the solutions from the second and third impingers to a 1-liter graduated cylinder. Rinse all connecting glassware (including back half of filter holder) between the filter and silica gel impinger with water, and add this rinse water to the cylinder. Dilute the contents of the cylinder to 950 ml with water. Transfer the solution to a storage container. Rinse the cylinder with 50 ml of water, and transfer the rinse to the storage container. Mark the level of liquid on the container. Seal and identify the sample container.
9.0 Quality Control
9.1 Miscellaneous Quality Control Measures.
| Section | Quality control measure | Effect |
|---|---|---|
| 7.1.3 | Isopropanol check | Ensure acceptable level of peroxide impurities in isopropanol. |
| 8.4, 8.5, 10.1 | Sampling equipment leak-check and calibration | Ensure accurate measurement of stack gas flow rate, sample volume. |
| 10.2 | Barium standard solution standardization | Ensure normality determination. |
| 11.2 | Replicate titrations | Ensure precision of titration determinations. |
9.2 Volume Metering System Checks. Same as Method 5, section 9.2.
10.0 Calibration and Standardization
10.1 Sampling Equipment. Same as Method 5, section 10.0.
10.2 Barium Standard Solution. Same as Method 6, section 10.5.
11.0 Analytical Procedure
11.1. Sample Loss. Same as Method 6, section 11.1.
11.2. Sample Analysis.
11.2.1 Container No. 1. Shake the container holding the isopropanol solution and the filter. If the filter breaks up, allow the fragments to settle for a few minutes before removing a sample aliquot. Pipette a 100-ml aliquot of this solution into a 250-ml Erlenmeyer flask, add 2 to 4 drops of thorin indicator, and titrate to a pink endpoint using 0.0100 N barium standard solution. Repeat the titration with a second aliquot of sample, and average the titration values. Replicate titrations must agree within 1 percent or 0.2 ml, whichever is greater.
11.2.2 Container No. 2. Thoroughly mix the solution in the container holding the contents of the second and third impingers. Pipette a 10-ml aliquot of sample into a 250-ml Erlenmeyer flask. Add 40 ml of isopropanol, 2 to 4 drops of thorin indicator, and titrate to a pink endpoint using 0.0100 N barium standard solution. Repeat the titration with a second aliquot of sample, and average the titration values. Replicate titrations must agree within 1 percent or 0.2 ml, whichever is greater.
11.2.3 Blanks. Prepare blanks by adding 2 to 4 drops of thorin indicator to 100 ml of 80 percent isopropanol. Titrate the blanks in the same manner as the samples.
12.0 Data Analysis and Calculations
Carry out calculations retaining at least one extra significant figure beyond that of the acquired data. Round off figures after final calculation.
12.1 Nomenclature. Same as Method 5, section 12.1, with the following additions and exceptions:
CH 2 SO 4 = Sulfuric acid (including SO3) concentration, g/dscm (lb/dscf).
CSO 2 = Sulfur dioxide concentration, g/dscm (lb/dscf).
N = Normality of barium perchlorate titrant, meq/ml.
Va = Volume of sample aliquot titrated, 100 ml for H2SO4 and 10 ml for SO2.
Vsoln = Total volume of solution in which the sample is contained, 1000 ml for the SO2 sample and 250 ml for the H2SO4 sample.
Vt = Volume of barium standard solution titrant used for the sample, ml.
Vtb = Volume of barium standard solution titrant used for the blank, ml.
12.2 Average Dry Gas Meter Temperature and Average Orifice Pressure Drop. See data sheet (Figure 8-2).
12.3 Dry Gas Volume. Same as Method 5, section 12.3.
12.4 Volume of Water Vapor Condensed and Moisture Content. Calculate the volume of water vapor using Equation 5-2 of Method 5; the weight of water collected in the impingers and silica gel can be converted directly to milliliters (the specific gravity of water is 1 g/ml). Calculate the moisture content of the stack gas (Bws) using Equation 5-3 of Method 5. The note in section 12.5 of Method 5 also applies to this method. Note that if the effluent gas stream can be considered dry, the volume of water vapor and moisture content need not be calculated.
12.5 Sulfuric Acid Mist (Including SO3) Concentration.

Where:
K3 = 0.04904 g/meq for metric units,
K3 = 1.081 × 10−4 lb/meq for English units.
12.6 Sulfur Dioxide Concentration.

Where:
K4 = 0.03203 g/meq for metric units,
K4 = 7.061 × 10−5 lb/meq for English units.
12.7 Isokinetic Variation. Same as Method 5, section 12.11.
12.8 Stack Gas Velocity and Volumetric Flow Rate. Calculate the average stack gas velocity and volumetric flow rate, if needed, using data obtained in this method and the equations in sections 12.6 and 12.7 of Method 2.
13.0 Method Performance
13.1 Analytical Range. Collaborative tests have shown that the minimum detectable limits of the method are 0.06 mg/m 3 (4 × 10−9 lb/ft 3) for H2SO4 and 1.2 mg/m 3 (74 × 10−9 lb/ft 3) for SO2. No upper limits have been established. Based on theoretical calculations for 200 ml of 3 percent H2O2 solution, the upper concentration limit for SO2 in a 1.0 m 3 (35.3 ft 3) gas sample is about 12,000 mg/m 3 (7.7 × 10−4 lb/ft 3). The upper limit can be extended by increasing the quantity of peroxide solution in the impingers.
14.0 Pollution Prevention [Reserved]
15.0 Waste Management [Reserved]
16.0 References
Same as section 17.0 of Methods 5 and 6.
17.0 Tables, Diagrams, Flowcharts, and Validation Data
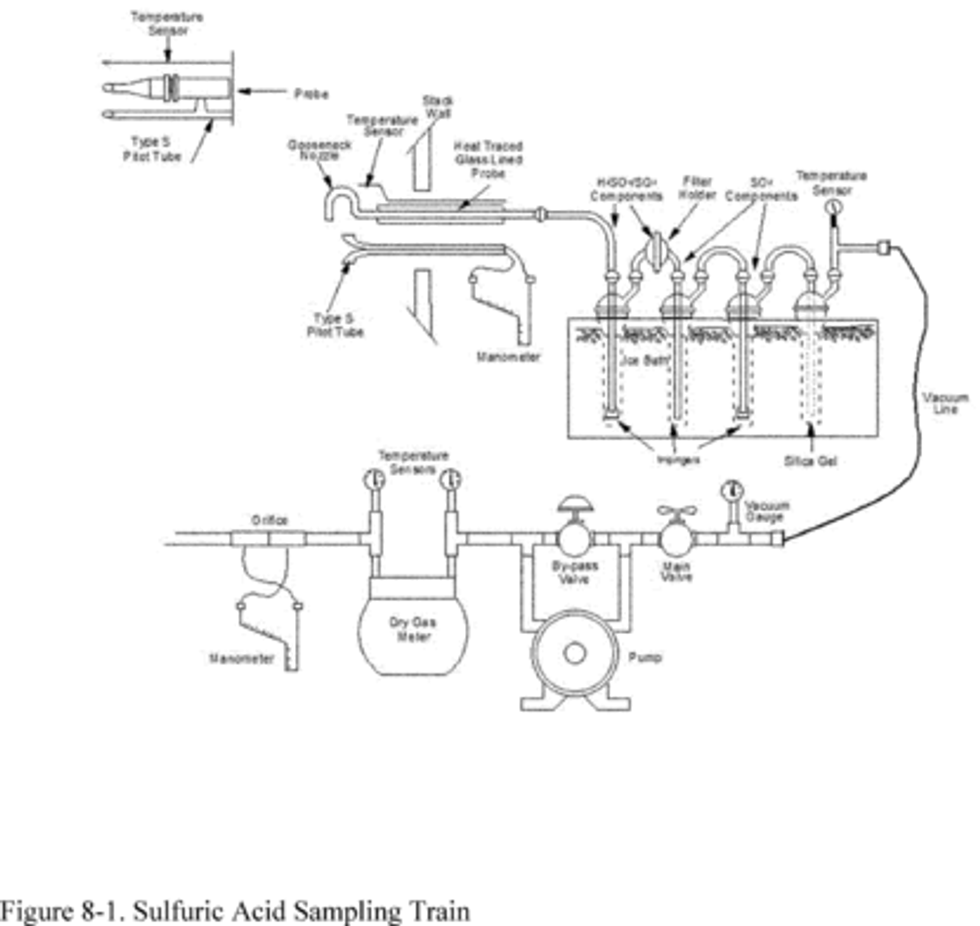
Method 9 - Visual Determination of the Opacity of Emissions From Stationary Sources
Many stationary sources discharge visible emissions into the atmosphere; these emissions are usually in the shape of a plume. This method involves the determination of plume opacity by qualified observers. The method includes procedures for the training and certification of observers, and procedures to be used in the field for determination of plume opacity. The appearance of a plume as viewed by an observer depends upon a number of variables, some of which may be controllable and some of which may not be controllable in the field. Variables which can be controlled to an extent to which they no longer exert a significant influence upon plume appearance include: Angle of the observer with respect to the plume; angle of the observer with respect to the sun; point of observation of attached and detached steam plume; and angle of the observer with respect to a plume emitted from a rectangular stack with a large length to width ratio. The method includes specific criteria applicable to these variables.
Other variables which may not be controllable in the field are luminescence and color contrast between the plume and the background against which the plume is viewed. These variables exert an influence upon the appearance of a plume as viewed by an observer, and can affect the ability of the observer to accurately assign opacity values to the observed plume. Studies of the theory of plume opacity and field studies have demonstrated that a plume is most visible and presents the greatest apparent opacity when viewed against a contrasting background. It follows from this, and is confirmed by field trials, that the opacity of a plume, viewed under conditions where a contrasting background is present can be assigned with the greatest degree of accuracy. However, the potential for a positive error is also the greatest when a plume is viewed under such contrasting conditions. Under conditions presenting a less contrasting background, the apparent opacity of a plume is less and approaches zero as the color and luminescence contrast decrease toward zero. As a result, significant negative bias and negative errors can be made when a plume is viewed under less contrasting conditions. A negative bias decreases rather than increases the possibility that a plant operator will be cited for a violation of opacity standards due to observer error.
Studies have been undertaken to determine the magnitude of positive errors which can be made by qualified observers while reading plumes under contrasting conditions and using the procedures set forth in this method. The results of these studies (field trials) which involve a total of 769 sets of 25 readings each are as follows:
(1) For black plumes (133 sets at a smoke generator), 100 percent of the sets were read with a positive error 1 of less than 7.5 percent opacity; 99 percent were read with a positive error of less than 5 percent opacity.
1 For a set, positive error = average opacity determined by observers' 25 observations - average opacity determined from transmissometer's 25 recordings.
(2) For white plumes (170 sets at a smoke generator, 168 sets at a coal-fired power plant, 298 sets at a sulfuric acid plant), 99 percent of the sets were read with a positive error of less than 7.5 percent opacity; 95 percent were read with a positive error of less than 5 percent opacity.
The positive observational error associated with an average of twenty-five readings is therefore established. The accuracy of the method must be taken into account when determining possible violations of applicable opacity standards.
1. Principle and Applicability
1.1 Principle. The opacity of emissions from stationary sources is determined visually by a qualified observer.
1.2 Applicability. This method is applicable for the determination of the opacity of emissions from stationary sources pursuant to §60.11(b) and for qualifying observers for visually determining opacity of emissions.
2. Procedures
The observer qualified in accordance with section 3 of this method shall use the following procedures for visually determining the opacity of emissions:
2.1 Position. The qualified observer shall stand at a distance sufficient to provide a clear view of the emissions with the sun oriented in the 140° sector to his back. Consistent with maintaining the above requirement, the observer shall, as much as possible, make his observations from a position such that his line of vision is approximately perpendicular to the plume direction, and when observing opacity of emissions from rectangular outlets (e.g., roof monitors, open baghouses, noncircular stacks), approximately perpendicular to the longer axis of the outlet. The observer's line of sight should not include more than one plume at a time when multiple stacks are involved, and in any case the observer should make his observations with his line of sight perpendicular to the longer axis of such a set of multiple stacks (e.g., stub stacks on baghouses).
2.2 Field Records. The observer shall record the name of the plant, emission location, type facility, observer's name and affiliation, a sketch of the observer's position relative to the source, and the date on a field data sheet (Figure 9-1). The time, estimated distance to the emission location, approximate wind direction, estimated wind speed, description of the sky condition (presence and color of clouds), and plume background are recorded on a field data sheet at the time opacity readings are initiated and completed.
2.3 Observations. Opacity observations shall be made at the point of greatest opacity in that portion of the plume where condensed water vapor is not present. The observer shall not look continuously at the plume, but instead shall observe the plume momentarily at 15-second intervals.
2.3.1 Attached Steam Plumes. When condensed water vapor is present within the plume as it emerges from the emission outlet, opacity observations shall be made beyond the point in the plume at which condensed water vapor is no longer visible. The observer shall record the approximate distance from the emission outlet to the point in the plume at which the observations are made.
2.3.2 Detached Steam Plume. When water vapor in the plume condenses and becomes visible at a distinct distance from the emission outlet, the opacity of emissions should be evaluated at the emission outlet prior to the condensation of water vapor and the formation of the steam plume.
2.4 Recording Observations. Opacity observations shall be recorded to the nearest 5 percent at 15-second intervals on an observational record sheet. (See Figure 9-2 for an example.) A minimum of 24 observations shall be recorded. Each momentary observation recorded shall be deemed to represent the average opacity of emissions for a 15-second period.
2.5 Data Reduction. Opacity shall be determined as an average of 24 consecutive observations recorded at 15-second intervals. Divide the observations recorded on the record sheet into sets of 24 consecutive observations. A set is composed of any 24 consecutive observations. Sets need not be consecutive in time and in no case shall two sets overlap. For each set of 24 observations, calculate the average by summing the opacity of the 24 observations and dividing this sum by 24. If an applicable standard specifies an averaging time requiring more than 24 observations, calculate the average for all observations made during the specified time period. Record the average opacity on a record sheet. (See Figure 9-1 for an example.)
3. Qualifications and Testing
3.1 Certification Requirements. To receive certification as a qualified observer, a candidate must be tested and demonstrate the ability to assign opacity readings in 5 percent increments to 25 different black plumes and 25 different white plumes, with an error not to exceed 15 percent opacity on any one reading and an average error not to exceed 7.5 percent opacity in each category. Candidates shall be tested according to the procedures described in section 3.2. Smoke generators used pursuant to section 3.2 shall be equipped with a smoke meter which meets the requirements of section 3.3.
The certification shall be valid for a period of 6 months, at which time the qualification procedure must be repeated by any observer in order to retain certification.
3.2 Certification Procedure. The certification test consists of showing the candidate a complete run of 50 plumes - 25 black plumes and 25 white plumes - generated by a smoke generator. Plumes within each set of 25 black and 25 white runs shall be presented in random order. The candidate assigns an opacity value to each plume and records his observation on a suitable form. At the completion of each run of 50 readings, the score of the candidate is determined. If a candidate fails to qualify, the complete run of 50 readings must be repeated in any retest. The smoke test may be administered as part of a smoke school or training program, and may be preceded by training or familiarization runs of the smoke generator during which candidates are shown black and white plumes of known opacity.
3.3 Smoke Generator Specifications. Any smoke generator used for the purposes of section 3.2 shall be equipped with a smoke meter installed to measure opacity across the diameter of the smoke generator stack. The smoke meter output shall display instack opacity based upon a pathlength equal to the stack exit diameter, on a full 0 to 100 percent chart recorder scale. The smoke meter optical design and performance shall meet the specifications shown in Table 9-1. The smoke meter shall be calibrated as prescribed in section 3.3.1 prior to the conduct of each smoke reading test. At the completion of each test, the zero and span drift shall be checked and if the drift exceeds ±1 percent opacity, the condition shall be corrected prior to conducting any subsequent test runs. The smoke meter shall be demonstrated, at the time of installation, to meet the specifications listed in Table 9-1. This demonstration shall be repeated following any subsequent repair or replacement of the photocell or associated electronic circuitry including the chart recorder or output meter, or every 6 months, whichever occurs first.
| Parameter | Specification |
|---|---|
| a. Light source | Incandescent lamp operated at nominal rated voltage. |
| b. Spectral response of photocell | Photopic (daylight spectral response of the human eye - Citation 3). |
| c. Angle of view | 15° maximum total angle. |
| d. Angle of projection | 15° maximum total angle. |
| e. Calibration error | ±3% opacity, maximum. |
| f. Zero and span drift | ±1% opacity, 30 minutes. |
| g. Response time | 5 seconds. |
3.3.1 Calibration. The smoke meter is calibrated after allowing a minimum of 30 minutes warmup by alternately producing simulated opacity of 0 percent and 100 percent. When stable response at 0 percent or 100 percent is noted, the smoke meter is adjusted to produce an output of 0 percent or 100 percent, as appropriate. This calibration shall be repeated until stable 0 percent and 100 percent readings are produced without adjustment. Simulated 0 percent and 100 percent opacity values may be produced by alternately switching the power to the light source on and off while the smoke generator is not producing smoke.
3.3.2 Smoke Meter Evaluation. The smoke meter design and performance are to be evaluated as follows:
3.3.2.1 Light Source. Verify from manufacturer's data and from voltage measurements made at the lamp, as installed, that the lamp is operated within ±5 percent of the nominal rated voltage.
3.3.2.2 Spectral Response of Photocell. Verify from manufacturer's data that the photocell has a photopic response; i.e., the spectral sensitivity of the cell shall closely approximate the standard spectral-luminosity curve for photopic vision which is referenced in (b) of Table 9-1.
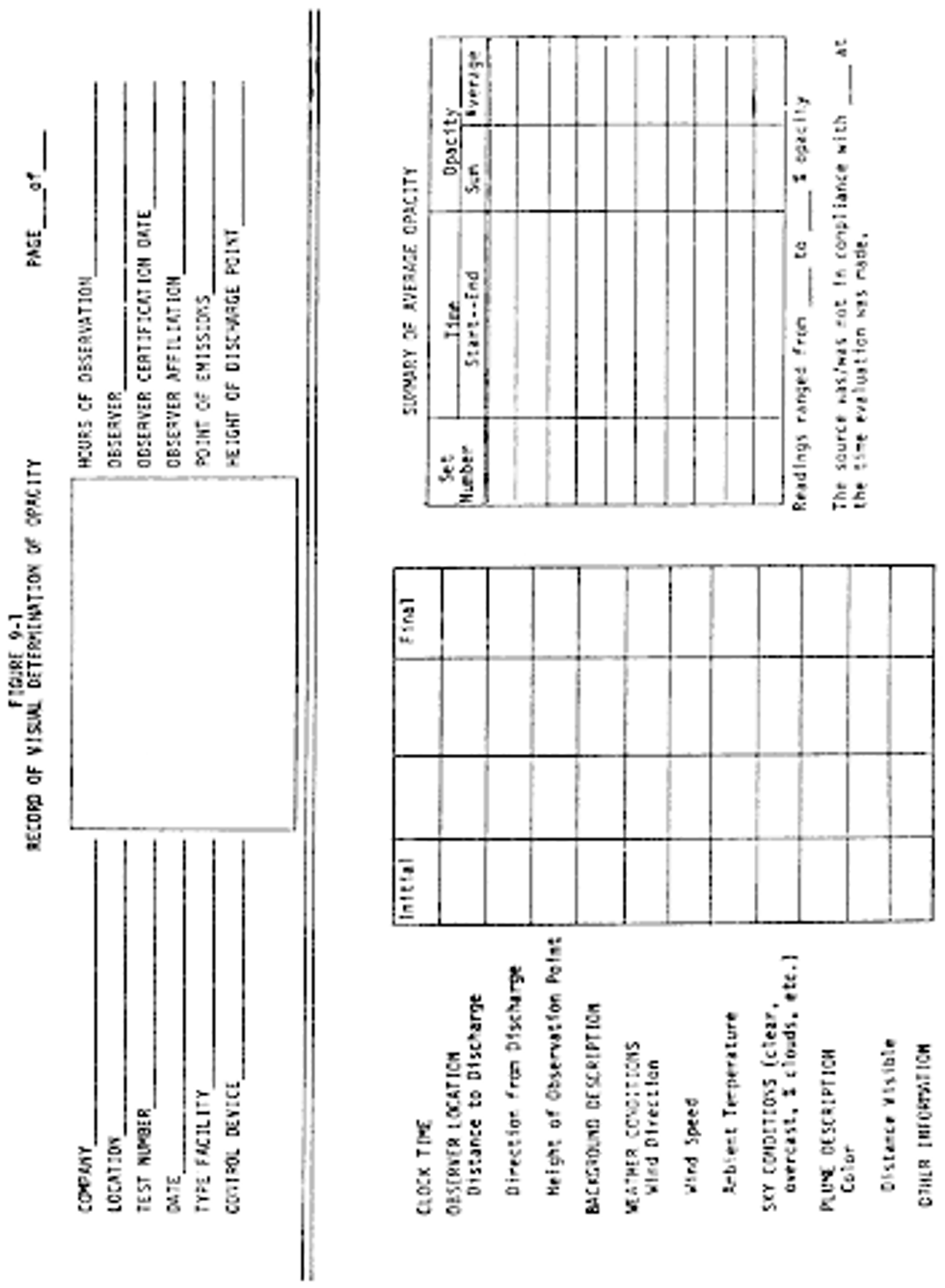
| Company | Observer | |
| Location | Type facility | |
| Test Number | Point of emissions | |
| Date |
| Hr. | Min. | Seconds | Steam plume (check if applicable) | Comments | ||||
|---|---|---|---|---|---|---|---|---|
| 0 | 15 | 30 | 45 | Attached | Detached | |||
| 0 | ||||||||
| 1 | ||||||||
| 2 | ||||||||
| 3 | ||||||||
| 4 | ||||||||
| 5 | ||||||||
| 6 | ||||||||
| 7 | ||||||||
| 8 | ||||||||
| 9 | ||||||||
| 10 | ||||||||
| 11 | ||||||||
| 12 | ||||||||
| 13 | ||||||||
| 14 | ||||||||
| 15 | ||||||||
| 16 | ||||||||
| 17 | ||||||||
| 18 | ||||||||
| 19 | ||||||||
| 20 | ||||||||
| 21 | ||||||||
| 22 | ||||||||
| 23 | ||||||||
| 24 | ||||||||
| 25 | ||||||||
| 26 | ||||||||
| 27 | ||||||||
| 28 | ||||||||
| 29 | ||||||||
| Company | Observer | |
| Location | Type facility | |
| Test Number | Point of emissions | |
| Date |
| Hr. | Min. | Seconds | Steam plume (check if applicable) | Comments | ||||
|---|---|---|---|---|---|---|---|---|
| 0 | 15 | 30 | 45 | Attached | Detached | |||
| 30 | ||||||||
| 31 | ||||||||
| 32 | ||||||||
| 33 | ||||||||
| 34 | ||||||||
| 35 | ||||||||
| 36 | ||||||||
| 37 | ||||||||
| 38 | ||||||||
| 39 | ||||||||
| 40 | ||||||||
| 41 | ||||||||
| 42 | ||||||||
| 43 | ||||||||
| 44 | ||||||||
| 45 | ||||||||
| 46 | ||||||||
| 47 | ||||||||
| 48 | ||||||||
| 49 | ||||||||
| 50 | ||||||||
| 51 | ||||||||
| 52 | ||||||||
| 53 | ||||||||
| 54 | ||||||||
| 55 | ||||||||
| 56 | ||||||||
| 57 | ||||||||
| 58 | ||||||||
| 59 | ||||||||
3.3.2.3 Angle of View. Check construction geometry to ensure that the total angle of view of the smoke plume, as seen by the photocell, does not exceed 15°. The total angle of view may be calculated from: θ = 2 tan−1d/2L, where θ = total angle of view; d = the sum of the photocell diameter the diameter of the limiting aperture; and L = the distance from the photocell to the limiting aperture. The limiting aperture is the point in the path between the photocell and the smoke plume where the angle of view is most restricted. In smoke generator smoke meters this is normally an orifice plate.
3.3.2.4 Angle of Projection. Check construction geometry to ensure that the total angle of projection of the lamp on the smoke plume does not exceed 15°. The total angle of projection may be calculated from: θ = 2 tan−1d/2L, where θ = total angle of projection; d = the sum of the length of the lamp filament the diameter of the limiting aperture; and L = the distance from the lamp to the limiting aperture.
3.3.2.5 Calibration Error. Using neutral-density filters of known opacity, check the error between the actual response and the theoretical linear response of the smoke meter. This check is accomplished by first calibrating the smoke meter according to 3.3.1 and then inserting a series of three neutral-density filters of nominal opacity of 20, 50, and 75 percent in the smoke meter pathlength. Filters calibrated within ±2 percent shall be used. Care should be taken when inserting the filters to prevent stray light from affecting the meter. Make a total of five nonconsecutive readings for each filter. The maximum error on any one reading shall be 3 percent opacity.
3.3.2.6 Zero and Span Drift. Determine the zero and span drift by calibrating and operating the smoke generator in a normal manner over a 1-hour period. The drift is measured by checking the zero and span at the end of this period.
3.3.2.7 Response Time. Determine the response time by producing the series of five simulated 0 percent and 100 percent opacity values and observing the time required to reach stable response. Opacity values of 0 percent and 100 percent may be simulated by alternately switching the power to the light source off and on while the smoke generator is not operating.
4. Bibliography
1. Air Pollution Control District Rules and Regulations, Los Angeles County Air Pollution Control District, Regulation IV, Prohibitions, Rule 50.
2. Weisburd, Melvin I., Field Operations and Enforcement Manual for Air, U.S. Environmental Protection Agency, Research Triangle Park, NC. APTD-1100, August 1972, pp. 4.1-4.36.
3. Condon, E.U., and Odishaw, H., Handbook of Physics, McGraw-Hill Co., New York, NY, 1958, Table 3.1, p. 6-52.
Alternate Method 1 - Determination of the Opacity of Emissions From Stationary Sources Remotely by Lidar
This alternate method provides the quantitative determination of the opacity of an emissions plume remotely by a mobile lidar system (laser radar; Light Detection and Ranging). The method includes procedures for the calibration of the lidar and procedures to be used in the field for the lidar determination of plume opacity. The lidar is used to measure plume opacity during either day or nighttime hours because it contains its own pulsed light source or transmitter. The operation of the lidar is not dependent upon ambient lighting conditions (light, dark, sunny or cloudy).
The lidar mechanism or technique is applicable to measuring plume opacity at numerous wavelengths of laser radiation. However, the performance evaluation and calibration test results given in support of this method apply only to a lidar that employs a ruby (red light) laser [Reference 5.1].
1. Principle and Applicability
1.1 Principle. The opacity of visible emissions from stationary sources (stacks, roof vents, etc.) is measured remotely by a mobile lidar (laser radar).
1.2 Applicability. This method is applicable for the remote measurement of the opacity of visible emissions from stationary sources during both nighttime and daylight conditions, pursuant to 40 CFR §60.11(b). It is also applicable for the calibration and performance verification of the mobile lidar for the measurement of the opacity of emissions. A performance/design specification for a basic lidar system is also incorporated into this method.
1.3 Definitions.
Azimuth angle: The angle in the horizontal plane that designates where the laser beam is pointed. It is measured from an arbitrary fixed reference line in that plane.
Backscatter: The scattering of laser light in a direction opposite to that of the incident laser beam due to reflection from particulates along the beam's atmospheric path which may include a smoke plume.
Backscatter signal: The general term for the lidar return signal which results from laser light being backscattered by atmospheric and smoke plume particulates.
Convergence distance: The distance from the lidar to the point of overlap of the lidar receiver's field-of-view and the laser beam.
Elevation angle: The angle of inclination of the laser beam referenced to the horizontal plane.
Far region: The region of the atmosphere's path along the lidar line-of-sight beyond or behind the plume being measured.
Lidar: Acronym for Light Detection and Ranging.
Lidar range: The range or distance from the lidar to a point of interest along the lidar line-of-sight.
Near region: The region of the atmospheric path along the lidar line-of-sight between the lidar's convergence distance and the plume being measured.
Opacity: One minus the optical transmittance of a smoke plume, screen target, etc.
Pick interval: The time or range intervals in the lidar backscatter signal whose minimum average amplitude is used to calculate opacity. Two pick intervals are required, one in the near region and one in the far region.
Plume: The plume being measured by lidar.
Plume signal: The backscatter signal resulting from the laser light pulse passing through a plume.
1/R 2Correction: The correction made for the systematic decrease in lidar backscatter signal amplitude with range.
Reference signal: The backscatter signal resulting from the laser light pulse passing through ambient air.
Sample interval: The time period between successive samples for a digital signal or between successive measurements for an analog signal.
Signal spike: An abrupt, momentary increase and decrease in signal amplitude.
Source: The source being tested by lidar.
Time reference: The time (to) when the laser pulse emerges from the laser, used as the reference in all lidar time or range measurements.
2. Procedures
The mobile lidar calibrated in accordance with Paragraph 3 of this method shall use the following procedures for remotely measuring the opacity of stationary source emissions:
2.1 Lidar Position. The lidar shall be positioned at a distance from the plume sufficient to provide an unobstructed view of the source emissions. The plume must be at a range of at least 50 meters or three consecutive pick intervals (whichever is greater) from the lidar's transmitter/receiver convergence distance along the line-of-sight. The maximum effective opacity measurement distance of the lidar is a function of local atmospheric conditions, laser beam diameter, and plume diameter. The test position of the lidar shall be selected so that the diameter of the laser beam at the measurement point within the plume shall be no larger than three-fourths the plume diameter. The beam diameter is calculated by Equation (AM1-1):
D(lidar) = A Rφ≤0.75 D(Plume) (AM1-1)
Where:
D(Plume) = diameter of the plume (cm),
φ = laser beam divergence measured in radians
R = range from the lidar to the source (cm)
D(Lidar) = diameter of the laser beam at range R (cm),
A = diameter of the laser beam or pulse where it leaves the laser.
The lidar range, R, is obtained by aiming and firing the laser at the emissions source structure immediately below the outlet. The range value is then determined from the backscatter signal which consists of a signal spike (return from source structure) and the atmospheric backscatter signal [Reference 5.1]. This backscatter signal should be recorded.
When there is more than one source of emissions in the immediate vicinity of the plume, the lidar shall be positioned so that the laser beam passes through only a single plume, free from any interference of the other plumes for a minimum of 50 meters or three consecutive pick intervals (whichever is greater) in each region before and beyond the plume along the line-of-sight (determined from the backscatter signals). The lidar shall initially be positioned so that its line-of-sight is approximately perpendicular to the plume.
When measuring the opacity of emissions from rectangular outlets (e.g., roof monitors, open baghouses, noncircular stacks, etc.), the lidar shall be placed in a position so that its line-of-sight is approximately perpendicular to the longer (major) axis of the outlet.
2.2 Lidar Operational Restrictions. The lidar receiver shall not be aimed within an angle of ±15° (cone angle) of the sun.
This method shall not be used to make opacity measurements if thunderstorms, snowstorms, hail storms, high wind, high-ambient dust levels, fog or other atmospheric conditions cause the reference signals to consistently exceed the limits specified in section 2.3.
2.3 Reference Signal Requirements. Once placed in its proper position for opacity measurement, the laser is aimed and fired with the line-of-sight near the outlet height and rotated horizontally to a position clear of the source structure and the associated plume. The backscatter signal obtained from this position is called the ambient-air or reference signal. The lidar operator shall inspect this signal [Section V of Reference 5.1] to: (1) determine if the lidar line-of-sight is free from interference from other plumes and from physical obstructions such as cables, power lines, etc., for a minimum of 50 meters or three consecutive pick intervals (whichever is greater) in each region before and beyond the plume, and (2) obtain a qualitative measure of the homogeneity of the ambient air by noting any signal spikes.
Should there be any signal spikes on the reference signal within a minimum of 50 meters or three consecutive pick intervals (whichever is greater) in each region before and beyond the plume, the laser shall be fired three more times and the operator shall inspect the reference signals on the display. If the spike(s) remains, the azimuth angle shall be changed and the above procedures conducted again. If the spike(s) disappears in all three reference signals, the lidar line-of-sight is acceptable if there is shot-to-shot consistency and there is no interference from other plumes.
Shot-to-shot consistency of a series of reference signals over a period of twenty seconds is verified in either of two ways. (1) The lidar operator shall observe the reference signal amplitudes. For shot-to-shot consistency the ratio of Rf to Rn [amplitudes of the near and far region pick intervals (Section 2.6.1)] shall vary by not more than ±6% between shots; or (2) the lidar operator shall accept any one of the reference signals and treat the other two as plume signals; then the opacity for each of the subsequent reference signals is calculated (Equation AM1-2). For shot-to-shot consistency, the opacity values shall be within ±3% of 0% opacity and the associated So values less than or equal to 8% (full scale) [Section 2.6].
If a set of reference signals fails to meet the requirements of this section, then all plume signals [Section 2.4] from the last set of acceptable reference signals to the failed set shall be discarded.
2.3.1 Initial and Final Reference Signals. Three reference signals shall be obtained within a 90-second time period prior to any data run. A final set of three reference signals shall be obtained within three (3) minutes after the completion of the same data run.
2.3.2 Temporal Criterion for Additional Reference Signals. An additional set of reference signals shall be obtained during a data run if there is a change in wind direction or plume drift of 30° or more from the direction that was prevalent when the last set of reference signals was obtained. An additional set of reference signals shall also be obtained if there is an increase in value of SIn (near region standard deviation, Equation AM1-5) or SIf (far region standard deviation, Equation AM1-6) that is greater than 6% (full scale) over the respective values calculated from the immediately previous plume signal, and this increase in value remains for 30 seconds or longer. An additional set of reference signals shall also be obtained if there is a change in amplitude in either the near or the far region of the plume signal, that is greater than 6% of the near signal amplitude and this change in amplitude remains for 30 seconds or more.
2.4 Plume Signal Requirements. Once properly aimed, the lidar is placed in operation with the nominal pulse or firing rate of six pulses/minute (1 pulse/10 seconds). The lidar operator shall observe the plume backscatter signals to determine the need for additional reference signals as required by section 2.3.2. The plume signals are recorded from lidar start to stop and are called a data run. The length of a data run is determined by operator discretion. Short-term stops of the lidar to record additional reference signals do not constitute the end of a data run if plume signals are resumed within 90 seconds after the reference signals have been recorded, and the total stop or interrupt time does not exceed 3 minutes.
2.4.1 Non-hydrated Plumes. The laser shall be aimed at the region of the plume which displays the greatest opacity. The lidar operator must visually verify that the laser is aimed clearly above the source exit structure.
2.4.2 Hydrated Plumes. The lidar will be used to measure the opacity of hydrated or so-called steam plumes. As listed in the reference method, there are two types, i.e., attached and detached steam plumes.
2.4.2.1 Attached Steam Plumes. When condensed water vapor is present within a plume, lidar opacity measurements shall be made at a point within the residual plume where the condensed water vapor is no longer visible. The laser shall be aimed into the most dense region (region of highest opacity) of the residual plume.
During daylight hours the lidar operator locates the most dense portion of the residual plume visually. During nighttime hours a high-intensity spotlight, night vision scope, or low light level TV, etc., can be used as an aid to locate the residual plume. If visual determination is ineffective, the lidar may be used to locate the most dense region of the residual plume by repeatedly measuring opacity, along the longitudinal axis or center of the plume from the emissions outlet to a point just beyond the steam plume. The lidar operator should also observe color differences and plume reflectivity to ensure that the lidar is aimed completely within the residual plume. If the operator does not obtain a clear indication of the location of the residual plume, this method shall not be used.
Once the region of highest opacity of the residual plume has been located, aiming adjustments shall be made to the laser line-of-sight to correct for the following: movement to the region of highest opacity out of the lidar line-of-sight (away from the laser beam) for more than 15 seconds, expansion of the steam plume (air temperature lowers and/or relative humidity increases) so that it just begins to encroach on the field-of-view of the lidar's optical telescope receiver, or a decrease in the size of the steam plume (air temperature higher and/or relative humidity decreases) so that regions within the residual plume whose opacity is higher than the one being monitored, are present.
2.4.2.2 Detached Steam Plumes. When the water vapor in a hydrated plume condenses and becomes visible at a finite distance from the stack or source emissions outlet, the opacity of the emissions shall be measured in the region of the plume clearly above the emissions outlet and below condensation of the water vapor.
During daylight hours the lidar operators can visually determine if the steam plume is detached from the stack outlet. During nighttime hours a high-intensity spotlight, night vision scope, low light level TV, etc., can be used as an aid in determining if the steam plume is detached. If visual determination is ineffective, the lidar may be used to determine if the steam plume is detached by repeatedly measuring plume opacity from the outlet to the steam plume along the plume's longitudinal axis or center line. The lidar operator should also observe color differences and plume reflectivity to detect a detached plume. If the operator does not obtain a clear indication of the location of the detached plume, this method shall not be used to make opacity measurements between the outlet and the detached plume.
Once the determination of a detached steam plume has been confirmed, the laser shall be aimed into the region of highest opacity in the plume between the outlet and the formation of the steam plume. Aiming adjustments shall be made to the lidar's line-of-sight within the plume to correct for changes in the location of the most dense region of the plume due to changes in wind direction and speed or if the detached steam plume moves closer to the source outlet encroaching on the most dense region of the plume. If the detached steam plume should move too close to the source outlet for the lidar to make interference-free opacity measurements, this method shall not be used.
2.5 Field Records. In addition to the recording recommendations listed in other sections of this method the following records should be maintained. Each plume measured should be uniquely identified. The name of the facility, type of facility, emission source type, geographic location of the lidar with respect to the plume, and plume characteristics should be recorded. The date of the test, the time period that a source was monitored, the time (to the nearest second) of each opacity measurement, and the sample interval should also be recorded. The wind speed, wind direction, air temperature, relative humidity, visibility (measured at the lidar's position), and cloud cover should be recorded at the beginning and end of each time period for a given source. A small sketch depicting the location of the laser beam within the plume should be recorded.
If a detached or attached steam plume is present at the emissions source, this fact should be recorded. Figures AM1-I and AM1-II are examples of logbook forms that may be used to record this type of data. Magnetic tape or paper tape may also be used to record data.


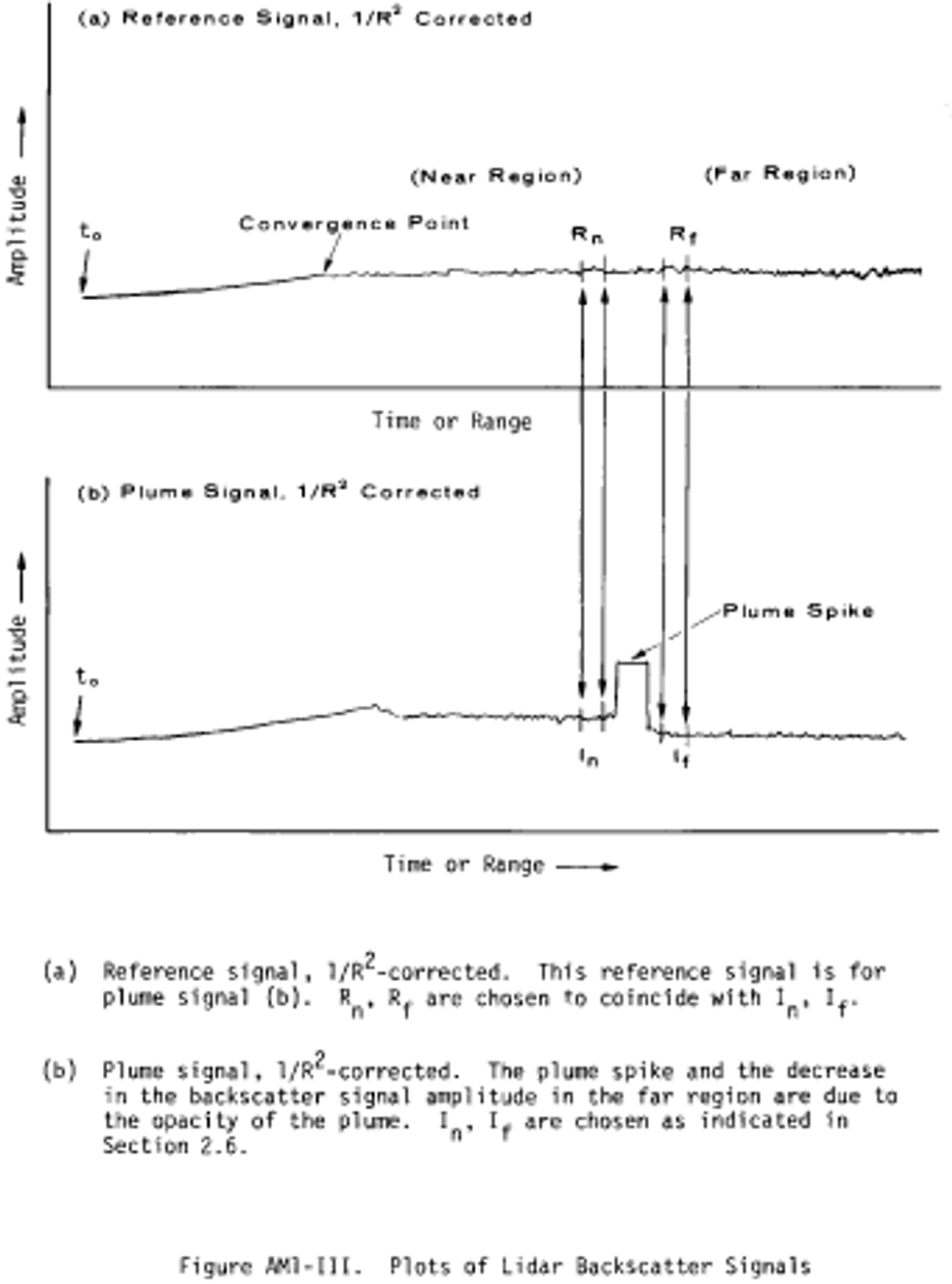
2.6 Opacity Calculation and Data Analysis. Referring to the reference signal and plume signal in Figure AM1-III, the measured opacity (Op) in percent for each lidar measurement is calculated using Equation AM1-2. (Op = 1−Tp; Tp is the plume transmittance.)
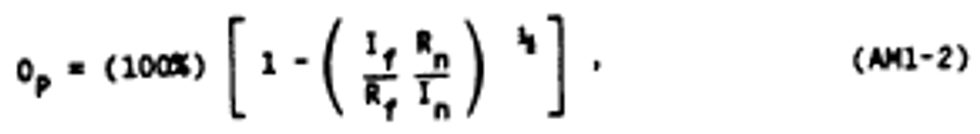
Where:
In = near-region pick interval signal amplitude, plume signal, 1/R 2 corrected,
If = far-region pick interval signal amplitude, plume signal, 1/R 2 corrected,
Rn = near-region pick interval signal amplitude, reference signal, 1/R 2 corrected, and
Rf = far-region pick interval signal amplitude, reference signal, 1/R 2 corrected.
The 1/R 2 correction to the plume and reference signal amplitudes is made by multiplying the amplitude for each successive sample interval from the time reference, by the square of the lidar time (or range) associated with that sample interval [Reference 5.1].
The first step in selecting the pick intervals for Equation AM1-2 is to divide the plume signal amplitude by the reference signal amplitude at the same respective ranges to obtain a “normalized” signal. The pick intervals selected using this normalized signal, are a minimum of 15 m (100 nanoseconds) in length and consist of at least 5 contiguous sample intervals. In addition, the following criteria, listed in order of importance, govern pick interval selection. (1) The intervals shall be in a region of the normalized signal where the reference signal meets the requirements of section 2.3 and is everywhere greater than zero. (2) The intervals (near and far) with the minimum average amplitude are chosen. (3) If more than one interval with the same minimum average amplitude is found, the interval closest to the plume is chosen. (4) The standard deviation, So, for the calculated opacity shall be 8% or less. (So is calculated by Equation AM1-7).
If So is greater than 8%, then the far pick interval shall be changed to the next interval of minimal average amplitude. If So is still greater than 8%, then this procedure is repeated for the far pick interval. This procedure may be repeated once again for the near pick interval, but if So remains greater than 8%, the plume signal shall be discarded.
The reference signal pick intervals, Rn and Rf, must be chosen over the same time interval as the plume signal pick intervals, In and If, respectively [Figure AM1-III]. Other methods of selecting pick intervals may be used if they give equivalent results. Field-oriented examples of pick interval selection are available in Reference 5.1.
The average amplitudes for each of the pick intervals, In, If, Rn, Rf, shall be calculated by averaging the respective individual amplitudes of the sample intervals from the plume signal and the associated reference signal each corrected for 1/R 2. The amplitude of In shall be calculated according to Equation (AM-3).
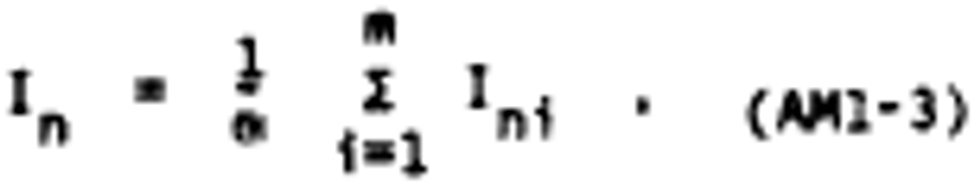
Where:
Ini = the amplitude of the ith sample interval (near-region),
Σ = sum of the individual amplitudes for the sample intervals,
m = number of sample intervals in the pick interval, and
In = average amplitude of the near-region pick interval.
Similarly, the amplitudes for If, Rn, and Rf are calculated with the three expressions in Equation (AM1-4).
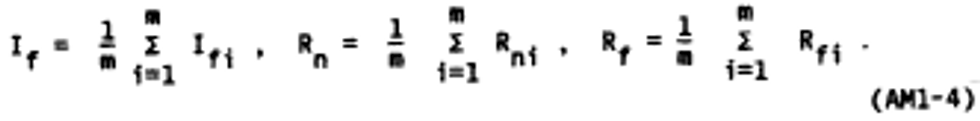
The standard deviation, SIn, of the set of amplitudes for the near-region pick interval, In, shall be calculated using Equation (AM1-5).
Similarly, the standard deviations SIf, SRn, and SRf are calculated with the three expressions in Equation (AM1-6).
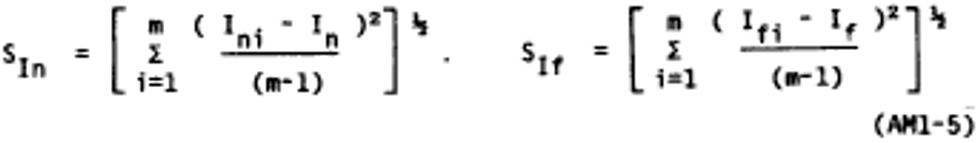
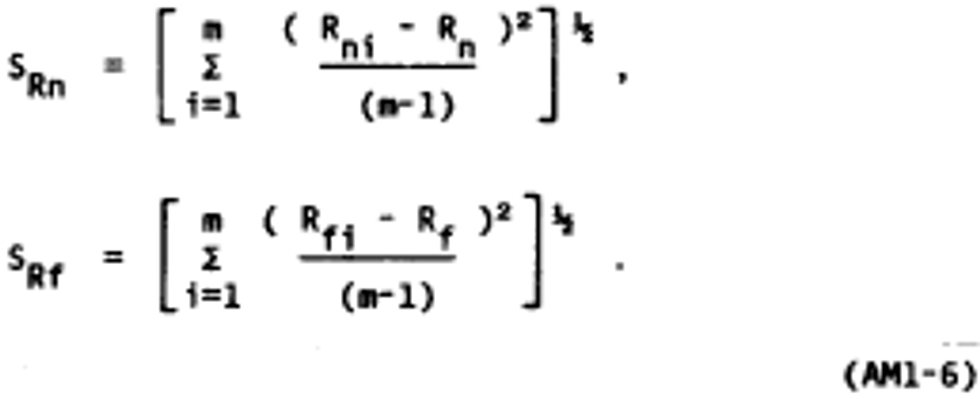
The standard deviation, So, for each associated opacity value, Op, shall be calculated using Equation (AM1-7).
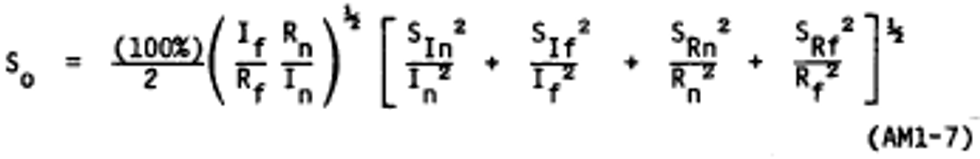
The calculated values of In, If, Rn, Rf, SIn, SIf, SRn, SRf, Op, and So should be recorded. Any plume signal with an So greater than 8% shall be discarded.
2.6.1 Azimuth Angle Correction. If the azimuth angle correction to opacity specified in this section is performed, then the elevation angle correction specified in section 2.6.2 shall not be performed. When opacity is measured in the residual region of an attached steam plume, and the lidar line-of-sight is not perpendicular to the plume, it may be necessary to correct the opacity measured by the lidar to obtain the opacity that would be measured on a path perpendicular to the plume. The following method, or any other method which produces equivalent results, shall be used to determine the need for a correction, to calculate the correction, and to document the point within the plume at which the opacity was measured.
Figure AM1-IV(b) shows the geometry of the opacity correction. L′ is the path through the plume along which the opacity measurement is made. P′ is the path perpendicular to the plume at the same point. The angle ε is the angle between L′ and the plume center line. The angle (π/2-ε), is the angle between the L′ and P′. The measured opacity, Op, measured along the path L′ shall be corrected to obtain the corrected opacity, Opc, for the path P′, using Equation (AM1-8).
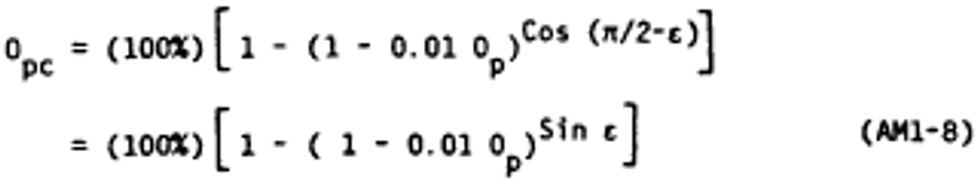
The correction in Equation (AM1-8) shall be performed if the inequality in Equation (AM1-9) is true.
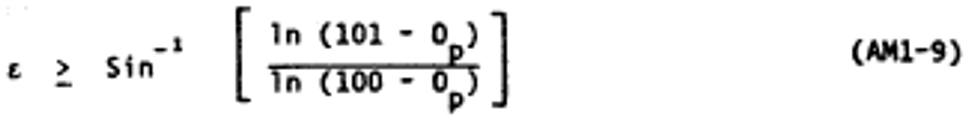
Figure AM1-IV(a) shows the geometry used to calculate ε and the position in the plume at which the lidar measurement is made. This analysis assumes that for a given lidar measurement, the range from the lidar to the plume, the elevation angle of the lidar from the horizontal plane, and the azimuth angle of the lidar from an arbitrary fixed reference in the horizontal plane can all be obtained directly.
Rs = range from lidar to source*
βs = elevation angle of Rs*
Rp = range from lidar to plume at the opacity measurement point*
βp = elevation angle of Rp*
Ra = range from lidar to plume at some arbitrary point, Pa, so the drift angle of the plume can be determined*
βa = elevation angle of Ra*
α = angle between Rp and Ra
R′s = projection of Rs in the horizontal plane
R′p = projection of Rp in the horizontal plane
R′a = projection of Ra in the horizontal plane
ψ′ = angle between R′s and R′p*
α′ = angle between R′p and R′a*
R≤ = distance from the source to the opacity measurement point projected in the horizontal plane
Rθ = distance from opacity measurement point Pp to the point in the plume Pa.
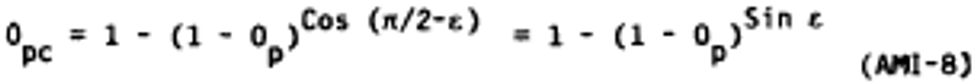
The correction angle ε shall be determined using Equation AM1-10.
*Obtained directly from lidar. These values should be recorded.
Where:
α = Cos−1 (Cosβp Cosβa Cosα′ Sinβp Sinβa),
and
Rθ = (Rp2 Ra2 − 2 Rp Ra Cosα) 1/2
R≤, the distance from the source to the opacity measurement point projected in the horizontal plane, shall be determined using Equation AM1-11.
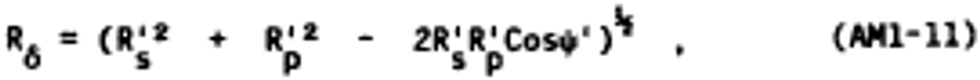
Where:
R′s = Rs Cos βs, and
R′p = Rp Cos βp.
In the special case where the plume centerline at the opacity measurement point is horizontal, parallel to the ground, Equation AM1-12 may be used to determine ε instead of Equation AM1-10.
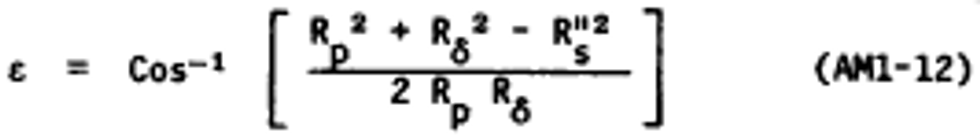
Where:
R″s = (R′ 2 s Rp 2Sin 2βp)1/2.
If the angle ε is such that ε≤30° or ε ≥150°, the azimuth angle correction shall not be performed and the associated opacity value shall be discarded.
2.6.2 Elevation Angle Correction. An individual lidar-measured opacity, Op, shall be corrected for elevation angle if the laser elevation or inclination angle, βp [Figure AM1-V], is greater than or equal to the value calculated in Equation AM1-13.
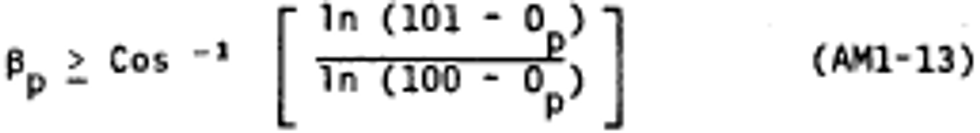
The measured opacity, Op, along the lidar path L, is adjusted to obtain the corrected opacity, Opc, for the actual plume (horizontal) path, P, by using Equation (AM1-14).

Where:
βp = lidar elevation or inclination angle,
Op = measured opacity along path L, and
Opc = corrected opacity for the actual plume thickness P.
The values for βp, Op and Opc should be recorded.
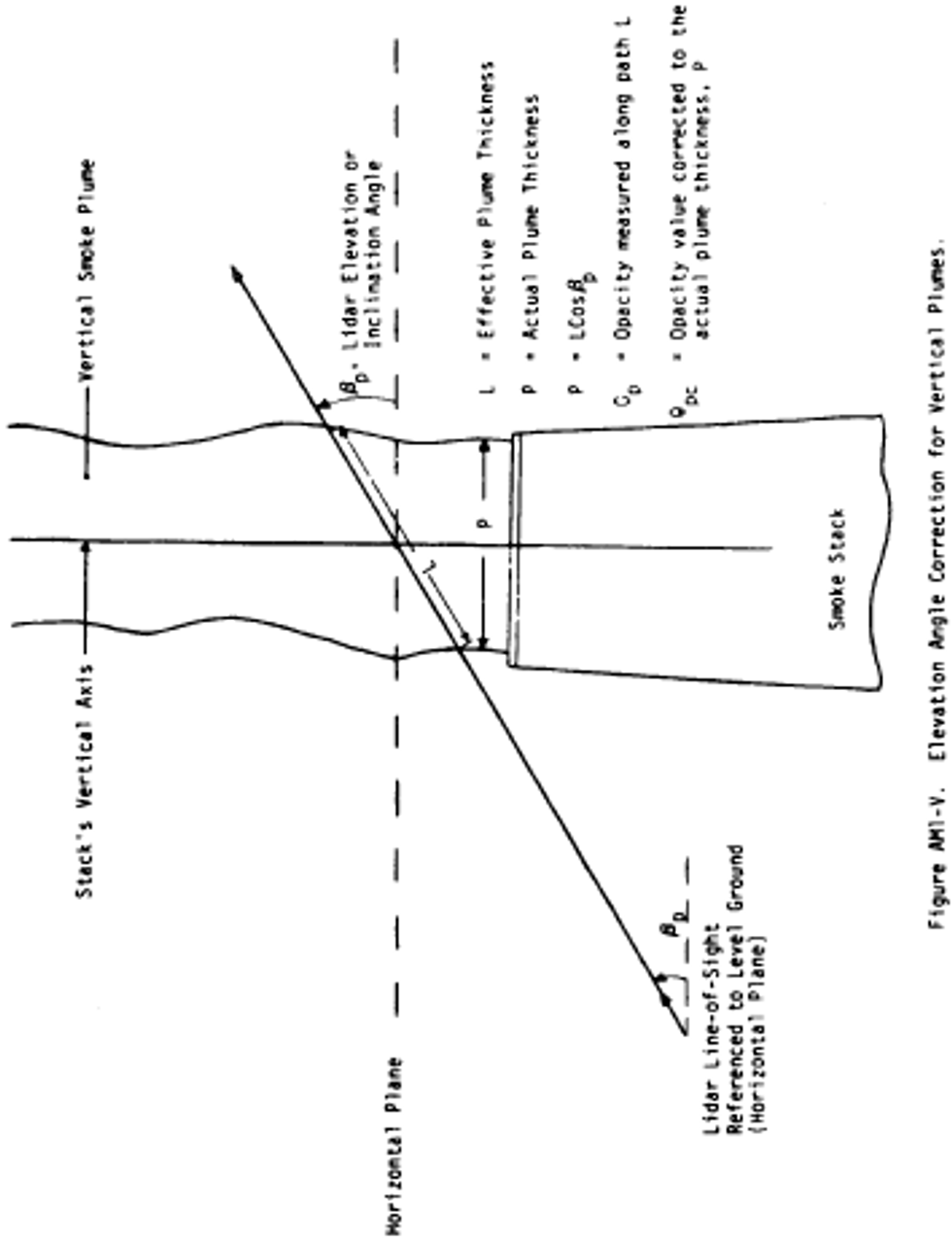
2.6.3 Determination of Actual Plume Opacity. Actual opacity of the plume shall be determined by Equation AM1-15.
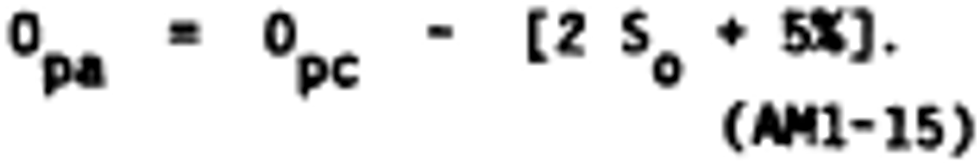
2.6.4 Calculation of Average Actual Plume Opacity. The average of the actual plume opacity, Opa, shall be calculated as the average of the consecutive individual actual opacity values, Opa, by Equation AM1-16.
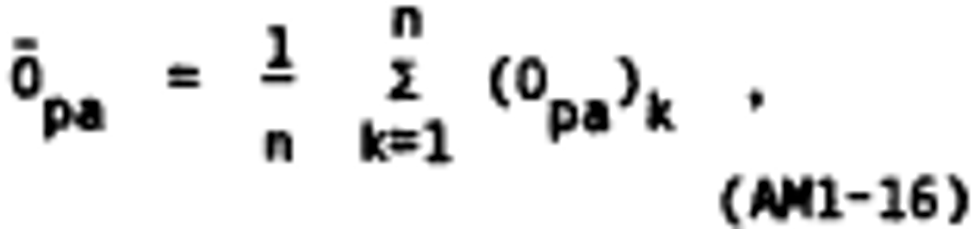
Where:
(Opa)k = the kth actual opacity value in an averaging interval containing n opacity values; k is a summing index.
Σ = the sum of the individual actual opacity values.
n = the number of individual actual opacity values contained in the averaging interval.
Opa = average actual opacity calculated over the averaging interval.
3. Lidar Performance Verification
The lidar shall be subjected to two types of performance verifications that shall be performed in the field. The annual calibration, conducted at least once a year, shall be used to directly verify operation and performance of the entire lidar system. The routine verification, conducted for each emission source measured, shall be used to insure proper performance of the optical receiver and associated electronics.
3.1 Annual Calibration Procedures. Either a plume from a smoke generator or screen targets shall be used to conduct this calibration.
If the screen target method is selected, five screens shall be fabricated by placing an opaque mesh material over a narrow frame (wood, metal extrusion, etc.). The screen shall have a surface area of at least one square meter. The screen material should be chosen for precise optical opacities of about 10, 20, 40, 60, and 80%. Opacity of each target shall be optically determined and should be recorded. If a smoke generator plume is selected, it shall meet the requirements of section 3.3 of Reference Method 9. This calibration shall be performed in the field during calm (as practical) atmospheric conditions. The lidar shall be positioned in accordance with section 2.1.
The screen targets must be placed perpendicular to and coincident with the lidar line-of-sight at sufficient height above the ground (suggest about 30 ft) to avoid ground-level dust contamination. Reference signals shall be obtained just prior to conducting the calibration test.
The lidar shall be aimed through the center of the plume within 1 stack diameter of the exit, or through the geometric center of the screen target selected. The lidar shall be set in operation for a 6-minute data run at a nominal pulse rate of 1 pulse every 10 seconds. Each backscatter return signal and each respective opacity value obtained from the smoke generator transmissometer, shall be obtained in temporal coincidence. The data shall be analyzed and reduced in accordance with section 2.6 of this method. This calibration shall be performed for 0% (clean air), and at least five other opacities (nominally 10, 20, 40, 60, and 80%).
The average of the lidar opacity values obtained during a 6-minute calibration run shall be calculated and should be recorded. Also the average of the opacity values obtained from the smoke generator transmissometer for the same 6-minute run shall be calculated and should be recorded.
Alternate calibration procedures that do not meet the above requirements but produce equivalent results may be used.
3.2 Routine Verification Procedures. Either one of two techniques shall be used to conduct this verification. It shall be performed at least once every 4 hours for each emission source measured. The following parameters shall be directly verified.
1) The opacity value of 0% plus a minimum of 5 (nominally 10, 20, 40, 60, and 80%) opacity values shall be verified through the PMT detector and data processing electronics.
2) The zero-signal level (receiver signal with no optical signal from the source present) shall be inspected to insure that no spurious noise is present in the signal. With the entire lidar receiver and analog/digital electronics turned on and adjusted for normal operating performance, the following procedures shall be used for Techniques 1 and 2, respectively.
3.2.1 Procedure for Technique 1. This test shall be performed with no ambient or stray light reaching the PMT detector. The narrow band filter (694.3 nanometers peak) shall be removed from its position in front of the PMT detector. Neutral density filters of nominal opacities of 10, 20, 40, 60, and 80% shall be used. The recommended test configuration is depicted in Figure AM1-VI.
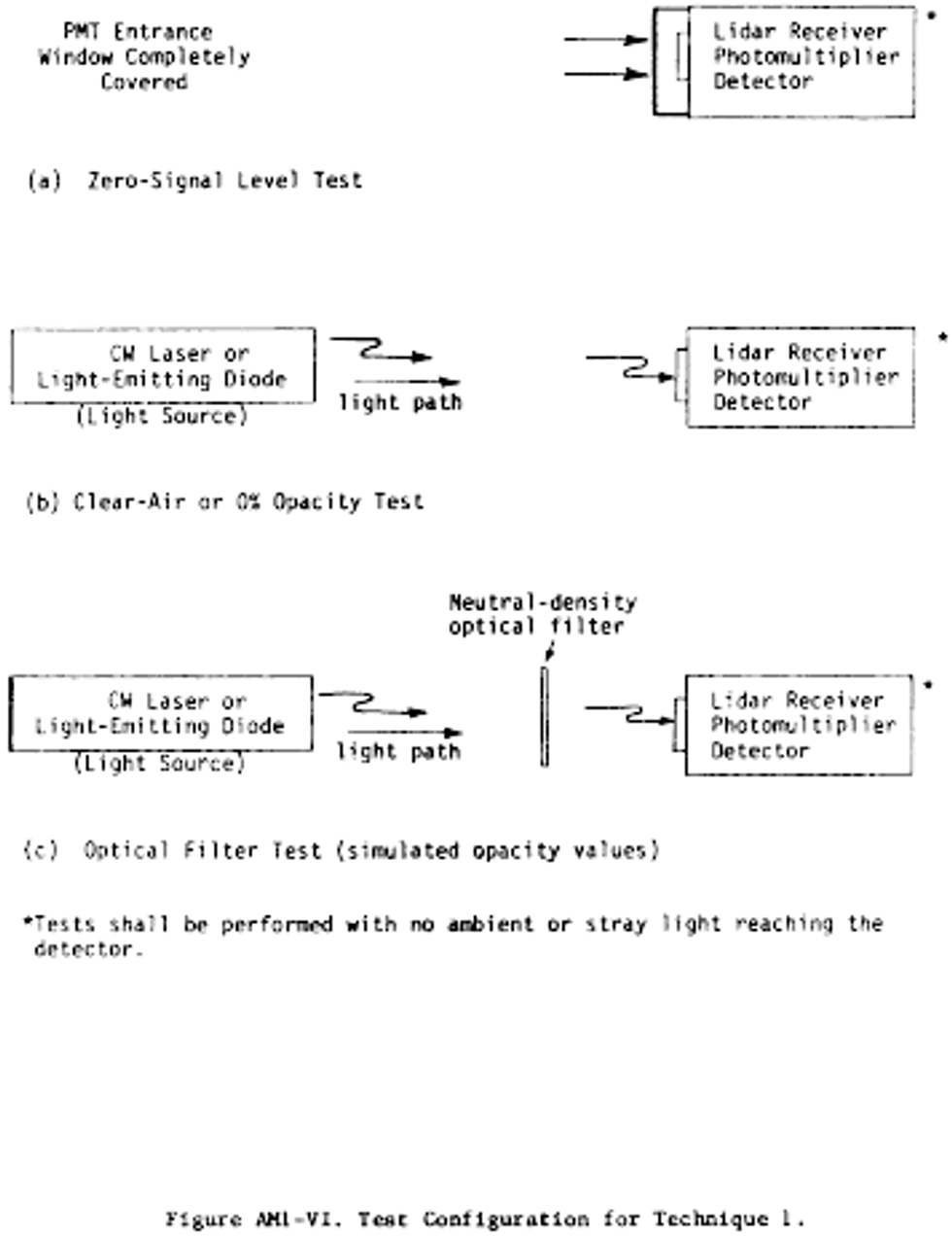
The zero-signal level shall be measured and should be recorded, as indicated in Figure AM1-VI(a). This simulated clear-air or 0% opacity value shall be tested in using the selected light source depicted in Figure AM1-VI(b).
The light source either shall be a continuous wave (CW) laser with the beam mechanically chopped or a light emitting diode controlled with a pulse generator (rectangular pulse). (A laser beam may have to be attenuated so as not to saturate the PMT detector). This signal level shall be measured and should be recorded. The opacity value is calculated by taking two pick intervals [Section 2.6] about 1 microsecond apart in time and using Equation (AM1-2) setting the ratio Rn/Rf = 1. This calculated value should be recorded.
The simulated clear-air signal level is also employed in the optical test using the neutral density filters. Using the test configuration in Figure AM1-VI(c), each neutral density filter shall be separately placed into the light path from the light source to the PMT detector. The signal level shall be measured and should be recorded. The opacity value for each filter is calculated by taking the signal level for that respective filter (If), dividing it by the 0% opacity signal level (In) and performing the remainder of the calculation by Equation (AM1-2) with Rn/Rf = 1. The calculated opacity value for each filter should be recorded.
The neutral density filters used for Technique 1 shall be calibrated for actual opacity with accuracy of ±2% or better. This calibration shall be done monthly while the filters are in use and the calibrated values should be recorded.
3.2.2 Procedure for Technique 2. An optical generator (built-in calibration mechanism) that contains a light-emitting diode (red light for a lidar containing a ruby laser) is used. By injecting an optical signal into the lidar receiver immediately ahead of the PMT detector, a backscatter signal is simulated. With the entire lidar receiver electronics turned on and adjusted for normal operating performance, the optical generator is turned on and the simulation signal (corrected for 1/R 2) is selected with no plume spike signal and with the opacity value equal to 0%. This simulated clear-air atmospheric return signal is displayed on the system's video display. The lidar operator then makes any fine adjustments that may be necessary to maintain the system's normal operating range.
The opacity values of 0% and the other five values are selected one at a time in any order. The simulated return signal data should be recorded. The opacity value shall be calculated. This measurement/calculation shall be performed at least three times for each selected opacity value. While the order is not important, each of the opacity values from the optical generator shall be verified. The calibrated optical generator opacity value for each selection should be recorded.
The optical generator used for Technique 2 shall be calibrated for actual opacity with an accuracy of ±1% or better. This calibration shall be done monthly while the generator is in use and calibrated value should be recorded.
Alternate verification procedures that do not meet the above requirements but produce equivalent results may be used.
3.3 Deviation. The permissible error for the annual calibration and routine verification are:
3.3.1 Annual Calibration Deviation.
3.3.1.1 Smoke Generator. If the lidar-measured average opacity for each data run is not within ±5% (full scale) of the respective smoke generator's average opacity over the range of 0% through 80%, then the lidar shall be considered out of calibration.
3.3.1.2 Screens. If the lidar-measured average opacity for each data run is not within ±3% (full scale) of the laboratory-determined opacity for each respective simulation screen target over the range of 0% through 80%, then the lidar shall be considered out of calibration.
3.3.2 Routine Verification Error. If the lidar-measured average opacity for each neutral density filter (Technique 1) or optical generator selection (Technique 2) is not within ±3% (full scale) of the respective laboratory calibration value then the lidar shall be considered non-operational.
4. Performance/Design Specification for Basic Lidar System
4.1 Lidar Design Specification. The essential components of the basic lidar system are a pulsed laser (transmitter), optical receiver, detector, signal processor, recorder, and an aiming device that is used in aiming the lidar transmitter and receiver. Figure AM1-VII shows a functional block diagram of a basic lidar system.
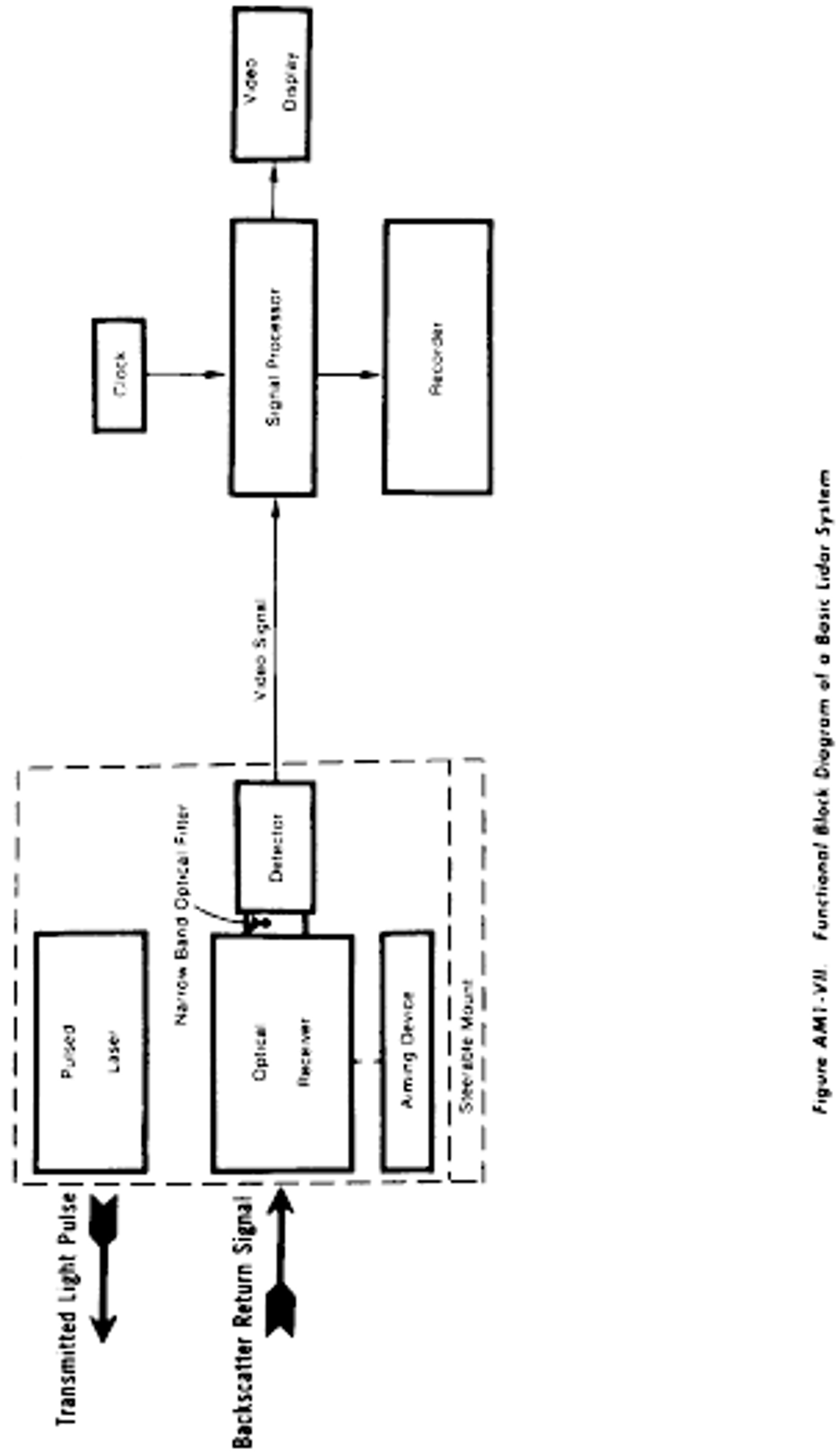
4.2 Performance Evaluation Tests. The owner of a lidar system shall subject such a lidar system to the performance verification tests described in section 3, prior to first use of this method. The annual calibration shall be performed for three separate, complete runs and the results of each should be recorded. The requirements of section 3.3.1 must be fulfilled for each of the three runs.
Once the conditions of the annual calibration are fulfilled the lidar shall be subjected to the routine verification for three separate complete runs. The requirements of section 3.3.2 must be fulfilled for each of the three runs and the results should be recorded. The Administrator may request that the results of the performance evaluation be submitted for review.
5. References
5.1 The Use of Lidar for Emissions Source Opacity Determination, U.S. Environmental Protection Agency, National Enforcement Investigations Center, Denver, CO. EPA-330/1-79-003-R, Arthur W. Dybdahl, current edition [NTIS No. PB81-246662].
5.2 Field Evaluation of Mobile Lidar for the Measurement of Smoke Plume Opacity, U.S. Environmental Protection Agency, National Enforcement Investigations Center, Denver, CO. EPA/NEIC-TS-128, February 1976.
5.3 Remote Measurement of Smoke Plume Transmittance Using Lidar, C. S. Cook, G. W. Bethke, W. D. Conner (EPA/RTP). Applied Optics 11, pg 1742. August 1972.
5.4 Lidar Studies of Stack Plumes in Rural and Urban Environments, EPA-650/4-73-002, October 1973.
5.5 American National Standard for the Safe Use of Lasers ANSI Z 136.1-176, March 8, 1976.
5.6 U.S. Army Technical Manual TB MED 279, Control of Hazards to Health from Laser Radiation, February 1969.
5.7 Laser Institute of America Laser Safety Manual, 4th Edition.
5.8 U.S. Department of Health, Education and Welfare, Regulations for the Administration and Enforcement of the Radiation Control for Health and Safety Act of 1968, January 1976.
5.9 Laser Safety Handbook, Alex Mallow, Leon Chabot, Van Nostrand Reinhold Co., 1978.
Method 10 - Determination of Carbon Monoxide Emissions From Stationary Sources (Instrumental Analyzer Procedure)
1.0 Scope and Application
What is Method 10?
Method 10 is a procedure for measuring carbon monoxide (CO) in stationary source emissions using a continuous instrumental analyzer. Quality assurance and quality control requirements are included to assure that you, the tester, collect data of known quality. You must document your adherence to these specific requirements for equipment, supplies, sample collection and analysis, calculations, and data analysis. This method does not completely describe all equipment, supplies, and sampling and analytical procedures you will need but refers to other methods for some of the details. Therefore, to obtain reliable results, you should also have a thorough knowledge of these additional test methods which are found in appendix A to this part:
(a) Method 1 - Sample and Velocity Traverses for Stationary Sources.
(b) Method 4 - Determination of Moisture Content in Stack Gases.
(c) Method 7E - Determination of Nitrogen Oxides Emissions from Stationary Sources (Instrumental Analyzer Procedure).
1.1 Analytes. What does this method determine? This method measures the concentration of carbon monoxide.
| Analyte | CAS No. | Sensitivity |
|---|---|---|
| CO | 630-08-0 | Typically <2% of Calibration Span. |
1.2 Applicability. When is this method required? The use of Method 10 may be required by specific New Source Performance Standards, State Implementation Plans, and permits where CO concentrations in stationary source emissions must be measured, either to determine compliance with an applicable emission standard or to conduct performance testing of a continuous emission monitoring system (CEMS). Other regulations may also require the use of Method 10.
1.3 Data Quality Objectives. Refer to section 1.3 of Method 7E.
2.0 Summary of Method
In this method, you continuously or intermittently sample the effluent gas and convey the sample to an analyzer that measures the concentration of CO. You must meet the performance requirements of this method to validate your data.
3.0 Definitions
Refer to section 3.0 of Method 7E for the applicable definitions.
4.0 Interferences
Substances having a strong absorption of infrared energy may interfere to some extent in some analyzers. Instrumental correction may be used to compensate for the interference. You may also use silica gel and ascarite traps to eliminate the interferences. If this option is used, correct the measured gas volume for the carbon dioxide (CO2) removed in the trap.
5.0 Safety
Refer to section 5.0 of Method 7E.
6.0 Equipment and Supplies
What do I need for the measurement system?
6.1 Continuous Sampling. Figure 7E-1 of Method 7E is a schematic diagram of an acceptable measurement system. The components are the same as those in sections 6.1 and 6.2 of Method 7E, except that the CO analyzer described in section 6.2 of this method must be used instead of the analyzer described in section 6.2 of Method 7E. You must follow the noted specifications in section 6.1 of Method 7E except that the requirements to use stainless steel, Teflon, or non-reactive glass filters do not apply. Also, a heated sample line is not required to transport dry gases or for systems that measure the CO concentration on a dry basis.
6.2 Integrated Sampling.
6.2.1 Air-Cooled Condenser or Equivalent. To remove any excess moisture.
6.2.2 Valve. Needle valve, or equivalent, to adjust flow rate.
6.2.3 Pump. Leak-free diaphragm type, or equivalent, to transport gas.
6.2.4 Rate Meter. Rotameter, or equivalent, to measure a flow range from 0 to 1.0 liter per minute (0.035 cfm).
6.2.5 Flexible Bag. Tedlar, or equivalent, with a capacity of 60 to 90 liters (2 to 3 ft 3). (Verify through the manufacturer that the Tedlar alternative is suitable for CO and make this verified information available for inspection.) Leak-test the bag in the laboratory before using by evacuating with a pump followed by a dry gas meter. When the evacuation is complete, there should be no flow through the meter.
6.2.6 Sample Tank. Stainless steel or aluminum tank equipped with a pressure indicator with a minimum volume of 4 liters.
6.3 What analyzer must I use? You must use an instrument that continuously measures CO in the gas stream and meets the specifications in section 13.0. The dual-range analyzer provisions in section 6.2.8.1 of Method 7E apply.
7.0 Reagents and Standards
7.1 Calibration Gas. What calibration gases do I need? Refer to section 7.1 of Method 7E for the calibration gas requirements.
7.2 Interference Check. What additional reagents do I need for the interference check? Use the appropriate test gases listed in Table 7E-3 of Method 7E (i.e., potential interferents, as identified by the instrument manufacturer) to conduct the interference check.
8.0 Sample Collection, Preservation, Storage, and Transport
Emission Test Procedure
8.1 Sampling Site and Sampling Points. You must follow section 8.1 of Method 7E.
8.2 Initial Measurement System Performance Tests. You must follow the procedures in section 8.2 of Method 7E. If a dilution-type measurement system is used, the special considerations in section 8.3 of Method 7E also apply.
8.3 Interference Check. You must follow the procedures of section 8.2.7 of Method 7E.
8.4 Sample Collection.
8.4.1 Continuous Sampling. You must follow the procedures of section 8.4 of Method 7E.
8.4.2 Integrated Sampling. Evacuate the flexible bag or sample tank. Set up the equipment as shown in Figure 10-1 with the bag disconnected. Place the probe in the stack and purge the sampling line. Connect the bag, making sure that all connections are leak-free. Sample at a rate proportional to the stack velocity. If needed, the CO2 content of the gas may be determined by using the Method 3 integrated sample procedures, or by weighing an ascarite CO2 removal tube used and computing CO2 concentration from the gas volume sampled and the weight gain of the tube. Data may be recorded on a form similar to Table 10-1. If a sample tank is used for sample collection, follow procedures similar to those in sections 8.1.2, 8.2.3, 8.3, and 12.4 of Method 25 as appropriate to prepare the tank, conduct the sampling, and correct the measured sample concentration.
8.5 Post-Run System Bias Check, Drift Assessment, and Alternative Dynamic Spike Procedure. You must follow the procedures in sections 8.5 and 8.6 of Method 7E.
9.0 Quality Control
Follow the quality control procedures in section 9.0 of Method 7E.
10.0 Calibration and Standardization
Follow the procedures for calibration and standardization in section 10.0 of Method 7E.
11.0 Analytical Procedures
Because sample collection and analysis are performed together (see section 8), additional discussion of the analytical procedure is not necessary.
12.0 Calculations and Data Analysis
You must follow the procedures for calculations and data analysis in section 12.0 of Method 7E, as applicable, substituting CO for NOX as applicable.
12.1 Concentration Correction for CO 2 Removal. Correct the CO concentration for CO2 removal (if applicable) using Eq. 10-1.

Where:
CAvg = Average gas concentration for the test run, ppm.
CCO stack = Average unadjusted stack gas CO concentration indicated by the data recorder for the test run, ppmv.
FCO2 = Volume fraction of CO2 in the sample, i.e., percent CO2 from Orsat analysis divided by 100.
13.0 Method Performance
The specifications for analyzer calibration error, system bias, drift, interference check, and alternative dynamic spike procedure are the same as in section 13.0 of Method 7E.
14.0 Pollution Prevention [Reserved]
15.0 Waste Management [Reserved]
16.0 Alternative Procedures
The dynamic spike procedure and the manufacturer stability test are the same as in sections 16.1 and 16.3 of Method 7E
17.0 References
1. “EPA Traceability Protocol for Assay and Certification of Gaseous Calibration Standards - September 1997 as amended, EPA-600/R-97/121
18.0 Tables, Diagrams, Flowcharts, and Validation Data
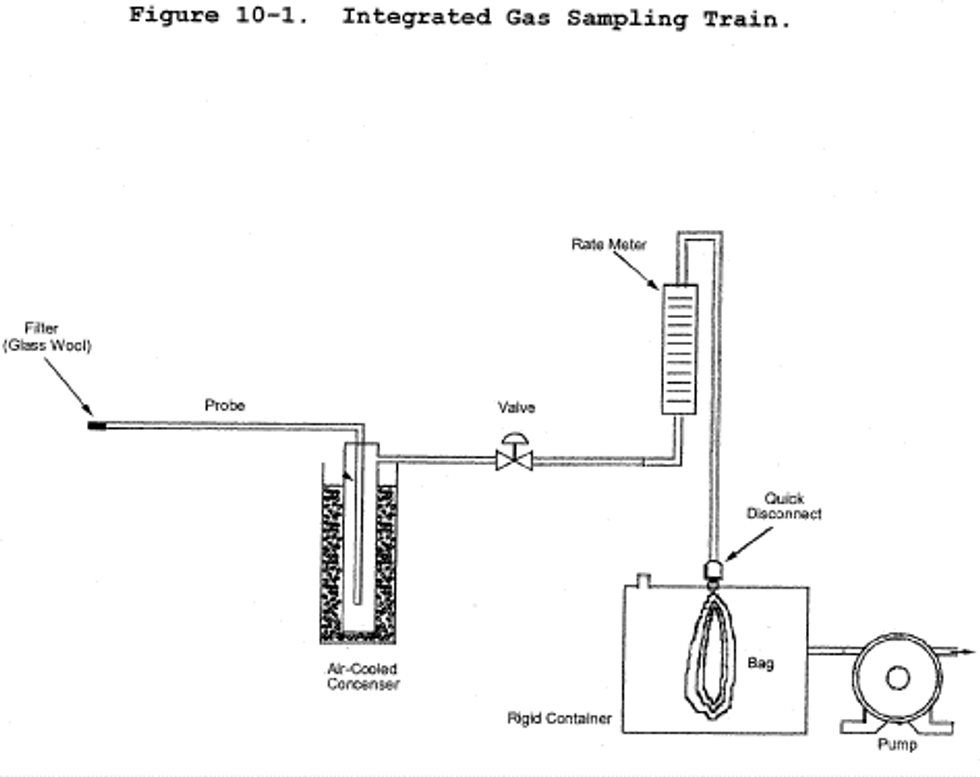
| Location: Date: | ||
| Test: Operator: | ||
| Clock Time | Rotameter Reading liters/min (cfm) | Comments |
Method 10A - Determination of Carbon Monoxide Emissions in Certifying Continuous Emission Monitoring Systems at Petroleum Refineries
Note:
This method does not include all of the specifications (e.g., equipment and supplies) and procedures (e.g., sampling and analytical) essential to its performance. Some material is incorporated by reference from other methods in this part. Therefore, to obtain reliable results, persons using this method should have a thorough knowledge of at least the following additional test methods: Method 1, Method 4, and Method 5.
1.0 Scope and Application
1.1 Analytes.
| Analyte | CAS No. | Sensitivity |
|---|---|---|
| Carbon monoxide (CO) | 630-08-0 | 3 ppmv |
1.2 Applicability. This method is applicable for the determination of CO emissions at petroleum refineries. This method serves as the reference method in the relative accuracy test for nondispersive infrared (NDIR) CO continuous emission monitoring systems (CEMS) that are required to be installed in petroleum refineries on fluid catalytic cracking unit catalyst regenerators (§60.105(a)(2) of this part).
1.3 Data Quality Objectives. Adherence to the requirements of this method will enhance the quality of the data obtained from air pollutant sampling methods.
2.0 Summary of Method
An integrated gas sample is extracted from the stack, passed through an alkaline permanganate solution to remove sulfur oxides and nitrogen oxides, and collected in a Tedlar or equivalent bag. (Verify through the manufacturer that the Tedlar alternative is suitable for NO and make this verified information available for inspection.) The CO concentration in the sample is measured spectrophotometrically using the reaction of CO with p-sulfaminobenzoic acid.
3.0 Definitions [Reserved]
4.0 Interferences
Sulfur oxides, nitric oxide, and other acid gases interfere with the colorimetric reaction. They are removed by passing the sampled gas through an alkaline potassium permanganate scrubbing solution. Carbon dioxide (CO2) does not interfere, but, because it is removed by the scrubbing solution, its concentration must be measured independently and an appropriate volume correction made to the sampled gas.
5.0 Safety
5.1 Disclaimer. This method may involve hazardous materials, operations, and equipment. This test method may not address all of the safety problems associated with its use. It is the responsibility of the user of this test method to establish appropriate safety and health practices and determine the applicability of regulatory limitations prior to performing this test method. The analyzer users manual should be consulted for specific precautions to be taken with regard to the analytical procedure.
5.2 Corrosive reagents. The following reagents are hazardous. Personal protective equipment and safe procedures are useful in preventing chemical splashes. If contact occurs, immediately flush with copious amounts of water for at least 15 minutes. Remove clothing under shower and decontaminate. Treat residual chemical burns as thermal burns.
5.2.1 Sodium Hydroxide (NaOH). Causes severe damage to eyes and skin. Inhalation causes irritation to nose, throat, and lungs. Reacts exothermically with limited amounts of water.
6.0 Equipment and Supplies
6.1 Sample Collection. The sampling train shown in Figure 10A-1 is required for sample collection. Component parts are described below:
6.1.1 Probe. Stainless steel, sheathed Pyrex glass, or equivalent, equipped with a glass wool plug to remove particulate matter.
6.1.2 Sample Conditioning System. Three Greenburg-Smith impingers connected in series with leak-free connections.
6.1.3 Pump. Leak-free pump with stainless steel and Teflon parts to transport sample at a flow rate of 300 ml/min (0.01 ft 3/min) to the flexible bag.
6.1.4 Surge Tank. Installed between the pump and the rate meter to eliminate the pulsation effect of the pump on the rate meter.
6.1.5 Rate Meter. Rotameter, or equivalent, to measure flow rate at 300 ml/min (0.01 ft 3/min). Calibrate according to section 10.2.
6.1.6 Flexible Bag. Tedlar, or equivalent, with a capacity of 10 liters (0.35 ft 3) and equipped with a sealing quick-connect plug. The bag must be leak-free according to section 8.1. For protection, it is recommended that the bag be enclosed within a rigid container.
6.1.7 Sample Tank. Stainless steel or aluminum tank equipped with a pressure indicator with a minimum volume of 10 liters.
6.1.8 Valves. Stainless-steel needle valve to adjust flow rate, and stainless-steel 3-way valve, or equivalent.
6.1.9 CO2 Analyzer. Fyrite, or equivalent, to measure CO2 concentration to within 0.5 percent.
6.1.10 Volume Meter. Dry gas meter, capable of measuring the sample volume under calibration conditions of 300 ml/min (0.01 ft 3/min) for 10 minutes.
6.1.11 Pressure Gauge. A water filled U-tube manometer, or equivalent, of about 30 cm (12 in.) to leak-check the flexible bag.
6.2 Sample Analysis.
6.2.1 Spectrophotometer. Single- or double-beam to measure absorbance at 425 and 600 nm. Slit width should not exceed 20 nm.
6.2.2 Spectrophotometer Cells. 1-cm pathlength.
6.2.3 Vacuum Gauge. U-tube mercury manometer, 1 meter (39 in.), with 1-mm divisions, or other gauge capable of measuring pressure to within 1 mm Hg.
6.2.4 Pump. Capable of evacuating the gas reaction bulb to a pressure equal to or less than 40 mm Hg absolute, equipped with coarse and fine flow control valves.
6.2.5 Barometer. Mercury, aneroid, or other barometer capable of measuring atmospheric pressure to within 1 mm Hg.
6.2.6 Reaction Bulbs. Pyrex glass, 100-ml with Teflon stopcock (Figure 10A-2), leak-free at 40 mm Hg, designed so that 10 ml of the colorimetric reagent can be added and removed easily and accurately. Commercially available gas sample bulbs such as Supelco Catalog No. 2-2161 may also be used.
6.2.7 Manifold. Stainless steel, with connections for three reaction bulbs and the appropriate connections for the manometer and sampling bag as shown in Figure 10A-3.
6.2.8 Pipets. Class A, 10-ml size.
6.2.9 Shaker Table. Reciprocating-stroke type such as Eberbach Corporation, Model 6015. A rocking arm or rotary-motion type shaker may also be used. The shaker must be large enough to accommodate at least six gas sample bulbs simultaneously. It may be necessary to construct a table top extension for most commercial shakers to provide sufficient space for the needed bulbs (Figure 10A-4).
6.2.10 Valve. Stainless steel shut-off valve.
6.2.11 Analytical Balance. Capable of weighing to 0.1 mg.
7.0 Reagents and Standards
Unless otherwise indicated, all reagents shall conform to the specifications established by the Committee on Analytical Reagents of the American Chemical Society, where such specifications are available; otherwise, the best available grade shall be used.
7.1 Sample Collection.
7.1.1 Water. Deionized distilled, to conform to ASTM D 1193-77 or 91, Type 3 (incorporated by reference - see §60.17). If high concentrations of organic matter are not expected to be present, the potassium permanganate test for oxidizable organic matter may be omitted.
7.1.2 Alkaline Permanganate Solution, 0.25 M KMnO4/1.5 M Sodium Hydroxide (NaOH). Dissolve 40 g KMnO4 and 60 g NaOH in approximately 900 ml water, cool, and dilute to 1 liter.
7.2 Sample Analysis.
7.2.1 Water. Same as in section 7.1.1.
7.2.2 1 M Sodium Hydroxide Solution. Dissolve 40 g NaOH in approximately 900 ml of water, cool, and dilute to 1 liter.
7.2.3 0.1 M NaOH Solution. Dilute 50 ml of the 1 M NaOH solution prepared in section 7.2.2 to 500 ml.
7.2.4 0.1 M Silver Nitrate (AgNO3) Solution. Dissolve 8.5 g AgNO3 in water, and dilute to 500 ml.
7.2.5 0.1 M Para-Sulfaminobenzoic Acid (p-SABA) Solution. Dissolve 10.0 g p-SABA in 0.1 M NaOH, and dilute to 500 ml with 0.1 M NaOH.
7.2.6 Colorimetric Solution. To a flask, add 100 ml of 0.1 M p-SABA solution and 100 ml of 0.1 M AgNO3 solution. Mix, and add 50 ml of 1 M NaOH with shaking. The resultant solution should be clear and colorless. This solution is acceptable for use for a period of 2 days.
7.2.7 Standard Gas Mixtures. Traceable to National Institute of Standards and Technology (NIST) standards and containing between 50 and 1000 ppm CO in nitrogen. At least two concentrations are needed to span each calibration range used (Section 10.3). The calibration gases must be certified by the manufacturer to be within 2 percent of the specified concentrations.
8.0 Sample Collection, Preservation, Storage, and Transport
8.1 Sample Bag or Tank Leak-Checks. While a leak-check is required after bag or sample tank use, it should also be done before the bag or sample tank is used for sample collection. The tank should be leak-checked according to the procedure specified in section 8.1.2 of Method 25. The bag should be leak-checked in the inflated and deflated condition according to the following procedure:
8.1.1 Connect the bag to a water manometer, and pressurize the bag to 5 to 10 cm H2O (2 to 4 in H2O). Allow the bag to stand for 60 minutes. Any displacement in the water manometer indicates a leak.
8.1.2 Evacuate the bag with a leakless pump that is connected to the downstream side of a flow indicating device such as a 0- to 100-ml/min rotameter or an impinger containing water. When the bag is completely evacuated, no flow should be evident if the bag is leak-free.
8.2 Sample Collection.
8.2.1 Evacuate and leak check the sample bag or tank as specified in section 8.1. Assemble the apparatus as shown in Figure 10A-1. Loosely pack glass wool in the tip of the probe. Place 400 ml of alkaline permanganate solution in the first two impingers and 250 ml in the third. Connect the pump to the third impinger, and follow this with the surge tank, rate meter, and 3-way valve. Do not connect the bag or sample tank to the system at this time.
8.2.2 Leak-check the sampling system by plugging the probe inlet, opening the 3-way valve, and pulling a vacuum of approximately 250 mm Hg on the system while observing the rate meter for flow. If flow is indicated on the rate meter, do not proceed further until the leak is found and corrected.
8.2.3 Purge the system with sample gas by inserting the probe into the stack and drawing the sample gas through the system at 300 ml/min ±10 percent for 5 minutes. Connect the evacuated bag or sample tank to the system, record the starting time, and sample at a rate of 300 ml/min for 30 minutes, or until the bag is nearly full, or the sample tank reaches ambient pressure. Record the sampling time, the barometric pressure, and the ambient temperature. Purge the system as described above immediately before each sample.
8.2.4 The scrubbing solution is adequate for removing sulfur oxides and nitrogen oxides from 50 liters (1.8 ft 3) of stack gas when the concentration of each is less than 1,000 ppm and the CO2 concentration is less than 15 percent. Replace the scrubber solution after every fifth sample.
8.3 Carbon Dioxide Measurement. Measure the CO2 content in the stack to the nearest 0.5 percent each time a CO sample is collected. A simultaneous grab sample analyzed by the Fyrite analyzer is acceptable.
9.0 Quality Control
9.1 Miscellaneous Quality Control Measures.
| Section | Quality control measure | Effect |
|---|---|---|
| 8.1 | Sampling equipment leak-checks and calibration | Ensure accuracy and precision of sampling measurements. |
| 10.3 | Spectrophotometer calibration | Ensure linearity of spectrophotometer response to standards. |
9.2 Volume Metering System Checks. Same as Method 5, section 9.2.
10.0 Calibration and Standardization
Note:
Maintain a laboratory log of all calibrations.
10.1 Gas Bulb Calibration. Weigh the empty bulb to the nearest 0.1 g. Fill the bulb to the stopcock with water, and again weigh to the nearest 0.1 g. Subtract the tare weight, and calculate the volume in liters to three significant figures using the density of water at the measurement temperature. Record the volume on the bulb. Alternatively, mark an identification number on the bulb, and record the volume in a notebook.
10.2 Rate Meter Calibration. Assemble the system as shown in Figure 10A-1 (the impingers may be removed), and attach a volume meter to the probe inlet. Set the rotameter at 300 ml/min, record the volume meter reading, start the pump, and pull ambient air through the system for 10 minutes. Record the final volume meter reading. Repeat the procedure and average the results to determine the volume of gas that passed through the system.
10.3 Spectrophotometer Calibration Curve.
10.3.1 Collect the standards as described in section 8.2. Prepare at least two sets of three bulbs as standards to span the 0 to 400 or 400 to 1000 ppm range. If any samples span both concentration ranges, prepare a calibration curve for each range using separate reagent blanks. Prepare a set of three bulbs containing colorimetric reagent but no CO to serve as a reagent blank. Analyze each standard and blank according to the sample analysis procedure of section 11.0 Reject the standard set where any of the individual bulb absorbances differs from the set mean by more than 10 percent.
10.3.2 Calculate the average absorbance for each set (3 bulbs) of standards using Equation 10A-1 and Table 10A-1. Construct a graph of average absorbance for each standard against its corresponding concentration. Draw a smooth curve through the points. The curve should be linear over the two concentration ranges discussed in section 13.3.
11.0 Analytical Procedure
11.1 Assemble the system shown in Figure 10A-3, and record the information required in Table 10A-1 as it is obtained. Pipet 10.0 ml of the colorimetric reagent into each gas reaction bulb, and attach the bulbs to the system. Open the stopcocks to the reaction bulbs, but leave the valve to the bag closed. Turn on the pump, fully open the coarse-adjust flow valve, and slowly open the fine-adjust valve until the pressure is reduced to at least 40 mm Hg. Now close the coarse adjust valve, and observe the manometer to be certain that the system is leak-free. Wait a minimum of 2 minutes. If the pressure has increased less than 1 mm Hg, proceed as described below. If a leak is present, find and correct it before proceeding further.
11.2 Record the vacuum pressure (Pv) to the nearest 1 mm Hg, and close the reaction bulb stopcocks. Open the bag valve, and allow the system to come to atmospheric pressure. Close the bag valve, open the pump coarse adjust valve, and evacuate the system again. Repeat this fill/evacuation procedure at least twice to flush the manifold completely. Close the pump coarse adjust valve, open the bag valve, and let the system fill to atmospheric pressure. Open the stopcocks to the reaction bulbs, and let the entire system come to atmospheric pressure. Close the bulb stopcocks, remove the bulbs, record the room temperature and barometric pressure (Pbar, to nearest mm Hg), and place the bulbs on the shaker table with their main axis either parallel to or perpendicular to the plane of the table top. Purge the bulb-filling system with ambient air for several minutes between samples. Shake the samples for exactly 2 hours.
11.3 Immediately after shaking, measure the absorbance (A) of each bulb sample at 425 nm if the concentration is less than or equal to 400 ppm CO or at 600 nm if the concentration is above 400 ppm.
Note:
This may be accomplished with multiple bulb sets by sequentially collecting sets and adding to the shaker at staggered intervals, followed by sequentially removing sets from the shaker for absorbance measurement after the two-hour designated intervals have elapsed.
11.4 Use a small portion of the sample to rinse a spectrophotometer cell several times before taking an aliquot for analysis. If one cell is used to analyze multiple samples, rinse the cell with deionized distilled water several times between samples. Prepare and analyze standards and a reagent blank as described in section 10.3. Use water as the reference. Reject the analysis if the blank absorbance is greater than 0.1. All conditions should be the same for analysis of samples and standards. Measure the absorbances as soon as possible after shaking is completed.
11.5 Determine the CO concentration of each bag sample using the calibration curve for the appropriate concentration range as discussed in section 10.3.
12.0 Calculations and Data Analysis
Carry out calculations retaining at least one extra decimal figure beyond that of the acquired data. Round off figures after final calculation.
12.1 Nomenclature.
A = Sample absorbance, uncorrected for the reagent blank.
Ar = Absorbance of the reagent blank.
As = Average sample absorbance per liter, units/liter.
Bw = Moisture content in the bag sample.
C = CO concentration in the stack gas, dry basis, ppm.
Cb = CO concentration of the bag sample, dry basis, ppm.
Cg = CO concentration from the calibration curve, ppm.
F = Volume fraction of CO2 in the stack.
n = Number of reaction bulbs used per bag sample.
Pb = Barometric pressure, mm Hg.
Pv = Residual pressure in the sample bulb after evacuation, mm Hg.
Pw = Vapor pressure of H2O in the bag (from Table 10A-2), mm Hg.
Vb = Volume of the sample bulb, liters.
Vr = Volume of reagent added to the sample bulb, 0.0100 liter.
12.2 Average Sample Absorbance per Liter. Calculate As for each gas bulb using Equation 10A-1, and record the value in Table 10A-1. Calculate the average As for each bag sample, and compare the three values to the average. If any single value differs by more than 10 percent from the average, reject this value, and calculate a new average using the two remaining values.
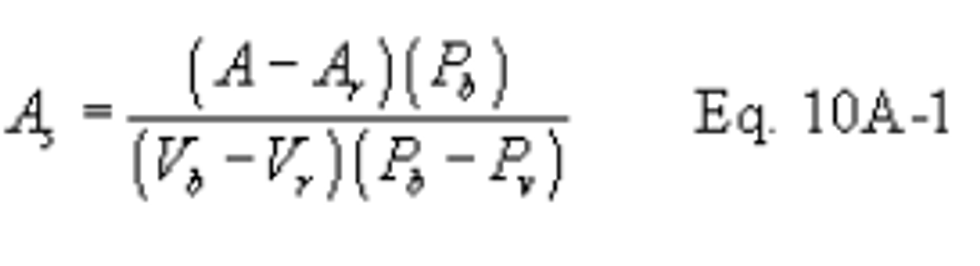
Note:
A and Ar must be at the same wavelength.
12.3 CO Concentration in the Bag. Calculate Cb using Equations 10A-2 and 10A-3. If condensate is visible in the bag, calculate Bw using Table 10A-2 and the temperature and barometric pressure in the analysis room. If condensate is not visible, calculate Bw using the temperature and barometric pressure at the sampling site.
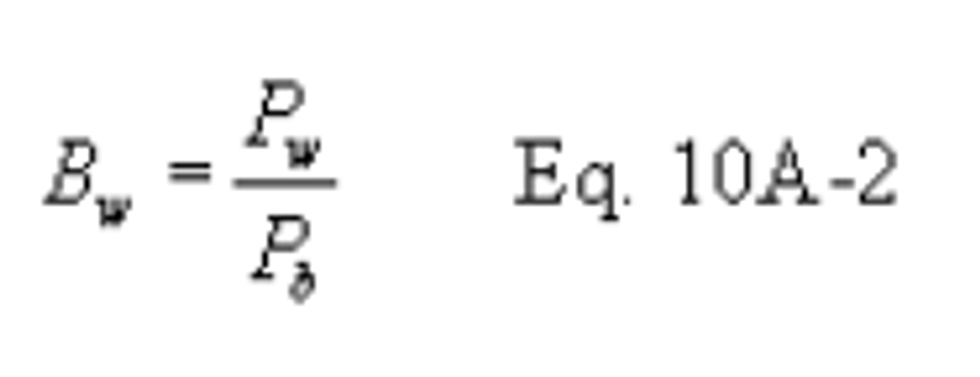
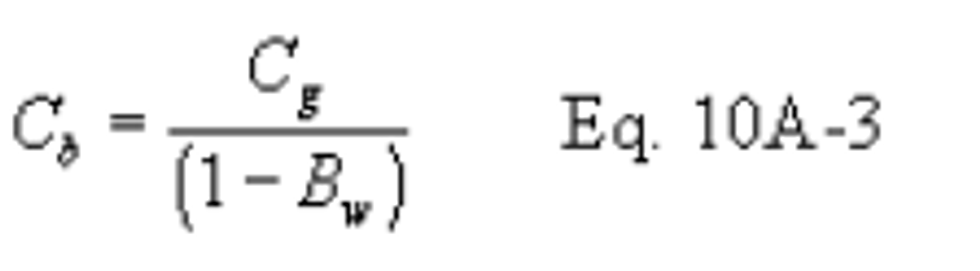
12.4 CO Concentration in the Stack.
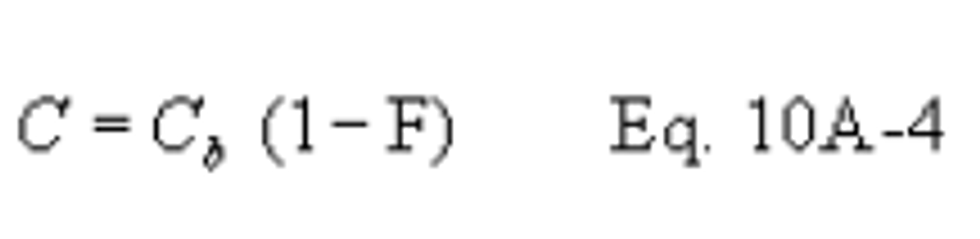
13.0 Method Performance
13.1 Precision. The estimated intralaboratory standard deviation of the method is 3 percent of the mean for gas samples analyzed in duplicate in the concentration range of 39 to 412 ppm. The interlaboratory precision has not been established.
13.2 Accuracy. The method contains no significant biases when compared to an NDIR analyzer calibrated with NIST standards.
13.3 Range. Approximately 3 to 1800 ppm CO. Samples having concentrations below 400 ppm are analyzed at 425 nm, and samples having concentrations above 400 ppm are analyzed at 600 nm.
13.4 Sensitivity. The detection limit is 3 ppmv based on a change in concentration equal to three times the standard deviation of the reagent blank solution.
13.5 Stability. The individual components of the colorimetric reagent are stable for at least one month. The colorimetric reagent must be used within two days after preparation to avoid excessive blank correction. The samples in the bag should be stable for at least one week if the bags are leak-free.
14.0 Pollution Prevention [Reserved]
15.0 Waste Management [Reserved]
16.0 References
1. Butler, F.E., J.E. Knoll, and M.R. Midgett. Development and Evaluation of Methods for Determining Carbon Monoxide Emissions. U.S. Environmental Protection Agency, Research Triangle Park, N.C. June 1985. 33 pp.
2. Ferguson, B.B., R.E. Lester, and W.J. Mitchell. Field Evaluation of Carbon Monoxide and Hydrogen Sulfide Continuous Emission Monitors at an Oil Refinery. U.S. Environmental Protection Agency, Research Triangle Park, N.C. Publication No. EPA-600/4-82-054. August 1982. 100 pp.
3. Lambert, J.L., and R.E. Weins. Induced Colorimetric Method for Carbon Monoxide. Analytical Chemistry. 46(7):929-930. June 1974.
4. Levaggi, D.A., and M. Feldstein. The Colorimetric Determination of Low Concentrations of Carbon Monoxide. Industrial Hygiene Journal. 25:64-66. January-February 1964.
5. Repp, M. Evaluation of Continuous Monitors For Carbon Monoxide in Stationary Sources. U.S. Environmental Protection Agency. Research Triangle Park, N.C. Publication No. EPA-600/2-77-063. March 1977. 155 pp.
6. Smith, F., D.E. Wagoner, and R.P. Donovan. Guidelines for Development of a Quality Assurance Program: Volume VIII - Determination of CO Emissions from Stationary Sources by NDIR Spectrometry. U.S. Environmental Protection Agency. Research Triangle Park, N.C. Publication No. EPA-650/4-74-005-h. February 1975. 96 pp.
17.0 Tables, Diagrams, Flowcharts, and Validation Data
| Sample No./type | Room temp °C | Stack %CO2 | Bulb No. | Bulb vol. liters | Reagent vol. in bulb, liter | Partial pressure of gas in bulb, mm Hg | Pb, mm Hg | Shaking time, min | Abs versus water | A-Ar | As | Avg As |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| blank | ||||||||||||
| Std. 1 | ||||||||||||
| Std. 2 | ||||||||||||
| Sample 1 | ||||||||||||
| Sample 2 | ||||||||||||
| Sample 3 |
| Temperature°C | Vapor pressure of H2O, mm Hg | Temperature°C | Vapor pressure of H2, mm Hg |
|---|---|---|---|
| 4 | 6.1 | 18 | 15.5 |
| 6 | 7.0 | 20 | 17.5 |
| 8 | 8.0 | 22 | 19.8 |
| 10 | 9.2 | 24 | 22.4 |
| 12 | 10.5 | 26 | 25.2 |
| 14 | 12.0 | 28 | 28.3 |
| 16 | 13.6 | 30 | 31.8 |
Method 10B - Determination of Carbon Monoxide Emissions From Stationary Sources
Note:
This method is not inclusive with respect to specifications (e.g., equipment and supplies) and procedures (e.g., sampling and analytical) essential to its performance. Some material is incorporated by reference from other methods in this part. Therefore, to obtain reliable results, persons using this method should have a thorough knowledge of at least the following additional test methods: Method 1, Method 4, Method 10A, and Method 25.
1.0 Scope and Application
1.1 Analytes.
| Analyte | CAS No. | Sensitivity |
|---|---|---|
| Carbon monoxide (CO) | 630-08-0 | Not determined. |
1.2 Applicability. This method applies to the measurement of CO emissions at petroleum refineries and from other sources when specified in an applicable subpart of the regulations.
1.3 Data Quality Objectives. Adherence to the requirements of this method will enhance the quality of the data obtained from air pollutant sampling methods.
2.0 Summary of Method
2.1 An integrated gas sample is extracted from the sampling point, passed through a conditioning system to remove interferences, and collected in a Tedlar or equivalent bag. (Verify through the manufacturer that the Tedlar alternative is suitable for NO and make this verifying information available for inspection.) The CO is separated from the sample by gas chromatography (GC) and catalytically reduced to methane (CH4) which is determined by flame ionization detection (FID). The analytical portion of this method is identical to applicable sections in Method 25 detailing CO measurement.
3.0 Definitions [Reserved]
4.0 Interferences
4.1 Carbon dioxide (CO2) and organics potentially can interfere with the analysis. Most of the CO2 is removed from the sample by the alkaline permanganate conditioning system; any residual CO2 and organics are separated from the CO by GC.
5.0 Safety
5.1 Disclaimer. This method may involve hazardous materials, operations, and equipment. This test method may not address all of the safety problems associated with its use. It is the responsibility of the user of this test method to establish appropriate safety and health practices and determine the applicability of regulatory limitations prior to performing this test method. The analyzer users manual should be consulted for specific precautions concerning the analytical procedure.
6.0 Equipment and Supplies
6.1 Sample Collection. Same as in Method 10A, section 6.1 (paragraphs 6.1.1 through 6.1.11).
6.2 Sample Analysis. A GC/FID analyzer, capable of quantifying CO in the sample and consisting of at least the following major components, is required for sample analysis. [Alternatively, complete Method 25 analytical systems (Method 25, section 6.3) are acceptable alternatives when calibrated for CO and operated in accordance with the Method 25 analytical procedures (Method 25, section 11.0).]
6.2.1 Separation Column. A column capable of separating CO from CO2 and organic compounds that may be present. A 3.2-mm ( 1/8-in.) OD stainless steel column packed with 1.7 m (5.5 ft.) of 60/80 mesh Carbosieve S-II (available from Supelco) has been used successfully for this purpose.
6.2.2 Reduction Catalyst. Same as in Method 25, section 6.3.1.2.
6.2.3 Sample Injection System. Same as in Method 25, Section 6.3.1.4, equipped to accept a sample line from the bag.
6.2.4 Flame Ionization Detector. Meeting the linearity specifications of section 10.3 and having a minimal instrument range of 10 to 1,000 ppm CO.
6.2.5 Data Recording System. Analog strip chart recorder or digital integration system, compatible with the FID, for permanently recording the analytical results.
7.0 Reagents and Standards
7.1 Sample Collection. Same as in Method 10A, section 7.1.
7.2 Sample Analysis.
7.2.1 Carrier, Fuel, and Combustion Gases. Same as in Method 25, sections 7.2.1, 7.2.2, and 7.2.3, respectively.
7.2.2 Calibration Gases. Three standard gases with nominal CO concentrations of 20, 200, and 1,000 ppm CO in nitrogen. The calibration gases shall be certified by the manufacturer to be ±2 percent of the specified concentrations.
7.2.3 Reduction Catalyst Efficiency Check Calibration Gas. Standard CH4 gas with a nominal concentration of 1,000 ppm in air.
8.0 Sample Collection, Preservation, Storage, and Transport
Same as in Method 10A, section 8.0.
9.0 Quality Control
| Section | Quality control measure | Effect |
|---|---|---|
| 8.0 | Sample bag/sampling system leak-checks | Ensures that negative bias introduced through leakage is minimized. |
| 10.1 | Carrier gas blank check | Ensures that positive bias introduced by contamination of carrier gas is less than 5 ppmv. |
| 10.2 | Reduction catalyst efficiency check | Ensures that negative bias introduced by inefficient reduction catalyst is less than 5 percent. |
| 10.3 | Analyzer calibration | Ensures linearity of analyzer response to standards. |
| 11.2 | Triplicate sample analyses | Ensures precision of analytical results. |
10.0 Calibration and Standardization
10.1 Carrier Gas Blank Check. Analyze each new tank of carrier gas with the GC analyzer according to section 11.2 to check for contamination. The corresponding concentration must be less than 5 ppm for the tank to be acceptable for use.
10.2 Reduction Catalyst Efficiency Check. Prior to initial use, the reduction catalyst shall be tested for reduction efficiency. With the heated reduction catalyst bypassed, make triplicate injections of the 1,000 ppm CH4 gas (Section 7.2.3) to calibrate the analyzer. Repeat the procedure using 1,000 ppm CO gas (Section 7.2.2) with the catalyst in operation. The reduction catalyst operation is acceptable if the CO response is within 5 percent of the certified gas value.
10.3 Analyzer Calibration. Perform this test before the system is first placed into operation. With the reduction catalyst in operation, conduct a linearity check of the analyzer using the standards specified in section 7.2.2. Make triplicate injections of each calibration gas, and then calculate the average response factor (area/ppm) for each gas, as well as the overall mean of the response factor values. The instrument linearity is acceptable if the average response factor of each calibration gas is within 2.5 percent of the overall mean value and if the relative standard deviation (calculated in section 12.8 of Method 25) for each set of triplicate injections is less than 2 percent. Record the overall mean of the response factor values as the calibration response factor (R).
11.0 Analytical Procedure
11.1 Preparation for Analysis. Before putting the GC analyzer into routine operation, conduct the calibration procedures listed in section 10.0. Establish an appropriate carrier flow rate and detector temperature for the specific instrument used.
11.2 Sample Analysis. Purge the sample loop with sample, and then inject the sample. Analyze each sample in triplicate, and calculate the average sample area (A). Determine the bag CO concentration according to section 12.2.
12.0 Calculations and Data Analysis
Carry out calculations retaining at least one extra significant figure beyond that of the acquired data. Round off results only after the final calculation.
12.1 Nomenclature.
A = Average sample area.
Bw = Moisture content in the bag sample, fraction.
C = CO concentration in the stack gas, dry basis, ppm.
Cb = CO concentration in the bag sample, dry basis, ppm.
F = Volume fraction of CO2 in the stack, fraction.
Pbar = Barometric pressure, mm Hg.
Pw = Vapor pressure of the H2O in the bag (from Table 10A-2, Method 10A), mm Hg.
R = Mean calibration response factor, area/ppm.
12.2 CO Concentration in the Bag. Calculate Cb using Equations 10B-1 and 10B-2. If condensate is visible in the bag, calculate Bw using Table 10A-2 of Method 10A and the temperature and barometric pressure in the analysis room. If condensate is not visible, calculate Bw using the temperature and barometric pressure at the sampling site.
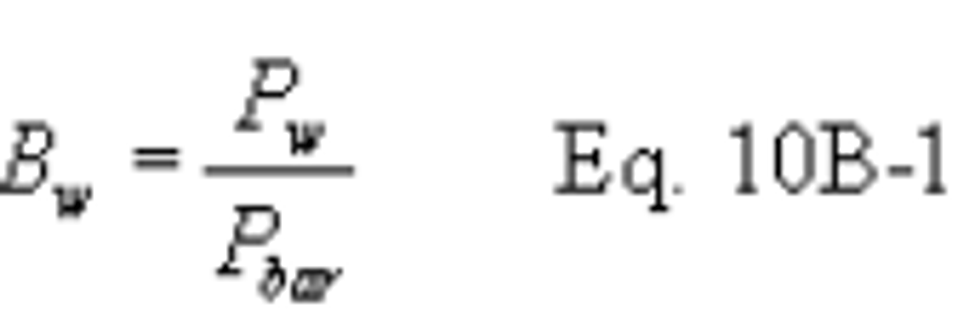
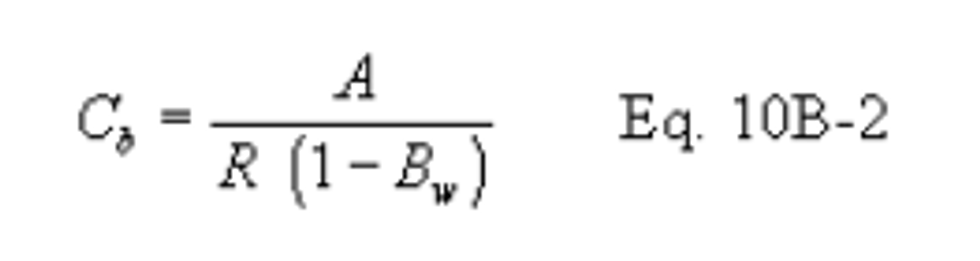
12.3 CO Concentration in the Stack
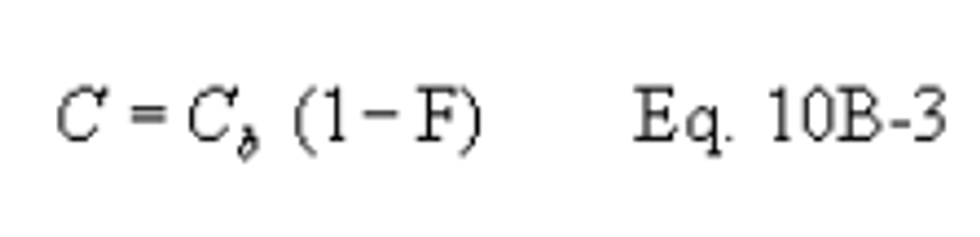
13.0 Method Performance [Reserved]
14.0 Pollution Prevention [Reserved]
15.0 Waste Management [Reserved]
16.0 References
Same as in Method 25, section 16.0, with the addition of the following:
1. Butler, F.E, J.E. Knoll, and M.R. Midgett. Development and Evaluation of Methods for Determining Carbon Monoxide Emissions. Quality Assurance Division, Environmental Monitoring Systems Laboratory, U.S. Environmental Protection Agency, Research Triangle Park, NC. June 1985. 33 pp.
17.0 Tables, Diagrams, Flowcharts, and Validation Data [Reserved]
Editorial Note: For Federal Register citations affecting appendix A-4 to part 60, see the List of CFR sections Affected, which appears in the Finding Aids section of the printed volume and at www.govinfo.gov.
[36 FR 24877, Dec. 23, 1971; 85 FR 63414, Oct. 7, 2020; 88 FR 18405, March 29, 2023]
['Air Programs']
['Air Quality']
UPGRADE TO CONTINUE READING