Compliance Just Got Easier: Stay ahead of regulatory changes with instant notifications on updates that matter.
['Air Programs']
['Air Quality']
11/20/2023

Copyright 2026 J. J. Keller & Associate, Inc. For re-use options please contact copyright@jjkeller.com or call 800-558-5011.
Method 19 - Determination of sulfur dioxide removal efficiency and particulate, sulfur dioxide and nitrogen oxides emission rates
Method 20 - Determination of nitrogen oxides, sulfur dioxide, and diluent emissions from stationary gas turbines
Method 21 - Determination of volatile organic compound leaks
Method 22 - Visual determination of fugitive emissions from material sources and smoke emissions from flares
Method 23 - Determination of Polychlorinated Dibenzo- -Dioxins, Polychlorinated Dibenzofurans, Polychlorinated Biphenyls, and Polycyclic Aromatic Hydrocarbons From Stationary Sources
Method 24 - Determination of volatile matter content, water content, density, volume solids, and weight solids of surface coatings
Method 24A - Determination of volatile matter content and density of printing inks and related coatings
Method 25 - Determination of total gaseous nonmethane organic emissions as carbon
Method 25A - Determination of total gaseous organic concentration using a flame ionization analyzer
Method 25B - Determination of total gaseous organic concentration using a nondispersive infrared analyzer
Method 25C - Determination of nonmethane organic compounds (NMOC) in MSW landfill gases
Method 25D - Determination of the Volatile Organic Concentration of Waste Samples
Method 25E - Determination of Vapor Phase Organic Concentration in Waste Samples
The test methods in this appendix are referred to in §60.8 (Performance Tests) and §60.11 (Compliance With Standards and Maintenance Requirements) of 40 CFR part 60, subpart A (General Provisions). Specific uses of these test methods are described in the standards of performance contained in the subparts, beginning with Subpart D.
Within each standard of performance, a section title “Test Methods and Procedures” is provided to: (1) Identify the test methods to be used as reference methods to the facility subject to the respective standard and (2) identify any special instructions or conditions to be followed when applying a method to the respective facility. Such instructions (for example, establish sampling rates, volumes, or temperatures) are to be used either in addition to, or as a substitute for procedures in a test method. Similarly, for sources subject to emission monitoring requirements, specific instructions pertaining to any use of a test method as a reference method are provided in the subpart or in Appendix B.
Inclusion of methods in this appendix is not intended as an endorsement or denial of their applicability to sources that are not subject to standards of performance. The methods are potentially applicable to other sources; however, applicability should be confirmed by careful and appropriate evaluation of the conditions prevalent at such sources.
The approach followed in the formulation of the test methods involves specifications for equipment, procedures, and performance. In concept, a performance specification approach would be preferable in all methods because this allows the greatest flexibility to the user. In practice, however, this approach is impractical in most cases because performance specifications cannot be established. Most of the methods described herein, therefore, involve specific equipment specifications and procedures, and only a few methods in this appendix rely on performance criteria.
Minor changes in the test methods should not necessarily affect the validity of the results and it is recognized that alternative and equivalent methods exist. section 60.8 provides authority for the Administrator to specify or approve (1) equivalent methods, (2) alternative methods, and (3) minor changes in the methodology of the test methods. It should be clearly understood that unless otherwise identified all such methods and changes must have prior approval of the Administrator. An owner employing such methods or deviations from the test methods without obtaining prior approval does so at the risk of subsequent disapproval and retesting with approved methods.
Within the test methods, certain specific equipment or procedures are recognized as being acceptable or potentially acceptable and are specifically identified in the methods. The items identified as acceptable options may be used without approval but must be identified in the test report. The potentially approvable options are cited as “subject to the approval of the Administrator” or as “or equivalent.” Such potentially approvable techniques or alternatives may be used at the discretion of the owner without prior approval. However, detailed descriptions for applying these potentially approvable techniques or alternatives are not provided in the test methods. Also, the potentially approvable options are not necessarily acceptable in all applications. Therefore, an owner electing to use such potentially approvable techniques or alternatives is responsible for: (1) assuring that the techniques or alternatives are in fact applicable and are properly executed; (2) including a written description of the alternative method in the test report (the written method must be clear and must be capable of being performed without additional instruction, and the degree of detail should be similar to the detail contained in the test methods); and (3) providing any rationale or supporting data necessary to show the validity of the alternative in the particular application. Failure to meet these requirements can result in the Administrator's disapproval of the alternative.
Method 19 - Determination of Sulfur Dioxide Removal Efficiency and Particulate Matter, Sulfur Dioxide, and Nitrogen Oxide Emission Rates
1.0 Scope and Application
1.1 Analytes. This method provides data reduction procedures relating to the following pollutants, but does not include any sample collection or analysis procedures.
| Analyte | CAS No. | Sensitivity |
|---|---|---|
| Nitrogen oxides (NOX), including: | ||
| Nitric oxide (NO) | 10102-43-9 | N/A |
| Nitrogen dioxide (NO2) | 10102-44-0 | |
| Particulate matter (PM) | None assigned | N/A |
| Sulfur dioxide (SO2) | 7499-09-05 | N/A |
1.2 Applicability. Where specified by an applicable subpart of the regulations, this method is applicable for the determination of (a) PM, SO2, and NOX emission rates; (b) sulfur removal efficiencies of fuel pretreatment and SO2 control devices; and (c) overall reduction of potential SO2 emissions.
2.0 Summary of Method
2.1 Emission Rates. Oxygen (O2) or carbon dioxide (CO2) concentrations and appropriate F factors (ratios of combustion gas volumes to heat inputs) are used to calculate pollutant emission rates from pollutant concentrations.
2.2 Sulfur Reduction Efficiency and SO2 Removal Efficiency. An overall SO2 emission reduction efficiency is computed from the efficiency of fuel pretreatment systems, where applicable, and the efficiency of SO2 control devices.
2.2.1 The sulfur removal efficiency of a fuel pretreatment system is determined by fuel sampling and analysis of the sulfur and heat contents of the fuel before and after the pretreatment system.
2.2.2 The SO2 removal efficiency of a control device is determined by measuring the SO2 rates before and after the control device.
2.2.2.1 The inlet rates to SO2 control systems (or, when SO2 control systems are not used, SO2 emission rates to the atmosphere) are determined by fuel sampling and analysis.
3.0 Definitions [Reserved]
4.0 Interferences [Reserved]
5.0 Safety [Reserved]
6.0 Equipment and Supplies [Reserved]
7.0 Reagents and Standards [Reserved]
8.0 Sample Collection, Preservation, Storage, and Transport [Reserved]
9.0 Quality Control [Reserved]
10.0 Calibration and Standardization [Reserved]
11.0 Analytical Procedures [Reserved]
12.0 Data Analysis and Calculations
12.1 Nomenclature
Bwa = Moisture fraction of ambient air, percent.
Bws = Moisture fraction of effluent gas, percent.
%C = Concentration of carbon from an ultimate analysis of fuel, weight percent.
Cd = Pollutant concentration, dry basis, ng/scm (lb/scf)
%CO2d,%CO2w = Concentration of carbon dioxide on a dry and wet basis, respectively, percent.
Cw = Pollutant concentration, wet basis, ng/scm (lb/scf).
D = Number of sampling periods during the performance test period.
E = Pollutant emission rate, ng/J (lb/million Btu).
Ea = Average pollutant rate for the specified performance test period, ng/J (lb/million Btu).
Eao, Eai = Average pollutant rate of the control device, outlet and inlet, respectively, for the performance test period, ng/J (lb/million Btu).
Ebi = Pollutant rate from the steam generating unit, ng/J (lb/million Btu)
Ebo = Pollutant emission rate from the steam generating unit, ng/J (lb/million Btu).
Eci = Pollutant rate in combined effluent, ng/J (lb/million Btu).
Eco = Pollutant emission rate in combined effluent, ng/J (lb/million Btu).
Ed = Average pollutant rate for each sampling period (e.g., 24-hr Method 6B sample or 24-hr fuel sample) or for each fuel lot (e.g., amount of fuel bunkered), ng/J (lb/million Btu).
Edi = Average inlet SO2 rate for each sampling period d, ng/J (lb/million Btu)
Eg = Pollutant rate from gas turbine, ng/J (lb/million Btu).
Ega = Daily geometric average pollutant rate, ng/J (lbs/million Btu) or ppm corrected to 7 percent O2.
Ejo,Eji = Matched pair hourly arithmetic average pollutant rate, outlet and inlet, respectively, ng/J (lb/million Btu) or ppm corrected to 7 percent O2.
Eh = Hourly average pollutant, ng/J (lb/million Btu).
Ehj = Hourly arithmetic average pollutant rate for hour “j,” ng/J (lb/million Btu) or ppm corrected to 7 percent O2.
EXP = Natural logarithmic base (2.718) raised to the value enclosed by brackets.
Fd, Fw, Fc = Volumes of combustion components per unit of heat content, scm/J (scf/million Btu).
GCV = Gross calorific value of the fuel consistent with the ultimate analysis, kJ/kg (Btu/lb).
GCVp, GCVr = Gross calorific value for the product and raw fuel lots, respectively, dry basis, kJ/kg (Btu/lb).
%H = Concentration of hydrogen from an ultimate analysis of fuel, weight percent.
H = Total number of operating hours for which pollutant rates are determined in the performance test period.
Hb = Heat input rate to the steam generating unit from fuels fired in the steam generating unit, J/hr (million Btu/hr).
Hg = Heat input rate to gas turbine from all fuels fired in the gas turbine, J/hr (million Btu/hr).
%H2O = Concentration of water from an ultimate analysis of fuel, weight percent.
Hr = Total numbers of hours in the performance test period (e.g., 720 hours for 30-day performance test period).
K = Conversion factor, 10−5 (kJ/J)/(%) [106 Btu/million Btu].
Kc = (9.57 scm/kg)/% [(1.53 scf/lb)/%].
Kcc = (2.0 scm/kg)/% [(0.321 scf/lb)/%].
Khd = (22.7 scm/kg)/% [(3.64 scf/lb)/%].
Khw = (34.74 scm/kg)/% [(5.57 scf/lb)/%].
Kn = (0.86 scm/kg)/% [(0.14 scf/lb)/%].
Ko = (2.85 scm/kg)/% [(0.46 scf/lb)/%].
Ks = (3.54 scm/kg)/% [(0.57 scf/lb)/%].
Kw = (1.30 scm/kg)/% [(0.21 scf/lb)/%].
ln = Natural log of indicated value.
Lp,Lr = Weight of the product and raw fuel lots, respectively, metric ton (ton).
%N = Concentration of nitrogen from an ultimate analysis of fuel, weight percent.
N = Number of fuel lots during the averaging period.
n = Number of fuels being burned in combination.
nd = Number of operating hours of the affected facility within the performance test period for each Ed determined.
nt = Total number of hourly averages for which paired inlet and outlet pollutant rates are available within the 24-hr midnight to midnight daily period.
%O = Concentration of oxygen from an ultimate analysis of fuel, weight percent.
%O2d, %O2w = Concentration of oxygen on a dry and wet basis, respectively, percent.
Ps = Potential SO2 emissions, percent.
%Rf = SO2 removal efficiency from fuel pretreatment, percent.
%Rg = SO2 removal efficiency of the control device, percent.
%Rga = Daily geometric average percent reduction.
%Ro = Overall SO2 reduction, percent.
%S = Sulfur content of as-fired fuel lot, dry basis, weight percent.
Se = Standard deviation of the hourly average pollutant rates for each performance test period, ng/J (lb/million Btu).
%Sf = Concentration of sulfur from an ultimate analysis of fuel, weight percent.
Si = Standard deviation of the hourly average inlet pollutant rates for each performance test period, ng/J (lb/million Btu).
So = Standard deviation of the hourly average emission rates for each performance test period, ng/J (lb/million Btu).
%Sp, %Sr = Sulfur content of the product and raw fuel lots respectively, dry basis, weight percent.
t0.95 = Values shown in Table 19-3 for the indicated number of data points n.
Xk = Fraction of total heat input from each type of fuel k.
12.2 Emission Rates of PM, SO2, and NOX. Select from the following sections the applicable procedure to compute the PM, SO2, or NOX emission rate (E) in ng/J (lb/million Btu). The pollutant concentration must be in ng/scm (lb/scf) and the F factor must be in scm/J (scf/million Btu). If the pollutant concentration (C) is not in the appropriate units, use Table 19-1 in section 17.0 to make the proper conversion. An F factor is the ratio of the gas volume of the products of combustion to the heat content of the fuel. The dry F factor (Fd) includes all components of combustion less water, the wet F factor (Fw) includes all components of combustion, and the carbon F factor (Fc) includes only carbon dioxide.
Note:
Since Fw factors include water resulting only from the combustion of hydrogen in the fuel, the procedures using Fw factors are not applicable for computing E from steam generating units with wet scrubbers or with other processes that add water (e.g., steam injection).
12.2.1 Oxygen-Based F Factor, Dry Basis. When measurements are on a dry basis for both O (%O2d) and pollutant (Cd) concentrations, use the following equation:
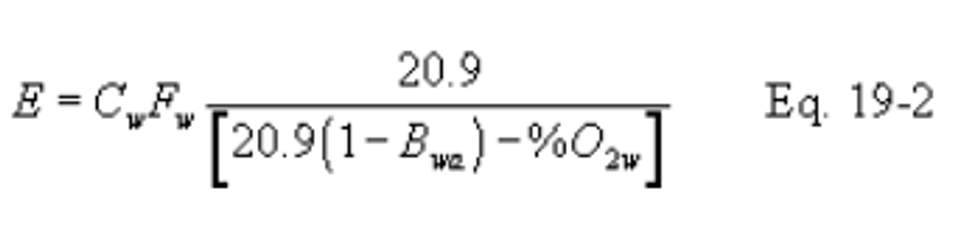
12.2.2 Oxygen-Based F Factor, Wet Basis. When measurements are on a wet basis for both O2 (%O2w) and pollutant (Cw) concentrations, use either of the following:
12.2.2.1 If the moisture fraction of ambient air (Bwa) is measured:
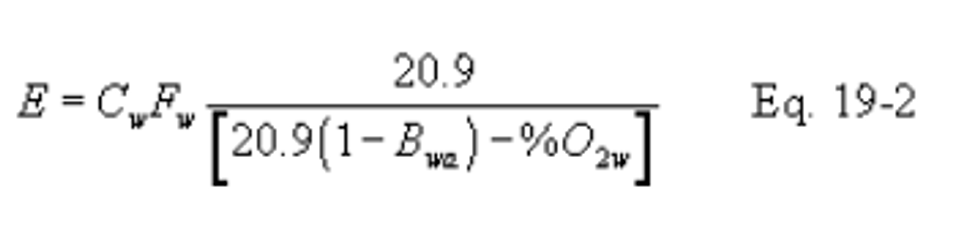
Instead of actual measurement, Bwa may be estimated according to the procedure below.
Note:
The estimates are selected to ensure that negative errors will not be larger than −1.5 percent. However, positive errors, or over-estimation of emissions by as much as 5 percent may be introduced depending upon the geographic location of the facility and the associated range of ambient moisture.
12.2.2.1.1 Bwa = 0.027. This value may be used at any location at all times.
12.2.2.1.2 Bwa = Highest monthly average of Bwa that occurred within the previous calendar year at the nearest Weather Service Station. This value shall be determined annually and may be used as an estimate for the entire current calendar year.
12.2.2.1.3 Bwa = Highest daily average of Bwa that occurred within a calendar month at the nearest Weather Service Station, calculated from the data from the past 3 years. This value shall be computed for each month and may be used as an estimate for the current respective calendar month.
12.2.2.2 If the moisture fraction (Bws) of the effluent gas is measured:
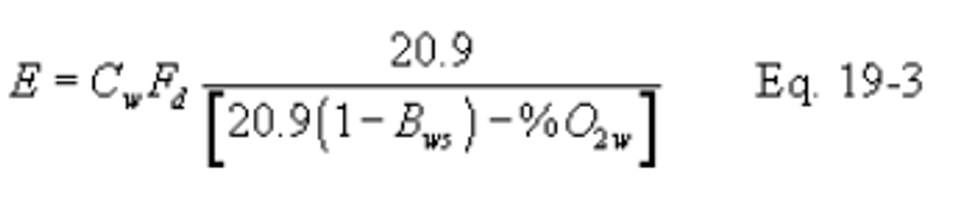
12.2.3 Oxygen-Based F Factor, Dry/Wet Basis.
12.2.3.1 When the pollutant concentration is measured on a wet basis (Cw) and O2 concentration is measured on a dry basis (%O2d), use the following equation:
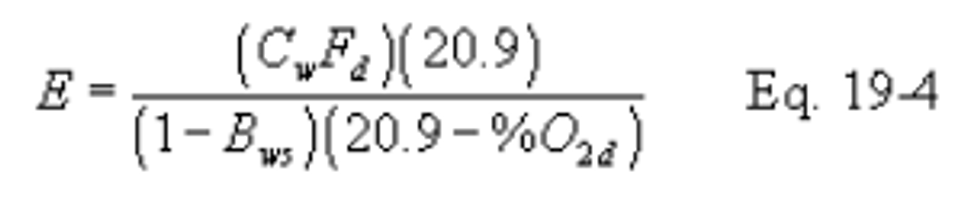
12.2.3.2 When the pollutant concentration is measured on a dry basis (Cd) and the O2 concentration is measured on a wet basis (%O2w), use the following equation:
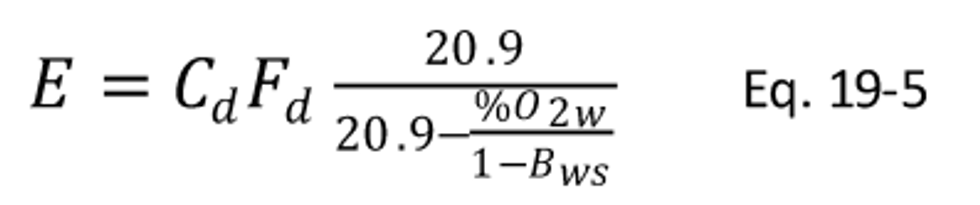
12.2.4 Carbon Dioxide-Based F Factor, Dry Basis. When measurements are on a dry basis for both CO2 (%CO2d) and pollutant (Cd) concentrations, use the following equation:
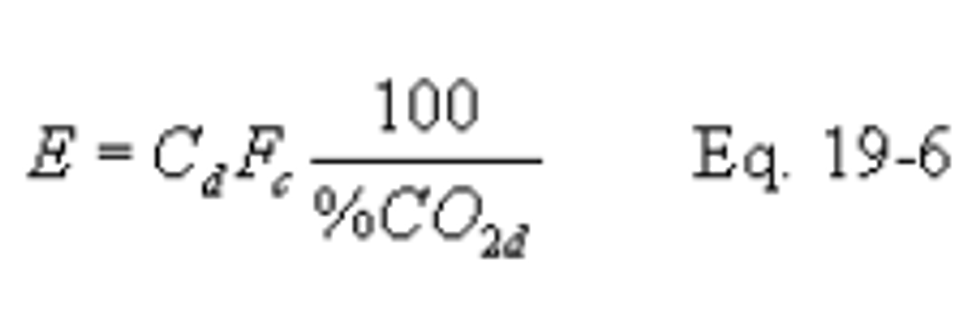
12.2.5 Carbon Dioxide-Based F Factor, Wet Basis. When measurements are on a wet basis for both CO2 (%CO2w) and pollutant (Cw) concentrations, use the following equation:
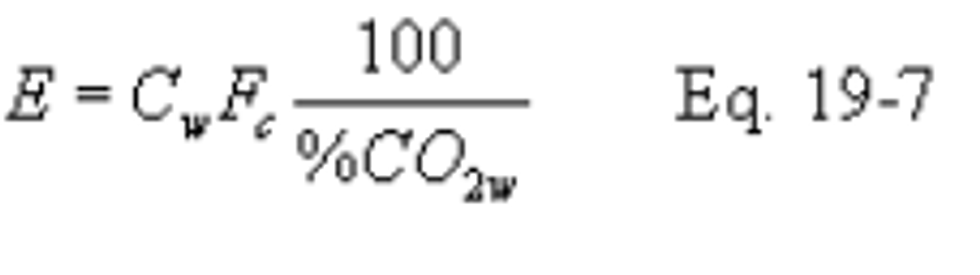
12.2.6 Carbon Dioxide-Based F Factor, Dry/Wet Basis.
12.2.6.1 When the pollutant concentration is measured on a wet basis (Cw) and CO2 concentration is measured on a dry basis (%CO2d), use the following equation:
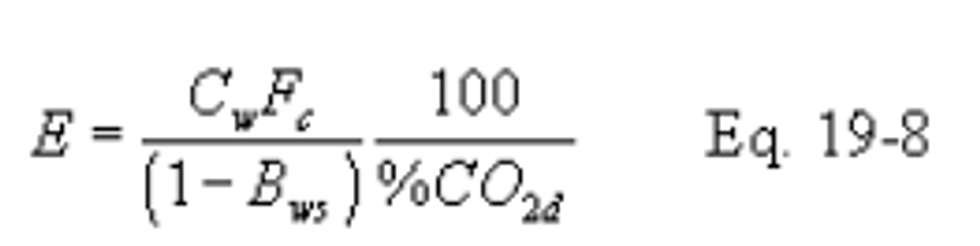
12.2.6.2 When the pollutant concentration is measured on a dry basis (Cd) and CO2 concentration is measured on a wet basis (%CO2w), use the following equation:
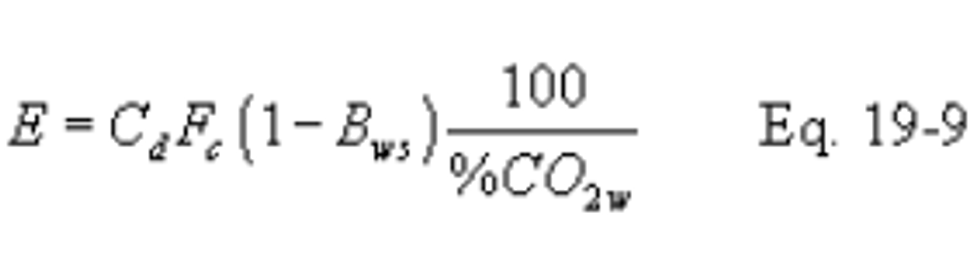
12.2.7 Direct-Fired Reheat Fuel Burning. The effect of direct-fired reheat fuel burning (for the purpose of raising the temperature of the exhaust effluent from wet scrubbers to above the moisture dew-point) on emission rates will be less than 1.0 percent and, therefore, may be ignored.
12.2.8 Combined Cycle-Gas Turbine Systems. For gas turbine-steam generator combined cycle systems, determine the emissions from the steam generating unit or the percent reduction in potential SO2 emissions as follows:
12.2.8.1 Compute the emission rate from the steam generating unit using the following equation:
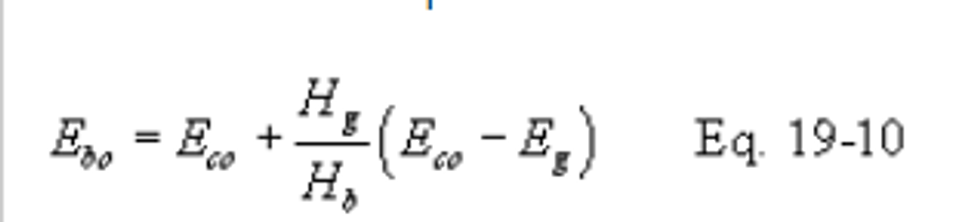
12.2.8.1.1 Use the test methods and procedures section of 40 CFR Part 60, Subpart GG to obtain Eco and Eg. Do not use Fw factors for determining Eg or Eco. If an SO2 control device is used, measure Eco after the control device.
12.2.8.1.2 Suitable methods shall be used to determine the heat input rates to the steam generating units (Hb) and the gas turbine (Hg).
12.2.8.2 If a control device is used, compute the percent of potential SO2 emissions (Ps) using the following equations:
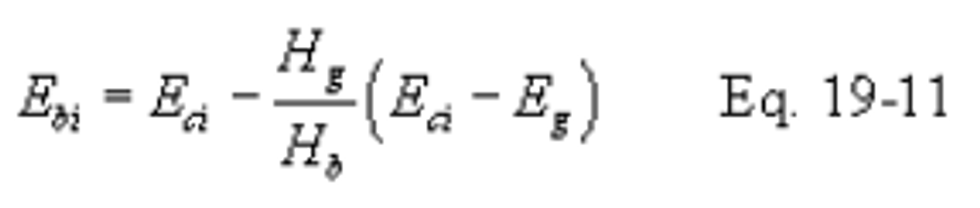
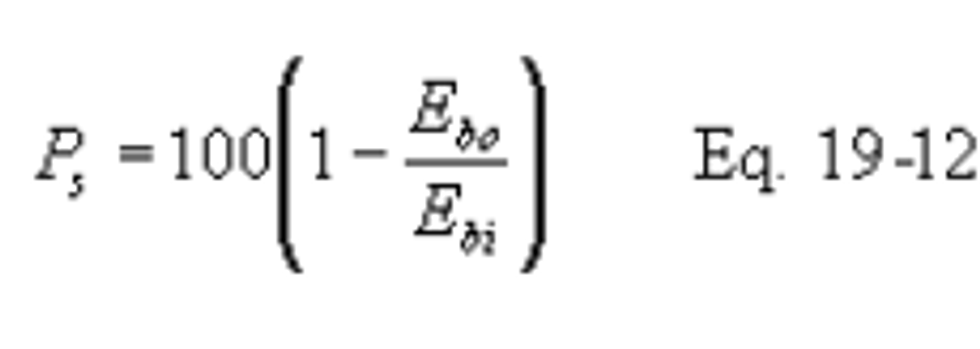
Note:
Use the test methods and procedures section of Subpart GG to obtain Eci and Eg. Do not use Fw factors for determining Eg or Eci.
12.3 F Factors. Use an average F factor according to section 12.3.1 or determine an applicable F factor according to section 12.3.2. If combined fuels are fired, prorate the applicable F factors using the procedure in section 12.3.3.
12.3.1 Average F Factors. Average F factors (Fd, Fw, or Fc) from Table 19-2 in section 17.0 may be used.
12.3.2 Determined F Factors. If the fuel burned is not listed in Table 19-2 or if the owner or operator chooses to determine an F factor rather than use the values in Table 19-2, use the procedure below:
12.3.2.1 Equations. Use the equations below, as appropriate, to compute the F factors:
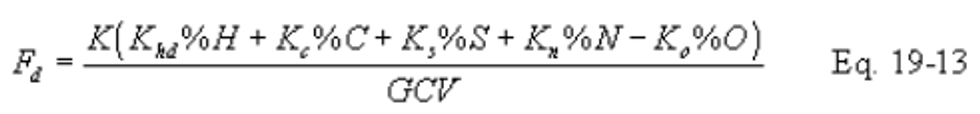
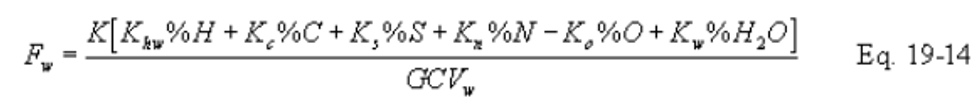
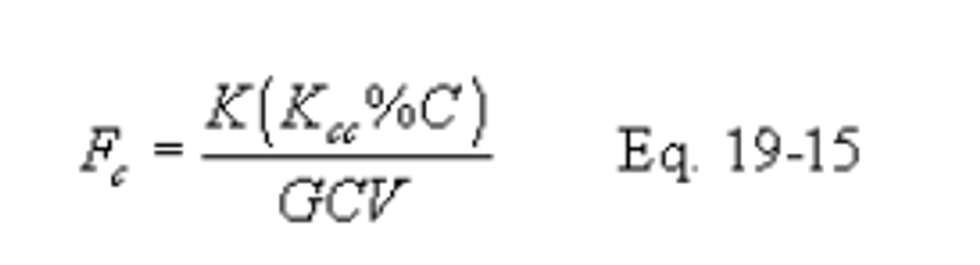
Note:
Omit the %H2O term in the equations for Fw if %H and %O include the unavailable hydrogen and oxygen in the form of H2O.)
12.3.2.2 Use applicable sampling procedures in section 12.5.2.1 or 12.5.2.2 to obtain samples for analyses.
12.3.2.3 Use ASTM D 3176-74 or 89 (all cited ASTM standards are incorporated by reference - see §60.17) for ultimate analysis of the fuel.
12.3.2.4 Use applicable methods in section 12.5.2.1 or 12.5.2.2 to determine the heat content of solid or liquid fuels. For gaseous fuels, use ASTM D 1826-77 or 94 (incorporated by reference - see §60.17) to determine the heat content.
12.3.3 F Factors for Combination of Fuels. If combinations of fuels are burned, use the following equations, as applicable unless otherwise specified in an applicable subpart:
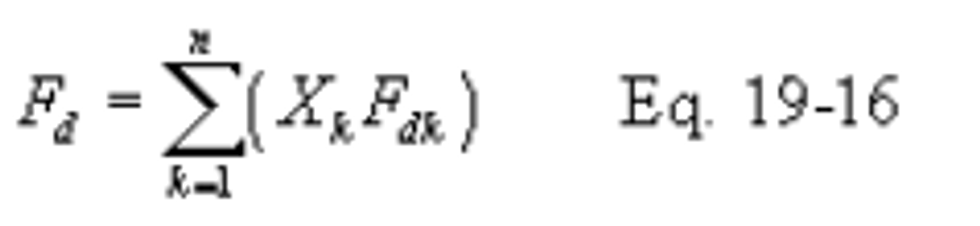
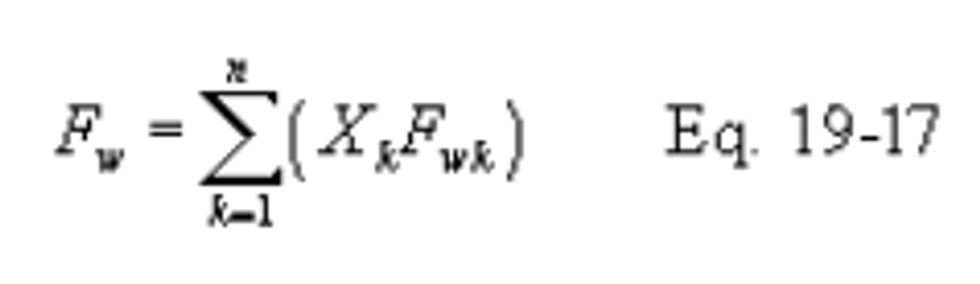
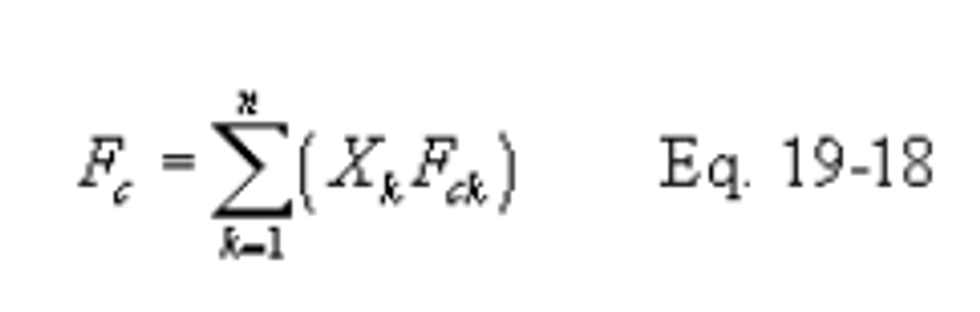
12.4 Determination of Average Pollutant Rates.
12.4.1 Average Pollutant Rates from Hourly Values. When hourly average pollutant rates (Eh), inlet or outlet, are obtained (e.g., CEMS values), compute the average pollutant rate (Ea) for the performance test period (e.g., 30 days) specified in the applicable regulation using the following equation:
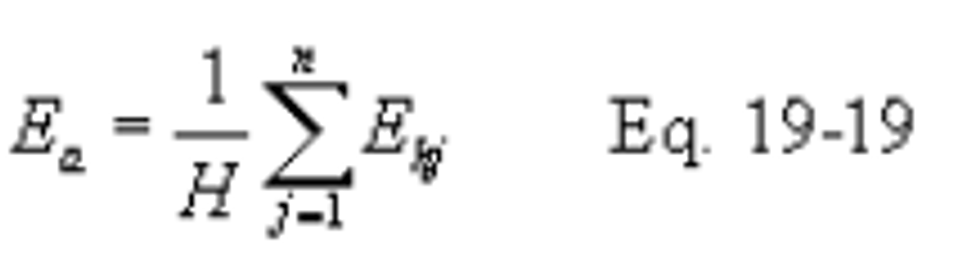
12.4.2 Average Pollutant Rates from Other than Hourly Averages. When pollutant rates are determined from measured values representing longer than 1-hour periods (e.g., daily fuel sampling and analyses or Method 6B values), or when pollutant rates are determined from combinations of 1-hour and longer than 1-hour periods (e.g., CEMS and Method 6B values), compute the average pollutant rate (Ea) for the performance test period (e.g., 30 days) specified in the applicable regulation using the following equation:
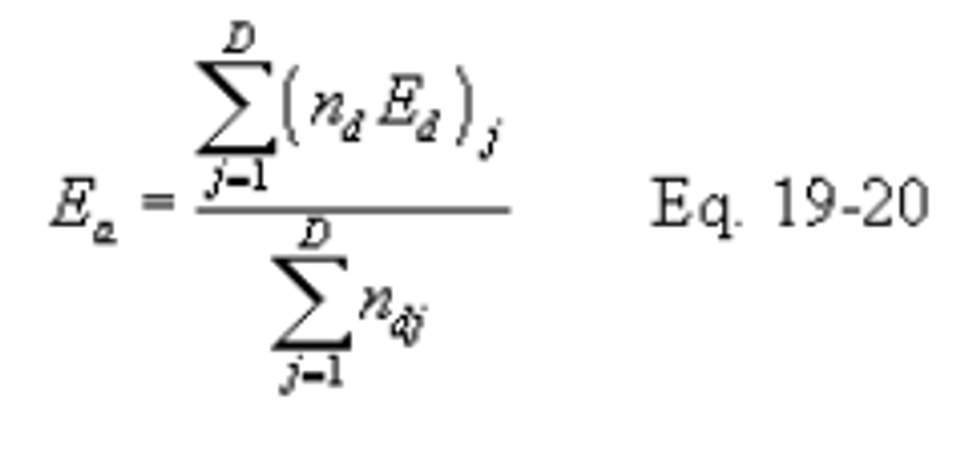
12.4.3 Daily Geometric Average Pollutant Rates from Hourly Values. The geometric average pollutant rate (Ega) is computed using the following equation:
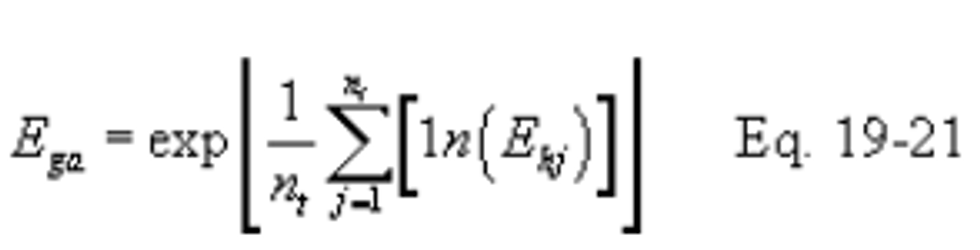
12.5 Determination of Overall Reduction in Potential Sulfur Dioxide Emission.
12.5.1 Overall Percent Reduction. Compute the overall percent SO2 reduction (%Ro) using the following equation:
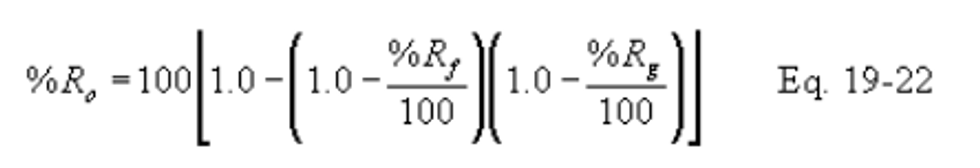
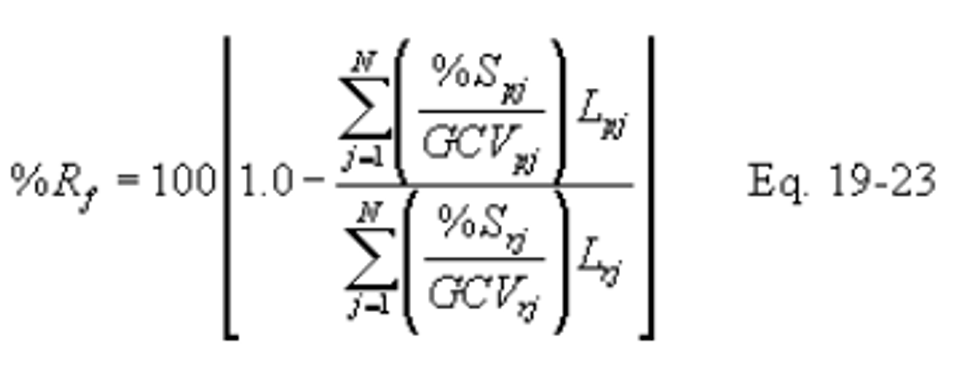
12.5.2 Pretreatment Removal Efficiency (Optional). Compute the SO2 removal efficiency from fuel pretreatment (%Rf) for the averaging period (e.g., 90 days) as specified in the applicable regulation using the following equation:
Note:
In calculating %Rf, include %S and GCV values for all fuel lots that are not pretreated and are used during the averaging period.
12.5.2.1 Solid Fossil (Including Waste) Fuel/Sampling and Analysis.
Note:
For the purposes of this method, raw fuel (coal or oil) is the fuel delivered to the desulfurization (pretreatment) facility. For oil, the input oil to the oil desulfurization process (e.g., hydrotreatment) is considered to be the raw fuel.
12.5.2.1.1 Sample Increment Collection. Use ASTM D 2234-76, 96, 97a, or 98 (incorporated by reference - see §60.17), Type I, Conditions A, B, or C, and systematic spacing. As used in this method, systematic spacing is intended to include evenly spaced increments in time or increments based on equal weights of coal passing the collection area. As a minimum, determine the number and weight of increments required per gross sample representing each coal lot according to Table 2 or Paragraph 7.1.5.2 of ASTM D 2234. Collect one gross sample for each lot of raw coal and one gross sample for each lot of product coal.
12.5.2.1.2 ASTM Lot Size. For the purpose of section 12.5.2 (fuel pretreatment), the lot size of product coal is the weight of product coal from one type of raw coal. The lot size of raw coal is the weight of raw coal used to produce one lot of product coal. Typically, the lot size is the weight of coal processed in a 1-day (24-hour) period. If more than one type of coal is treated and produced in 1 day, then gross samples must be collected and analyzed for each type of coal. A coal lot size equaling the 90-day quarterly fuel quantity for a steam generating unit may be used if representative sampling can be conducted for each raw coal and product coal.
Note:
Alternative definitions of lot sizes may be used, subject to prior approval of the Administrator.
12.5.2.1.3 Gross Sample Analysis. Use ASTM D 2013-72 or 86 to prepare the sample, ASTM D 3177-75 or 89 or ASTM D 4239-85, 94, or 97 to determine sulfur content (%S), ASTM D 3173-73 or 87 to determine moisture content, and ASTM D 2015-77 (Reapproved 1978) or 96, D 3286-85 or 96, or D 5865-98 or 10 to determine gross calorific value (GCV) (all standards cited are incorporated by reference - see §60.17 for acceptable versions of the standards) on a dry basis for each gross sample.
12.5.2.2 Liquid Fossil Fuel-Sampling and Analysis. See Note under section 12.5.2.1.
12.5.2.2.1 Sample Collection. Follow the procedures for continuous sampling in ASTM D 270 or D 4177-95 (incorporated by reference - see §60.17) for each gross sample from each fuel lot.
12.5.2.2.2 Lot Size. For the purpose of section 12.5.2 (fuel pretreatment), the lot size of a product oil is the weight of product oil from one pretreatment facility and intended as one shipment (ship load, barge load, etc.). The lot size of raw oil is the weight of each crude liquid fuel type used to produce a lot of product oil.
Note:
Alternative definitions of lot sizes may be used, subject to prior approval of the Administrator.
12.5.2.2.3 Sample Analysis. Use ASTM D 129-64, 78, or 95, ASTM D 1552-83 or 95, or ASTM D 4057-81 or 95 to determine the sulfur content (%S) and ASTM D 240-76 or 92 (all standards cited are incorporated by reference - see §60.17) to determine the GCV of each gross sample. These values may be assumed to be on a dry basis. The owner or operator of an affected facility may elect to determine the GCV by sampling the oil combusted on the first steam generating unit operating day of each calendar month and then using the lowest GCV value of the three GCV values per quarter for the GCV of all oil combusted in that calendar quarter.
12.5.2.3 Use appropriate procedures, subject to the approval of the Administrator, to determine the fraction of total mass input derived from each type of fuel.
12.5.3 Control Device Removal Efficiency. Compute the percent removal efficiency (%Rg) of the control device using the following equation:
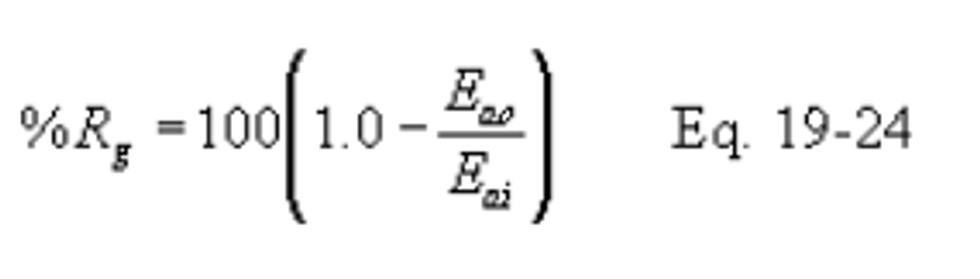
12.5.3.1 Use continuous emission monitoring systems or test methods, as appropriate, to determine the outlet SO2 rates and, if appropriate, the inlet SO2 rates. The rates may be determined as hourly (Eh) or other sampling period averages (Ed). Then, compute the average pollutant rates for the performance test period (Eao and Eai) using the procedures in section 12.4.
12.5.3.2 As an alternative, as-fired fuel sampling and analysis may be used to determine inlet SO2 rates as follows:
12.5.3.2.1 Compute the average inlet SO2 rate (Edi) for each sampling period using the following equation:
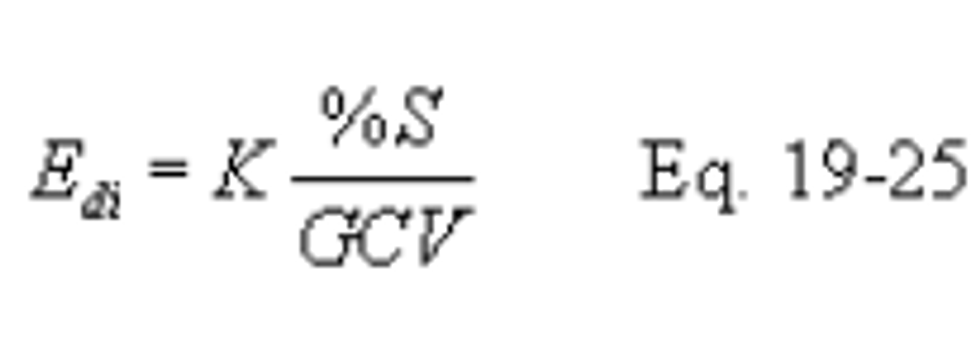
Where:
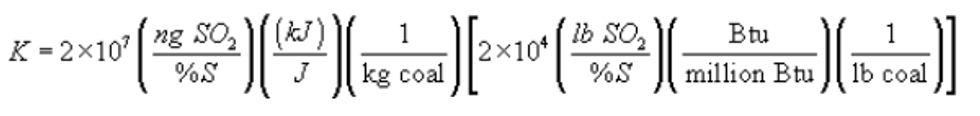
After calculating Edi, use the procedures in section 12.4 to determine the average inlet SO2 rate for the performance test period (Eai).
12.5.3.2.2 Collect the fuel samples from a location in the fuel handling system that provides a sample representative of the fuel bunkered or consumed during a steam generating unit operating day. For the purpose of as-fired fuel sampling under section 12.5.3.2 or section 12.6, the lot size for coal is the weight of coal bunkered or consumed during each steam generating unit operating day. The lot size for oil is the weight of oil supplied to the “day” tank or consumed during each steam generating unit operating day. For reporting and calculation purposes, the gross sample shall be identified with the calendar day on which sampling began. For steam generating unit operating days when a coal-fired steam generating unit is operated without coal being added to the bunkers, the coal analysis from the previous “as bunkered” coal sample shall be used until coal is bunkered again. For steam generating unit operating days when an oil-fired steam generating unit is operated without oil being added to the oil “day” tank, the oil analysis from the previous day shall be used until the “day” tank is filled again. Alternative definitions of fuel lot size may be used, subject to prior approval of the Administrator.
12.5.3.2.3 Use ASTM procedures specified in section 12.5.2.1 or 12.5.2.2 to determine %S and GCV.
12.5.4 Daily Geometric Average Percent Reduction from Hourly Values. The geometric average percent reduction (%Rga) is computed using the following equation:
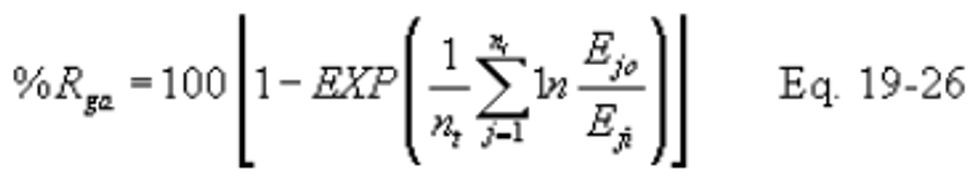
Note:
The calculation includes only paired data sets (hourly average) for the inlet and outlet pollutant measurements.
12.6 Sulfur Retention Credit for Compliance Fuel. If fuel sampling and analysis procedures in section 12.5.2.1 are being used to determine average SO2 emission rates (Eas) to the atmosphere from a coal-fired steam generating unit when there is no SO2 control device, the following equation may be used to adjust the emission rate for sulfur retention credits (no credits are allowed for oil-fired systems) (Edi) for each sampling period using the following equation:
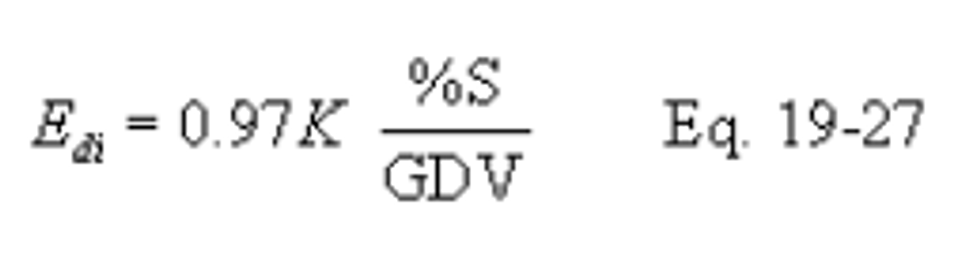
Where:
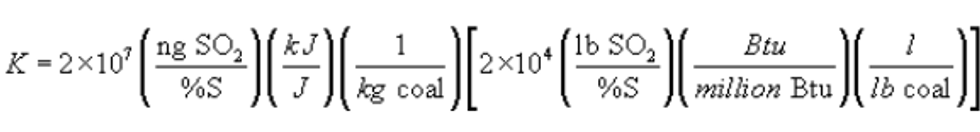
After calculating Edi, use the procedures in section 12.4.2 to determine the average SO2 emission rate to the atmosphere for the performance test period (Eao).
12.7 Determination of Compliance When Minimum Data Requirement Is Not Met.
12.7.1 Adjusted Emission Rates and Control Device Removal Efficiency. When the minimum data requirement is not met, the Administrator may use the following adjusted emission rates or control device removal efficiencies to determine compliance with the applicable standards.
12.7.1.1 Emission Rate. Compliance with the emission rate standard may be determined by using the lower confidence limit of the emission rate (Eao*) as follows:
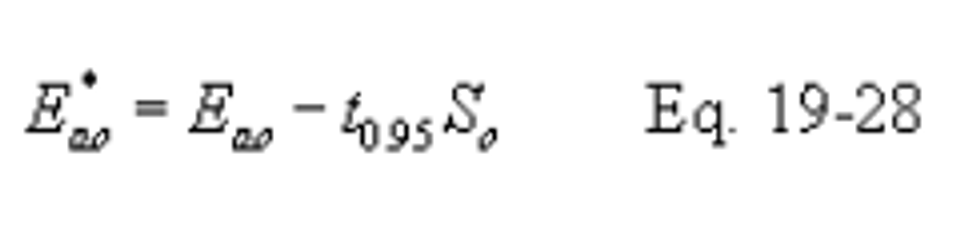
12.7.1.2 Control Device Removal Efficiency. Compliance with the overall emission reduction (%Ro) may be determined by using the lower confidence limit of the emission rate (Eao*) and the upper confidence limit of the inlet pollutant rate (Eai*) in calculating the control device removal efficiency (%Rg) as follows:
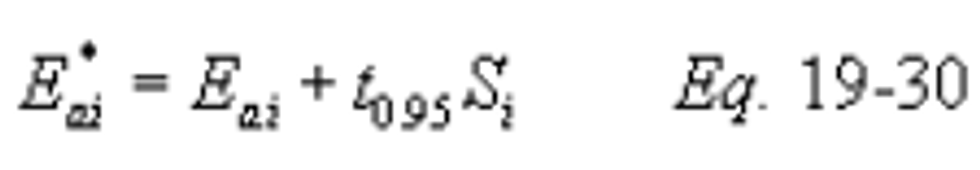

12.7.2 Standard Deviation of Hourly Average Pollutant Rates. Compute the standard deviation (Se) of the hourly average pollutant rates using the following equation:
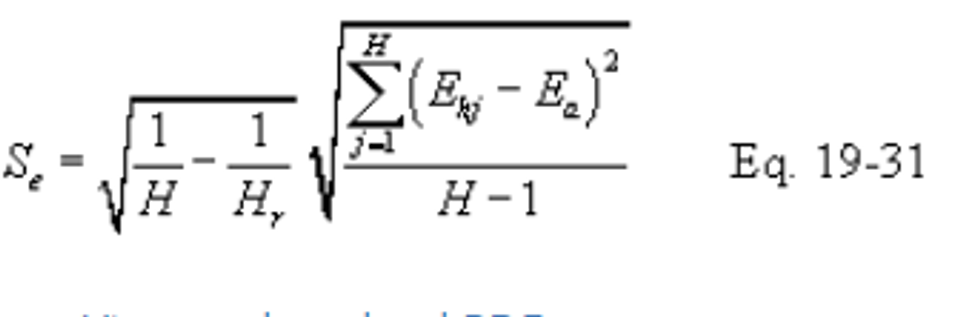
Equation 19-19 through 19-31 may be used to compute the standard deviation for both the outlet (So) and, if applicable, inlet (Si) pollutant rates.
13.0 Method Performance [Reserved]
14.0 Pollution Prevention [Reserved]
15.0 Waste Management [Reserved]
16.0 References [Reserved]
17.0 Tables, Diagrams, Flowcharts, and Validation Data
| From | To | Multiply by |
|---|---|---|
| g/scm | ng/scm | 10 9 |
| mg/scm | ng/scm | 10 6 |
| lb/scf | ng/scm | 1.602 × 10 13 |
| ppm SO2 | ng/scm | 2.66 × 10 6 |
| ppm NOX | ng/scm | 1.912 × 10 6 |
| ppm SO2 | lb/scf | 1.660 × 10−7 |
| ppm NOX | lb/scf | 1.194 × 10−7 |
| Fuel Type | Fd | Fw | Fc | |||
|---|---|---|---|---|---|---|
| dscm/J | dscf/10 6 Btu | wscm/J | wscf/10 6 Btu | scm/J | scf/10 6 Btu | |
| Coal: | ||||||
| Anthracite 2 | 2.71 × 10−7 | 10,100 | 2.83 × 10−7 | 10,540 | 0.530 × 10−7 | 1,970 |
| Bituminus 2 | 2.63 × 10−7 | 9,780 | 2.86 × 10−7 | 10,640 | 0.484 × 10−7 | 1,800 |
| Lignite | 2.65 × 10−7 | 9,860 | 3.21 × 10−7 | 11,950 | 0.513 × 10−7 | 1,910 |
| Oil 3 | 2.47 × 10−7 | 9,190 | 2.77 × 10−7 | 10,320 | 0.383 × 10−7 | 1,420 |
| Gas: | ||||||
| Natural | 2.34 × 10−7 | 8,710 | 2.85 × 10−7 | 10,610 | 0.287 × 10−7 | 1,040 |
| Propane | 2.34 × 10−7 | 8,710 | 2.74 × 10−7 | 10,200 | 0.321 × 10−7 | 1,190 |
| Butane | 2.34 × 10−7 | 8,710 | 2.79 × 10−7 | 10,390 | 0.337 × 10−7 | 1,250 |
| Wood | 2.48 × 10−7 | 9,240 | 0.492 × 10−7 | 1,830 | ||
| Wood Bark | 2.58 × 10−7 | 9,600 | 0.516 × 10−7 | 1,920 | ||
| Municipal | 2.57 × 10−7 | 9,570 | 0.488 × 10−7 | 1,820 | ||
| Solid Waste | ||||||
| 1 Determined at standard conditions: 20°C (68°F) and 760 mm Hg (29.92 in Hg) 2 As classified according to ASTM D 388. 3 Crude, residual, or distillate. | ||||||
| n 1 | t0.95 | n 1 | t0.95 | n 1 | t0.95 |
|---|---|---|---|---|---|
| 2 | 6.31 | 8 | 1.89 | 22-26 | 1.71 |
| 3 | 2.42 | 9 | 1.86 | 27-31 | 1.70 |
| 4 | 2.35 | 10 | 1.83 | 32-51 | 1.68 |
| 5 | 2.13 | 11 | 1.81 | 52-91 | 1.67 |
| 6 | 2.02 | 12-16 | 1.77 | 92-151 | 1.66 |
| 7 | 1.94 | 17-21 | 1.73 | 152 or more | 1.65 |
| 1The values of this table are corrected for n-1 degrees of freedom. Use n equal to the number (H) of hourly average data points. | |||||
Method 20 - Determination of Nitrogen Oxides, Sulfur Dioxide, and Diluent Emissions From Stationary Gas Turbines
1.0 Scope and Application
What is Method 20?
Method 20 contains the details you must follow when using an instrumental analyzer to determine concentrations of nitrogen oxides, oxygen, carbon dioxide, and sulfur dioxide in the emissions from stationary gas turbines. This method follows the specific instructions for equipment and performance requirements, supplies, sample collection and analysis, calculations, and data analysis in the methods listed in section 2.0.
1.1 Analytes. What does this method determine?
| Analyte | CAS No. | Sensitivity |
|---|---|---|
| Nitrogen oxides (NOX) as nitrogen dioxide: | 10102-43-9 | Typically <2% of Calibration Span. |
| Nitric oxide (NO) | 10102-44-0 | |
| Nitrogen dioxide NO2 | ||
| Diluent oxygen (O2) or carbon dioxide (CO2) | Typically <2% of Calibration Span. | |
| Sulfur dioxide (SOX) | 7446-09-5 | Typically <2% of Calibration Span. |
1.2 Applicability. When is this method required? The use of Method 20 may be required by specific New Source Performance Standards, Clean Air Marketing rules, and State Implementation Plans and permits where measuring SO2, NOX, CO2, and/or O2 concentrations in stationary gas turbines emissions are required. Other regulations may also require its use.
1.3 Data Quality Objectives. How good must my collected data be? Refer to section 1.3 of Method 7E.
2.0 Summary of Method
In this method, NOX, O2 (or CO2), and SOX are measured using the following methods found in appendix A to this part:
(a) Method 1 - Sample and Velocity Traverses for Stationary Sources.
(b) Method 3A - Determination of Oxygen and Carbon Dioxide Emissions From Stationary Sources (Instrumental Analyzer Procedure).
(c) Method 6C - Determination of Sulfur Dioxide Emissions From Stationary Sources (Instrumental Analyzer Procedure).
(d) Method 7E - Determination of Nitrogen Oxides Emissions From Stationary Sources (Instrumental Analyzer Procedure).
(e) Method 19 - Determination of Sulfur Dioxide Removal Efficiency and Particulate Matter, Sulfur Dioxide, and Nitrogen Oxide Emission Rates.
3.0 Definitions
Refer to section 3.0 of Method 7E for the applicable definitions.
4.0 Interferences
Refer to section 4.0 of Methods 3A, 6C, and 7E as applicable.
5.0 Safety
Refer to section 5.0 of Method 7E.
6.0 Equipment and Supplies
The measurement system design is shown in Figure 7E-1 of Method 7E. Refer to the appropriate methods listed in section 2.0 for equipment and supplies.
7.0 Reagents and Standards
Refer to the appropriate methods listed in section 2.0 for reagents and standards.
8.0 Sample Collection, Preservation, Storage, and Transport
8.1 Sampling Site and Sampling Points. Follow the procedures of section 8.1 of Method 7E. For the stratification test in section 8.1.2, determine the diluent-corrected pollutant concentration at each traverse point.
8.2 Initial Measurement System Performance Tests. You must refer to the appropriate methods listed in section 2.0 for the measurement system performance tests as applicable.
8.3 Interference Check. You must follow the procedures in section 8.3 of Method 3A or 6C, or section 8.2.7 of Method 7E (as appropriate).
8.4 Sample Collection. You must follow the procedures of section 8.4 of the appropriate methods listed in section 2.0. A test run must have a duration of at least 21 minutes.
8.5 Post-Run System Bias Check, Drift Assessment, and Alternative Dynamic Spike Procedure. You must follow the procedures of sections 8.5 and 8.6 of the appropriate methods listed in section 2.0. A test run must have a duration of at least 21 minutes.
9.0 Quality Control
Follow quality control procedures in section 9.0 of Method 7E.
10.0 Calibration and Standardization
Follow the procedures for calibration and standardization in section 10.0 of Method 7E.
11.0 Analytical Procedures
Because sample collection and analysis are performed together (see section 8), additional discussion of the analytical procedure is not necessary.
12.0 Calculations and Data Analysis
You must follow the procedures for calculations and data analysis in section 12.0 of the appropriate method listed in section 2.0. Follow the procedures in section 12.0 of Method 19 for calculating fuel-specific F factors, diluent-corrected pollutant concentrations, and emission rates.
13.0 Method Performance
The specifications for the applicable performance checks are the same as in section 13.0 of Method 7E.
14.0 Pollution Prevention [Reserved]
15.0 Waste Management [Reserved]
16.0 Alternative Procedures
Refer to section 16.0 of the appropriate method listed in section 2.0 for alternative procedures.
17.0 References
Refer to section 17.0 of the appropriate method listed in section 2.0 for references.
18.0 Tables, Diagrams, Flowcharts, and Validation Data
Refer to section 18.0 of the appropriate method listed in section 2.0 for tables, diagrams, flowcharts, and validation data.
Method 21 - Determination of Volatile Organic Compound Leaks
1.0 Scope and Application
1.1 Analytes.
| Analyte | CAS No. |
|---|---|
| Volatile Organic Compounds (VOC) | No CAS number assigned. |
1.2 Scope. This method is applicable for the determination of VOC leaks from process equipment. These sources include, but are not limited to, valves, flanges and other connections, pumps and compressors, pressure relief devices, process drains, open-ended valves, pump and compressor seal system degassing vents, accumulator vessel vents, agitator seals, and access door seals.
1.3 Data Quality Objectives. Adherence to the requirements of this method will enhance the quality of the data obtained from air pollutant sampling methods.
2.0 Summary of Method
2.1 A portable instrument is used to detect VOC leaks from individual sources. The instrument detector type is not specified, but it must meet the specifications and performance criteria contained in section 6.0. A leak definition concentration based on a reference compound is specified in each applicable regulation. This method is intended to locate and classify leaks only, and is not to be used as a direct measure of mass emission rate from individual sources.
3.0 Definitions
3.1 Calibration gas means the VOC compound used to adjust the instrument meter reading to a known value. The calibration gas is usually the reference compound at a known concentration approximately equal to the leak definition concentration.
3.2 Calibration precision means the degree of agreement between measurements of the same known value, expressed as the relative percentage of the average difference between the meter readings and the known concentration to the known concentration.
3.3 Leak definition concentration means the local VOC concentration at the surface of a leak source that indicates that a VOC emission (leak) is present. The leak definition is an instrument meter reading based on a reference compound.
3.4 No detectable emission means a local VOC concentration at the surface of a leak source, adjusted for local VOC ambient concentration, that is less than 2.5 percent of the specified leak definition concentration. that indicates that a VOC emission (leak) is not present.
3.5 Reference compound means the VOC species selected as the instrument calibration basis for specification of the leak definition concentration. (For example, if a leak definition concentration is 10,000 ppm as methane, then any source emission that results in a local concentration that yields a meter reading of 10,000 on an instrument meter calibrated with methane would be classified as a leak. In this example, the leak definition concentration is 10,000 ppm and the reference compound is methane.)
3.6 Response factor means the ratio of the known concentration of a VOC compound to the observed meter reading when measured using an instrument calibrated with the reference compound specified in the applicable regulation.
3.7 Response time means the time interval from a step change in VOC concentration at the input of the sampling system to the time at which 90 percent of the corresponding final value is reached as displayed on the instrument readout meter.
4.0 Interferences [Reserved]
5.0 Safety
5.1 Disclaimer. This method may involve hazardous materials, operations, and equipment. This test method may not address all of the safety problems associated with its use. It is the responsibility of the user of this test method to establish appropriate safety and health practices and determine the applicability of regulatory limitations prior to performing this test method.
5.2 Hazardous Pollutants. Several of the compounds, leaks of which may be determined by this method, may be irritating or corrosive to tissues (e.g., heptane) or may be toxic (e.g., benzene, methyl alcohol). Nearly all are fire hazards. Compounds in emissions should be determined through familiarity with the source. Appropriate precautions can be found in reference documents, such as reference No. 4 in section 16.0.
6.0 Equipment and Supplies
A VOC monitoring instrument meeting the following specifications is required:
6.1 The VOC instrument detector shall respond to the compounds being processed. Detector types that may meet this requirement include, but are not limited to, catalytic oxidation, flame ionization, infrared absorption, and photoionization.
6.2 The instrument shall be capable of measuring the leak definition concentration specified in the regulation.
6.3 The scale of the instrument meter shall be readable to ±2.5 percent of the specified leak definition concentration.
6.4 The instrument shall be equipped with an electrically driven pump to ensure that a sample is provided to the detector at a constant flow rate. The nominal sample flow rate, as measured at the sample probe tip, shall be 0.10 to 3.0 l/min (0.004 to 0.1 ft 3/min) when the probe is fitted with a glass wool plug or filter that may be used to prevent plugging of the instrument.
6.5 The instrument shall be equipped with a probe or probe extension or sampling not to exceed 6.4 mm ( 1/4 in) in outside diameter, with a single end opening for admission of sample.
6.6 The instrument shall be intrinsically safe for operation in explosive atmospheres as defined by the National Electrical Code by the National Fire Prevention Association or other applicable regulatory code for operation in any explosive atmospheres that may be encountered in its use. The instrument shall, at a minimum, be intrinsically safe for Class 1, Division 1 conditions, and/or Class 2, Division 1 conditions, as appropriate, as defined by the example code. The instrument shall not be operated with any safety device, such as an exhaust flame arrestor, removed.
7.0 Reagents and Standards
7.1 Two gas mixtures are required for instrument calibration and performance evaluation:
7.1.1 Zero Gas. Air, less than 10 parts per million by volume (ppmv) VOC.
7.1.2 Calibration Gas. For each organic species that is to be measured during individual source surveys, obtain or prepare a known standard in air at a concentration approximately equal to the applicable leak definition specified in the regulation.
7.2 Cylinder Gases. If cylinder calibration gas mixtures are used, they must be analyzed and certified by the manufacturer to be within 2 percent accuracy, and a shelf life must be specified. Cylinder standards must be either reanalyzed or replaced at the end of the specified shelf life.
7.3 Prepared Gases. Calibration gases may be prepared by the user according to any accepted gaseous preparation procedure that will yield a mixture accurate to within 2 percent. Prepared standards must be replaced each day of use unless it is demonstrated that degradation does not occur during storage.
7.4 Mixtures with non-Reference Compound Gases. Calibrations may be performed using a compound other than the reference compound. In this case, a conversion factor must be determined for the alternative compound such that the resulting meter readings during source surveys can be converted to reference compound results.
8.0 Sample Collection, Preservation, Storage, and Transport
8.1 Instrument Performance Evaluation. Assemble and start up the instrument according to the manufacturer's instructions for recommended warmup period and preliminary adjustments.
8.1.1 Response Factor. A response factor must be determined for each compound that is to be measured, either by testing or from reference sources. The response factor tests are required before placing the analyzer into service, but do not have to be repeated at subsequent intervals.
8.1.1.1 Calibrate the instrument with the reference compound as specified in the applicable regulation. Introduce the calibration gas mixture to the analyzer and record the observed meter reading. Introduce zero gas until a stable reading is obtained. Make a total of three measurements by alternating between the calibration gas and zero gas. Calculate the response factor for each repetition and the average response factor.
8.1.1.2 The instrument response factors for each of the individual VOC to be measured shall be less than 10 unless otherwise specified in the applicable regulation. When no instrument is available that meets this specification when calibrated with the reference VOC specified in the applicable regulation, the available instrument may be calibrated with one of the VOC to be measured, or any other VOC, so long as the instrument then has a response factor of less than 10 for each of the individual VOC to be measured.
8.1.1.3 Alternatively, if response factors have been published for the compounds of interest for the instrument or detector type, the response factor determination is not required, and existing results may be referenced. Examples of published response factors for flame ionization and catalytic oxidation detectors are included in References 1-3 of section 17.0.
8.1.2 Calibration Precision. The calibration precision test must be completed prior to placing the analyzer into service and at subsequent 3-month intervals or at the next use, whichever is later.
8.1.2.1 Make a total of three measurements by alternately using zero gas and the specified calibration gas. Record the meter readings. Calculate the average algebraic difference between the meter readings and the known value. Divide this average difference by the known calibration value and multiply by 100 to express the resulting calibration precision as a percentage.
8.1.2.2 The calibration precision shall be equal to or less than 10 percent of the calibration gas value.
8.1.3 Response Time. The response time test is required before placing the instrument into service. If a modification to the sample pumping system or flow configuration is made that would change the response time, a new test is required before further use.
8.1.3.1 Introduce zero gas into the instrument sample probe. When the meter reading has stabilized, switch quickly to the specified calibration gas. After switching, measure the time required to attain 90 percent of the final stable reading. Perform this test sequence three times and record the results. Calculate the average response time.
8.1.3.2 The instrument response time shall be equal to or less than 30 seconds. The instrument pump, dilution probe (if any), sample probe, and probe filter that will be used during testing shall all be in place during the response time determination.
8.2 Instrument Calibration. Calibrate the VOC monitoring instrument according to section 10.0.
8.3 Individual Source Surveys.
8.3.1 Type I - Leak Definition Based on Concentration. Place the probe inlet at the surface of the component interface where leakage could occur. Move the probe along the interface periphery while observing the instrument readout. If an increased meter reading is observed, slowly sample the interface where leakage is indicated until the maximum meter reading is obtained. Leave the probe inlet at this maximum reading location for approximately two times the instrument response time. If the maximum observed meter reading is greater than the leak definition in the applicable regulation, record and report the results as specified in the regulation reporting requirements. Examples of the application of this general technique to specific equipment types are:
8.3.1.1 Valves. The most common source of leaks from valves is the seal between the stem and housing. Place the probe at the interface where the stem exits the packing gland and sample the stem circumference. Also, place the probe at the interface of the packing gland take-up flange seat and sample the periphery. In addition, survey valve housings of multipart assembly at the surface of all interfaces where a leak could occur.
8.3.1.2 Flanges and Other Connections. For welded flanges, place the probe at the outer edge of the flange-gasket interface and sample the circumference of the flange. Sample other types of nonpermanent joints (such as threaded connections) with a similar traverse.
8.3.1.3 Pumps and Compressors. Conduct a circumferential traverse at the outer surface of the pump or compressor shaft and seal interface. If the source is a rotating shaft, position the probe inlet within 1 cm of the shaft-seal interface for the survey. If the housing configuration prevents a complete traverse of the shaft periphery, sample all accessible portions. Sample all other joints on the pump or compressor housing where leakage could occur.
8.3.1.4 Pressure Relief Devices. The configuration of most pressure relief devices prevents sampling at the sealing seat interface. For those devices equipped with an enclosed extension, or horn, place the probe inlet at approximately the center of the exhaust area to the atmosphere.
8.3.1.5 Process Drains. For open drains, place the probe inlet at approximately the center of the area open to the atmosphere. For covered drains, place the probe at the surface of the cover interface and conduct a peripheral traverse.
8.3.1.6 Open-ended Lines or Valves. Place the probe inlet at approximately the center of the opening to the atmosphere.
8.3.1.7 Seal System Degassing Vents and Accumulator Vents. Place the probe inlet at approximately the center of the opening to the atmosphere.
8.3.1.8 Access door seals. Place the probe inlet at the surface of the door seal interface and conduct a peripheral traverse.
8.3.2 Type II - “No Detectable Emission”. Determine the local ambient VOC concentration around the source by moving the probe randomly upwind and downwind at a distance of one to two meters from the source. If an interference exists with this determination due to a nearby emission or leak, the local ambient concentration may be determined at distances closer to the source, but in no case shall the distance be less than 25 centimeters. Then move the probe inlet to the surface of the source and determine the concentration as outlined in section 8.3.1. The difference between these concentrations determines whether there are no detectable emissions. Record and report the results as specified by the regulation. For those cases where the regulation requires a specific device installation, or that specified vents be ducted or piped to a control device, the existence of these conditions shall be visually confirmed. When the regulation also requires that no detectable emissions exist, visual observations and sampling surveys are required. Examples of this technique are:
8.3.2.1 Pump or Compressor Seals. If applicable, determine the type of shaft seal. Perform a survey of the local area ambient VOC concentration and determine if detectable emissions exist as described in section 8.3.2.
8.3.2.2 Seal System Degassing Vents, Accumulator Vessel Vents, Pressure Relief Devices. If applicable, observe whether or not the applicable ducting or piping exists. Also, determine if any sources exist in the ducting or piping where emissions could occur upstream of the control device. If the required ducting or piping exists and there are no sources where the emissions could be vented to the atmosphere upstream of the control device, then it is presumed that no detectable emissions are present. If there are sources in the ducting or piping where emissions could be vented or sources where leaks could occur, the sampling surveys described in section 8.3.2 shall be used to determine if detectable emissions exist.
8.3.3 Alternative Screening Procedure.
8.3.3.1 A screening procedure based on the formation of bubbles in a soap solution that is sprayed on a potential leak source may be used for those sources that do not have continuously moving parts, that do not have surface temperatures greater than the boiling point or less than the freezing point of the soap solution, that do not have open areas to the atmosphere that the soap solution cannot bridge, or that do not exhibit evidence of liquid leakage. Sources that have these conditions present must be surveyed using the instrument technique of section 8.3.1 or 8.3.2.
8.3.3.2 Spray a soap solution over all potential leak sources. The soap solution may be a commercially available leak detection solution or may be prepared using concentrated detergent and water. A pressure sprayer or squeeze bottle may be used to dispense the solution. Observe the potential leak sites to determine if any bubbles are formed. If no bubbles are observed, the source is presumed to have no detectable emissions or leaks as applicable. If any bubbles are observed, the instrument techniques of section 8.3.1 or 8.3.2 shall be used to determine if a leak exists, or if the source has detectable emissions, as applicable.
9.0 Quality Control
| Section | Quality control measure | Effect |
|---|---|---|
| 8.1.2 | Instrument calibration precision check | Ensure precision and accuracy, respectively, of instrument response to standard. |
| 10.0 | Instrument calibration |
10.0 Calibration and Standardization
10.1 Calibrate the VOC monitoring instrument as follows. After the appropriate warmup period and zero internal calibration procedure, introduce the calibration gas into the instrument sample probe. Adjust the instrument meter readout to correspond to the calibration gas value.
Note:
If the meter readout cannot be adjusted to the proper value, a malfunction of the analyzer is indicated and corrective actions are necessary before use.
11.0 Analytical Procedures [Reserved]
12.0 Data Analyses and Calculations [Reserved]
13.0 Method Performance [Reserved]
14.0 Pollution Prevention [Reserved]
15.0 Waste Management [Reserved]
16.0 References
1. Dubose, D.A., and G.E. Harris. Response Factors of VOC Analyzers at a Meter Reading of 10,000 ppmv for Selected Organic Compounds. U.S. Environmental Protection Agency, Research Triangle Park, NC. Publication No. EPA 600/2-81051. September 1981.
2. Brown, G.E., et al. Response Factors of VOC Analyzers Calibrated with Methane for Selected Organic Compounds. U.S. Environmental Protection Agency, Research Triangle Park, NC. Publication No. EPA 600/2-81-022. May 1981.
3. DuBose, D.A. et al. Response of Portable VOC Analyzers to Chemical Mixtures. U.S. Environmental Protection Agency, Research Triangle Park, NC. Publication No. EPA 600/2-81-110. September 1981.
4. Handbook of Hazardous Materials: Fire, Safety, Health. Alliance of American Insurers. Schaumberg, IL. 1983.
17.0 Tables, Diagrams, Flowcharts, and Validation Data [Reserved]
Method 22 - Visual Determination of Fugitive Emissions From Material Sources and Smoke Emissions From Flares
Note:
This method is not inclusive with respect to observer certification. Some material is incorporated by reference from Method 9.
1.0 Scope and Application
This method is applicable for the determination of the frequency of fugitive emissions from stationary sources, only as specified in an applicable subpart of the regulations. This method also is applicable for the determination of the frequency of visible smoke emissions from flares.
2.0 Summary of Method
2.1 Fugitive emissions produced during material processing, handling, and transfer operations or smoke emissions from flares are visually determined by an observer without the aid of instruments.
2.2 This method is used also to determine visible smoke emissions from flares used for combustion of waste process materials.
2.3 This method determines the amount of time that visible emissions occur during the observation period (i.e., the accumulated emission time). This method does not require that the opacity of emissions be determined. Since this procedure requires only the determination of whether visible emissions occur and does not require the determination of opacity levels, observer certification according to the procedures of Method 9 is not required. However, it is necessary that the observer is knowledgeable with respect to the general procedures for determining the presence of visible emissions. At a minimum, the observer must be trained and knowledgeable regarding the effects of background contrast, ambient lighting, observer position relative to lighting, wind, and the presence of uncombined water (condensing water vapor) on the visibility of emissions. This training is to be obtained from written materials found in References 1 and 2 or from the lecture portion of the Method 9 certification course.
3.0 Definitions
3.1 Emission frequency means the percentage of time that emissions are visible during the observation period.
3.2 Emission time means the accumulated amount of time that emissions are visible during the observation period.
3.3 Fugitive emissions means emissions generated by an affected facility which is not collected by a capture system and is released to the atmosphere. This includes emissions that (1) escape capture by process equipment exhaust hoods; (2) are emitted during material transfer; (3) are emitted from buildings housing material processing or handling equipment; or (4) are emitted directly from process equipment.
3.4 Observation period means the accumulated time period during which observations are conducted, not to be less than the period specified in the applicable regulation.
3.5 Smoke emissions means a pollutant generated by combustion in a flare and occurring immediately downstream of the flame. Smoke occurring within the flame, but not downstream of the flame, is not considered a smoke emission.
4.0 Interferences
4.1 Occasionally, fugitive emissions from sources other than the affected facility (e.g., road dust) may prevent a clear view of the affected facility. This may particularly be a problem during periods of high wind. If the view of the potential emission points is obscured to such a degree that the observer questions the validity of continuing observations, then the observations shall be terminated, and the observer shall clearly note this fact on the data form.
5.0 Safety
5.1 Disclaimer. This method may involve hazardous materials, operations, and equipment. This test method may not address all of the safety problems associated with its use. It is the responsibility of the user of this test method to establish appropriate safety and health practices and determine the applicability of regulatory limitations prior to performing this test method.
6.0 Equipment
6.1 Stopwatches (two). Accumulative type with unit divisions of at least 0.5 seconds.
6.2 Light Meter. Light meter capable of measuring illuminance in the 50 to 200 lux range, required for indoor observations only.
7.0 Reagents and Supplies [Reserved]
8.0 Sample Collection, Preservation, Storage, and Transfer [Reserved]
9.0 Quality Control [Reserved]
10.0 Calibration and Standardization [Reserved]
11.0 Analytical Procedure
11.1 Selection of Observation Location. Survey the affected facility, or the building or structure housing the process to be observed, and determine the locations of potential emissions. If the affected facility is located inside a building, determine an observation location that is consistent with the requirements of the applicable regulation (i.e., outside observation of emissions escaping the building/structure or inside observation of emissions directly emitted from the affected facility process unit). Then select a position that enables a clear view of the potential emission point(s) of the affected facility or of the building or structure housing the affected facility, as appropriate for the applicable subpart. A position at least 4.6 m (15 feet), but not more than 400 m (0.25 miles), from the emission source is recommended. For outdoor locations, select a position where the sunlight is not shining directly in the observer's eyes.
11.2 Field Records.
11.2.1 Outdoor Location. Record the following information on the field data sheet (Figure 22-1): Company name, industry, process unit, observer's name, observer's affiliation, and date. Record also the estimated wind speed, wind direction, and sky condition. Sketch the process unit being observed, and note the observer location relative to the source and the sun. Indicate the potential and actual emission points on the sketch. Alternatively, digital photography as described in section 11.2.3 may be used for a subset of the recordkeeping requirements of this section.
11.2.2 Indoor Location. Record the following information on the field data sheet (Figure 22-2): Company name, industry, process unit, observer's name, observer's affiliation, and date. Record as appropriate the type, location, and intensity of lighting on the data sheet. Sketch the process unit being observed, and note the observer location relative to the source. Indicate the potential and actual fugitive emission points on the sketch. Alternatively, digital photography as described in section 11.2.3 may be used for a subset of the recordkeeping requirements of this section.
11.2.3 Digital Photographic Records. Digital photographs, annotated or unaltered, may be used to record and report sky conditions, observer's location relative to the source, observer's location relative to the sun, process unit being observed, potential emission points and actual emission points for the requirements in sections 11.2.1 and 11.2.2. The image must have the proper lighting, field of view and depth of field to properly distinguish the sky condition (if applicable), process unit, potential emission point and actual emission point. At least one digital photograph must be from the point of the view of the observer. The photograph(s) representing the environmental conditions including the sky conditions and the position of the sun relative to the observer and the emission point must be taken within a reasonable time of the observation (i.e., 15 minutes). When observations are taken from exactly the same observation point on a routine basis (i.e., daily) and as long as there are no modifications to the units depicted, only a single photograph each is necessary to document the observer's location relative to the emissions source, the process unit being observed, and the location of potential and actual emission points. Any photographs altered or annotated must be retained in an unaltered format for recordkeeping purposes.
11.3 Indoor Lighting Requirements. For indoor locations, use a light meter to measure the level of illumination at a location as close to the emission source(s) as is feasible. An illumination of greater than 100 lux (10 foot candles) is considered necessary for proper application of this method.
11.4 Observations.
11.4.1 Procedure. Record the clock time when observations begin. Use one stopwatch to monitor the duration of the observation period. Start this stopwatch when the observation period begins. If the observation period is divided into two or more segments by process shutdowns or observer rest breaks (see section 11.4.3), stop the stopwatch when a break begins and restart the stopwatch without resetting it when the break ends. Stop the stopwatch at the end of the observation period. The accumulated time indicated by this stopwatch is the duration of observation period. When the observation period is completed, record the clock time. During the observation period, continuously watch the emission source. Upon observing an emission (condensed water vapor is not considered an emission), start the second accumulative stopwatch; stop the watch when the emission stops. Continue this procedure for the entire observation period. The accumulated elapsed time on this stopwatch is the total time emissions were visible during the observation period (i.e., the emission time.)
11.4.2 Observation Period. Choose an observation period of sufficient length to meet the requirements for determining compliance with the emission standard in the applicable subpart of the regulations. When the length of the observation period is specifically stated in the applicable subpart, it may not be necessary to observe the source for this entire period if the emission time required to indicate noncompliance (based on the specified observation period) is observed in a shorter time period. In other words, if the regulation prohibits emissions for more than 6 minutes in any hour, then observations may (optional) be stopped after an emission time of 6 minutes is exceeded. Similarly, when the regulation is expressed as an emission frequency and the regulation prohibits emissions for greater than 10 percent of the time in any hour, then observations may (optional) be terminated after 6 minutes of emission are observed since 6 minutes is 10 percent of an hour. In any case, the observation period shall not be less than 6 minutes in duration. In some cases, the process operation may be intermittent or cyclic. In such cases, it may be convenient for the observation period to coincide with the length of the process cycle.
11.4.3 Observer Rest Breaks. Do not observe emissions continuously for a period of more than 15 to 20 minutes without taking a rest break. For sources requiring observation periods of greater than 20 minutes, the observer shall take a break of not less than 5 minutes and not more than 10 minutes after every 15 to 20 minutes of observation. If continuous observations are desired for extended time periods, two observers can alternate between making observations and taking breaks.
11.5 Recording Observations. Record the accumulated time of the observation period on the data sheet as the observation period duration. Record the accumulated time emissions were observed on the data sheet as the emission time. Record the clock time the observation period began and ended, as well as the clock time any observer breaks began and ended.
12.0 Data Analysis and Calculations
If the applicable subpart requires that the emission rate be expressed as an emission frequency (in percent), determine this value as follows: Divide the accumulated emission time (in seconds) by the duration of the observation period (in seconds) or by any minimum observation period required in the applicable subpart, if the actual observation period is less than the required period, and multiply this quotient by 100.
13.0 Method Performance [Reserved]
14.0 Pollution Prevention [Reserved]
15.0 Waste Management [Reserved]
16.0 References
1. Missan, R., and A. Stein. Guidelines for Evaluation of Visible Emissions Certification, Field Procedures, Legal Aspects, and Background Material. EPA Publication No. EPA-340/1-75-007. April 1975.
2. Wohlschlegel, P., and D.E. Wagoner. Guideline for Development of a Quality Assurance Program: Volume IX - Visual Determination of Opacity Emissions from Stationary Sources. EPA Publication No. EPA-650/4-74-005i. November 1975.
17.0 Tables, Diagrams, Flowcharts, and Validation Data


Method 23 - Determination of Polychlorinated Dibenzo- -Dioxins, Polychlorinated Dibenzofurans, Polychlorinated Biphenyls, and Polycyclic Aromatic Hydrocarbons From Stationary Sources
[Change Notice] [Previous Text]
1.0 Scope and Application
1.1 Applicability. This method applies to the measurement of polychlorinated dibenzo- p -dioxins and polychlorinated dibenzofurans (PCDD/PCDF), polychlorinated biphenyls (PCB), and/or polycyclic aromatic hydrocarbons (PAH) in emissions from stationary sources. Using this method, you can measure these analyte groups individually or in any combination using a single sample acquisition unless otherwise specified in a rule, regulation, or permit. Tables 23-1 through 23-3 of this method list the applicable target analytes for Method 23. If all 209 PCB are analyzed, the 17 toxic PCB congeners should be resolved and reported while the other PCB can be reported as totals by homolog, for example, total trichlorobiphenyl (TrCB).
1.2 Scope. This method describes the sampling and analytical procedures used to measure selected PCDD and PCDF in stationary sources when required in an applicable subpart. This method also describes how the same sampling and analysis technology can be used to measure selected PCB and PAH from stationary source in combination or as each individual compound class when required in an applicable subpart. However, Method 23 incorporates by reference some of the specifications ( e.g., equipment and supplies) and procedures ( e.g., sampling and analytical) from other methods in this part that are essential to conducting Method 23. To obtain reliable samples, source sampling teams should be trained and experienced with the following additional EPA test methods: Method 1, Method 2, Method 3, Method 4, and Method 5 of Appendices A-1, A-2, and A-3 to 40 CFR part 60. Laboratory analysis teams should be trained and experienced with Method 1668C (found at: https://www.epa.gov/sites/production/files/2015-09/documents/method_1668c_2010.pdf) and Method 1613B of 40 CFR part 136 Appendix A and have a working knowledge of isotope dilution and the use of high-resolution gas chromatography/high-resolution mass spectrometry (HRGC/HRMS).
1.3 The HRGC/HRMS portions of this method are for use by laboratory analysts experienced with HRGC/HRMS analysis of PCDD, PCDF, PCB, and PAH or under the close supervision of such qualified persons. Each source testing team, including the sampling and laboratory organization(s) that use this method, must demonstrate the ability to generate acceptable results that meet the performance criteria in Section 13 of this method.
1.4 This method is “performance-based” and includes acceptability criteria for assessing sampling and analytical procedures. Users may modify the method to overcome interferences or to substitute superior materials and equipment, provided that they meet all performance criteria in this method. Section 13 of this method presents requirements for method performance.
2.0 Summary of Method
This method identifies and determines the concentration of specific PCDD, PCDF, PCB, and PAH compounds. Gaseous and particulate bound target pollutants are withdrawn from the gas stream isokinetically and collected in the sample probe, on a glass fiber or quartz filter, and on a packed column of adsorbent material. This method is not intended to differentiate between target compounds in particulate or vapor fractions. The target compounds are extracted from the combined sample collection media. Portions of the extract are chromatographically fractionated to remove interferences, separated into individual compounds or simple mixtures by HRGC, and measured with HRMS. This method uses isotopically labeled standards to improve method accuracy and precision through isotope dilution quantitation.
3.0 Definitions
3.1 Alternate Recovery Standards. A group of isotopically labeled compounds that is not otherwise designated in this method for quality control (QC) purposes. Alternate recovery standards can be used to assess the recovery of a compound class relative to any step in the sampling and analysis procedure that is not already assessed as a mandatory part of this method, such as the cleanup step.
3.2 Benzo[a]pyrene Toxic Equivalency Quotient (B[a]P-TEQ). One of several schemes that express the toxicity for PAH compounds in terms of the most toxic form of PAH, benzo[ a ]pyrene, as specified in applicable regulations, permits, or other requirements.
3.3 Continuing Calibration Verification (CCV) Standard. A standard prepared at the mid-point concentration of the calibration used to verify the initial calibration. Prepare the CCV standard at the same time as the batch of field samples using the same labeled standards.
3.4 Congener. An individual compound with a common structure (dioxin, furan, or biphenyl), only differing by the number of chlorine or other substituent attached to the structure.
3.5 Estimated Detection Limit (EDL). The minimum qualitatively recognizable signal above background for a target compound. The EDL is a detection limit specific to each sample analysis based on the noise signal measured near the retention time of a target compound or target isomer group. Being sample specific, the EDL is affected by sample size, dilution, recoveries of pre-extraction standard, chemical noise from sample extract, electronic noise from instrument, extract aliquot, relative response of instrument, etc.
3.6 Estimated Maximum Possible Concentration (EMPC). An EMPC is a worst-case estimate of the target compound concentration. Report the results as EMPC when the ion abundance ratio for a target analyte is outside the performance criteria. Calculate the EMPC using both quantitation ions.
3.7 Field Train Proof Blank. A field train proof blank train is a QC sample to evaluate equipment preparation and potential contamination during sample recovery and consists of a fully assembled train at the sampling site, without actual sampling. The field train proof blank train uses glassware from the same preparation batch as the field samples.
3.8 Homolog. A compound belonging to a series of compounds with the same general molecular formula, differing from each other by the number of repeating units of chlorine.
3.9 Isomer. An individual compound with a common structure (dioxin, furan, or biphenyl), only differing by the position of chlorine atoms attached to the structure.
3.10 Isotope Dilution. A means of determining a naturally occurring (native) compound by reference to the same compound in which one or more atoms has been isotopically enriched.
3.11 Laboratory Method Blank (LMB). A quality control sample to assess background contamination or interference from media, reagents, equipment, etc. An LMB is prepared in the laboratory, composed of clean sampling media (filter and XAD-2), using same labeled standards, media, reagents, and materials (sodium sulfate, glass wool, etc.) and processed (extraction, fractionations, cleanup) and analyzed using the same procedures as a field sample.
3.12 Polychlorinated Biphenyl (PCB) congeners. Any or all 209 chlorinated biphenyl congeners. Table 23-3 of this method lists the primary target compounds and Appendix A to this method provides the full list of 209 PCB congeners and isomers.
3.12.1 Monochlorobiphenyl (MoCB). Any or all three monochlorinated biphenyl isomers.
3.12.2 Dichlorobiphenyl (DiCB). Any or all 12 dichlorinated biphenyl isomers.
3.12.3 Trichlorobiphenyl (TrCB). Any or all 24 trichlorinated biphenyl isomers.
3.12.4 Tetrachlorobiphenyl (TeCB). Any or all 42 tetrachlorinated biphenyl isomers.
3.12.5 Pentachlorobiphenyl (PeCB). Any or all 46 pentachlorinated biphenyl isomers.
3.12.6 Hexachlorobiphenyl (HxCB). Any or all 42 hexachlorinated biphenyl isomers.
3.12.7 Heptachlorobiphenyl (HpCB). Any or all 24 heptachlorinated biphenyl isomers.
3.12.8 Octachlorobiphenyl (OcCB). Any or all 12 octachlorinated biphenyl isomers.
3.12.9 Nonachlorobiphenyl (NoCB). Any or all three nonachlorinated biphenyl isomers.
3.12.10 Decachlorobiphenyl (DeCB). Biphenyl fully chlorinated with 10 chlorine atom substituents replacing hydrogen in the parent compound.
3.13 Polychlorinated dibenzo-p-dioxin (PCDD) congeners. Any or all 75 chlorinated dibenzo- p -dioxin congeners. There are seven 2,3,7,8 substituted PCDD congeners and four PCDD homolog groups listed in Table 23-1 of this method. This method does not measure mono- through tri-PCDD and includes non-2,3,7,8 substituted congeners in the total homolog categories.
3.13.1 Tetrachlorodibenzo- p -dioxin (TeCDD ). Any or all 22 tetrachlorinated dibenzo- p -dioxin isomers.
3.13.2 Pentachlorodibenzo- p -dioxin (PeCDD). Any or all 14 pentachlorinated dibenzo- p -dioxin isomers.
3.13.3 Hexachlorodibenzo- p -dioxin (HxCDD). Any or all 10 hexachlorinated dibenzo- p -dioxin isomers.
3.13.4 Heptachlorodibenzo- p -dioxin (HpCDD). Any or all two heptachlorinated dibenzo- p -dioxin isomers.
3.13.5 Octachlorodibenzo- p -dioxin (OCDD). Dibenzodioxin fully chlorinated with eight chlorine atom substituents replacing hydrogen in the parent compound.
3.14 Polychlorinated dibenzofuran (PCDF) congeners. Any or all chlorinated dibenzofuran congeners. There are ten 2,3,7,8 substituted PCDF congeners and four PCDF homolog groups listed in Table 23-1 of this method. This method does not measure mono- through tri-PCDF and includes non-2,3,7,8 substituted congeners in the total homolog categories.
3.14.1 Tetrachlorodibenzofuran (TeCDF). Any or all 38 tetrachlorinated dibenzofuran isomers.
3.14.2 Pentachlorodibenzofuran (PeCDF). Any or all 28 pentachlorinated dibenzofuran isomers.
3.14.3 Hexachlorodibenzofuran (HxCDF). Any or all 16 hexachlorinated dibenzofuran isomers.
3.14.4 Heptachlordibenzofuran (HpCDF). Any or all four heptachlorinated dibenzofuran isomers.
3.14.5 Octachlorodibenzofuran (OCDF). Dibenzofuran fully chlorinated with eight chlorine atom substituents replacing hydrogen in the parent compound.
3.15 Polychlorinated diphenyl ethers (PCDPE). Any or all chlorinated substituted diphenyl ethers.
3.15.1 Hexachlorodiphenyl ether (HxCDPE). Any or all 42 hexachlorinated diphenyl ether isomers.
3.15.2 Heptachlorodiphenyl ether (HpCDPE). Any or all 24 heptachlorinated diphenyl ether isomers.
3.15.3 Octachlorodiphenyl ether (OCDPE). Any or all 12 octachlorinated diphenyl ether isomers.
3.15.4 Nonachlorodiphenyl ether (NCDPE). Any or all three nonachlorinated diphenyl ether isomers.
3.15.5 Decachlorodiphenyl ether (DCDPE).
3.16 Polycyclic Aromatic Hydrocarbons (PAH). Any or all aromatic compounds with two or more fused six-member rings. Table 23-2 of this method lists the target PAH compounds for this method. You may add and analyze additional PAH compounds by adding the appropriate 13 C isotopically labeled compound to the pre-extraction standard mixture and by following the other requirements for target PAH compounds in this method.
3.17 Pre-analysis Standard. A group of isotopically labeled compounds added at a known amount immediately prior to analysis and used to monitor instrument response, injection errors, instrument drift and to determine the recovery of the pre-extraction standard compounds. Add pre-analysis standard to every sample (including blank, QC samples, and calibration solutions) at a known amount.
3.18 Pre-extraction Filter Recovery Standard. A group of isotopically labeled compounds added at a known amount to the filter used to indicate the extraction efficiency of the filter media. Add pre-extraction filter recovery standard to the filter samples just prior extraction. The pre-extraction filter recovery standard is not used for quantitating or recovery correction.
3.19 Pre-extraction Standard. A group of isotopically labeled compounds added in a known amount to the XAD-2 adsorbent resin of each sample immediately before extraction and used for quantitation of target and other labeled compounds to correct for extraction, cleanup, and concentration recovery. These isotopically labeled compounds constitute a matrix spike of the resin. Add pre-extraction standard to every sample at the same level (including blank, QC samples, and calibration solutions).
3.20 Pre-sampling Adsorbent Standard. A group of isotopically labeled compounds added in a known amount to the XAD-2 adsorbent prior to sampling used to monitor sampling aspects of the method.
3.21 Pre-transport Standard. Spiking compound from the list of alternative recovery standards that can be added by the laboratory to the sample shipping containers used to transport field equipment rinse and recovery samples prior to sampling. The measured concentration of the pre-transport recovery standard provides a quality check on potential probe rinse sample spillage or mishandling after sample collection and during shipping.
3.22 Quality Control Sample (QCS). A mid-level standard prepared from a second source standard or prepared from a source of standards different from the source of calibration standards. The purpose of the QCS is to verify the integrity of the primary calibration standards. A QCS is analyzed during the initial demonstration of capability (IDC) and following each initial calibration (at a minimum quarterly) thereafter.
3.23 Relative Response Factor (RRF). The response of the mass spectrometer (MS) to a known amount of an analyte relative to a known amount of an isotopically labeled standard.
3.24 2,3,7,8-Tetrachlorodibenzo-p-dioxin Toxic Equivalency Quotient (2,3,7,8-TeCDD TEQ). A procedure that expresses the toxicity of PCDD, PCDF, and PCB in terms of the most toxic dioxin, as specified in applicable regulations, permits, or other requirements.
4.0 Interferences
Despite interferences, confidence of the data is based on the enhanced selectivity of fractionation, gas chromatograph (GC) separation and detector resolving power, the QC check ions, and monitoring PCDPE.
4.1 PCB and PCDPE have similar molecular weight and chromatographic properties to PCDD and PCDF. PCB may produce fragment ions at interfering mass-to-charge ratios (m/z) when losing chlorine (Cl 2 ) or 2 Cl 2 during ionization processes. With HRMS, GC separation, and fractionation, PCB should not pose a problem for PCDD/PCDF identification and quantitation. PCDPE, when losing Cl 2, also produce interfering m/z values in the PCDF homolog group with two fewer chlorine atoms ( i.e., an octachlorinated PCDPE can interfere with a hexachlorinated PCDF). The latter interferences are potentially detected by monitoring an m/z corresponding to the potentially interfering PCDPE; however, the fragmentation patterns of all PCDPE may not be known, complicating any attempt to quantify the extent of ether interference.
Note: Consider monitoring 328 m/z if high levels of PCB are expected.
4.2 Very high amounts of other organic compounds in the matrix may interfere with the analysis. This method provides examples of column-chromatographic cleanup as procedures to reduce, but not necessarily eliminate, matrix effects due to high concentrations of organic compounds (International Agency for Research on Cancer 1991).
4.3 Target compound contaminants or related organics in solvents, reagents, glassware, isotopically labeled spiking standards, and other sample processing hardware are potential method interferences. Routinely evaluate all these materials to demonstrate that they are either free from interferences under the conditions of the analysis, or that the interference does not compromise the quality of the analysis results. Evaluate chemical interference through the preparation and analysis of an LMB. Use high purity reagents, solvents, and standards to minimize interferences in sample analysis.
4.4 PAH are subject to degradation when exposed to ultraviolet light. Take precautions to shield samples from sunlight or fluorescent light sources during sample collection, recovery, extraction, cleanup, and concentration.
5.0 Safety
Note: Develop a strict laboratory safety program for the handling of PCDD, PCDF, PCB, and/or PAH.
5.1 Compounds in the PCDD and PCDF classes such as 2,3,7,8-TeCDD are aneugenic, carcinogenic, and teratogenic in laboratory animal studies. Other PCDD and PCDF containing chlorine atoms in positions 2,3,7,8 have toxicities comparable to that of 2,3,7,8-TeCDD.
5.2 PCB and benzo[a]pyrene are classified as known or suspected human or mammalian carcinogens. Be aware of the potential for inhalation and ingestion exposure to laboratory analysts.
5.3 This method recommends that the laboratory purchase dilute standard solutions of the analytes required for this method. However, if preparing primary solutions, use a hood or glove box. Laboratory personnel handling primary solutions should wear personal protective equipment including a toxic gas respirator mask fitted with charcoal filters approved by the National Institute for Occupational Safety and Health (NIOSH)/Mine Safety Health Administration (MSHA) to prevent the inhalation of airborne particulates if not working in an approved hood or glove box.
5.4 The toxicity or carcinogenicity of other reagents or chemicals used in this method is not precisely defined. However, treat each chemical as a potential health hazard and minimize exposure to these chemicals. The laboratory is responsible for maintaining a current awareness file of Occupational Safety and Health Administration (OSHA) regulations regarding the safe handling of the chemicals specified in this method. Ensure that a reference file or list of internet sites that contain safety data sheets (SDS) is available to all personnel involved in the sampling and chemical analysis of samples known or suspected to contain PCDD, PCDF, PCB, and PAH.
6.0 Equipment and Supplies
Note: Brand names, suppliers, and part numbers are for illustration purposes only and no endorsement is implied. Apparatus and materials other than those specified in this method may achieve equivalent performance. Meeting the performance requirements of this method is the responsibility of the source testing team and laboratory team.
6.1 Sampling Apparatus. Figure 23-1 of this method shows a schematic of the Method 23 sampling train. Do not use sealing greases or brominated flame retardant-coated tape in assembling the train. Do not use silicon tubing in direct contact with flue gases. The train is identical to that described in Section 6.1.1 of Method 5 of Appendix A-3 to 40 CFR part 60 with the following additions:
6.1.1 Nozzle. The nozzle must be made of quartz, borosilicate glass, or titanium. Stainless steel nozzles should not be used.
6.1.2 Probe Liner. Use either polytetrafluoroethylene (PTFE), borosilicate, or quartz glass probe liners with a heating system capable of maintaining a probe gas temperature of 120 ± 14 °C (248 ± 25 °F) during sampling, or such other temperature as specified by an applicable subpart of the standards or as approved by the Administrator. Use a PTFE ferrule or single-use PTFE coated O-ring to achieve the seal at the nozzle end of the probe for stack temperatures up to about 300 °C (572 °F). Use a quartz glass liner and integrated quartz nozzle for stack temperatures between 300 and 1,200 °C (572 and 2,192 °F).
6.1.3 Filter Holder. Use a filter holder of borosilicate glass with a PTFE frit or PTFE-coated wire filter support. The holder design should provide a positive seal against leakage from the outside or around the filter. The holder should be durable, easy to load, leak-free in normal applications, and positioned immediately following the probe and cyclone bypass (or cyclone, if used) with the active side of the filter perpendicular to the source of the flow.
6.1.4 Filter Heating System. Use any heating system capable of monitoring and maintaining the temperature around the filter to ensure that the sample gas temperature exiting the filter is 120 ± 14 °C (248 ± 25 °F) during sampling or such other temperature as specified by an applicable subpart of the standards or approved by the Administrator for a particular application.
6.1.5 Filter Temperature Sensor. Install a temperature sensor capable of measuring temperature to within ±3 °C (5.4 °F) so that the sensing tip protrudes at least 1.3 centimeters (cm) (1-2 in.) into the sample gas exiting the filter. Encase the sensing tip of the sensor in glass or PTFE, if needed.
6.1.6 Sample Transfer Line. The sample transfer line transports gaseous emissions from the heated filter holder to the condenser and must be heat traced and constructed of glass or PTFE with connecting fittings that form leak-free, vacuum-tight connections without using sealing greases or tapes. Keep the sample transfer lines as short as possible and maintain the lines at a temperature of 120 °C ± 14 °C (248 °F ± 25 °F) using active heating when necessary. Orient the sample transfer lines with the downstream end lower than the upstream end so that any condensate will flow away from the filter and into the condenser.
6.1.7 Condenser. Glass, water-jacketed, coil-type with compatible fittings. Orient the condenser to cause moisture to flow down to the adsorbent module to facilitate condensate drainage. Figure 23-2 of this method shows a schematic diagram of the condenser.
6.1.8 Water Circulating Bath. Use a bath pump circulating system capable of providing chilled water flow to the condenser and adsorbent module water jackets. Typically, a submersible pump is placed in the impinger ice water bath to circulate the ice water contained in the bath. Verify the function of this system by measuring the gas temperature at the entrance to the adsorbent module. Maintain this temperature at <20 °C (68 °F).
6.1.9 Adsorbent Module. Use a water-jacketed glass container to hold up to 40 grams (g) of the solid adsorbent. Figure 23-2 of this method shows a schematic diagram of the adsorbent module. Other physical configurations of the adsorbent resin module/condenser assembly are acceptable if the configuration contains the requisite amount of solid adsorbent and maintains the minimum length-to-width adsorbent bed ratio of two-to-one. Orient the adsorbent module vertically to facilitate condensate drainage. The connecting fittings must form leak-free, vacuum-tight seals. Include a coarse glass frit in the adsorbent module to retain the adsorbent.
6.1.10 Impingers. Use five impingers connected in series with leak-free ground glass fittings or any similar leak-free noncontaminating fittings. The first impinger must be a short-stem (water-dropout) design or equivalent. The second, fourth, and fifth impingers must be of the Greenburg-Smith design, modified by replacing the tip with a 1.3 cm ( 1/2 in.) inside diameter (ID) glass tube extending to approximately 1.3 cm ( 1/2 in.) from the bottom of the flask. The third impinger must be of the Greenburg-Smith design with the standard tip. The second and third impingers must contain known quantities of water, and the fifth impinger must contain a known weight of silica gel or equivalent desiccant. Alternatively, you may omit the first impinger if you do not expect excess moisture in the sample gas.
6.2 Sample Recovery Equipment.
6.2.1 Fitting Caps. Use leak-free ground glass fittings or any similar leak-free non-contaminating fitting to cap the sections of the sampling train exposed to the sample gas. Alternatively, use PTFE tape or contaminant-free aluminum foil for this purpose (see Section 6.2.6 of this method).
6.2.2 Wash Bottles. Use PTFE bottles.
6.2.3 Probe-Liner, Probe-Nozzle, and Filter-Holder Brushes. Use inert bristle brushes with precleaned stainless steel or PTFE handles. Extensions of the probe brush must be made of stainless steel or PTFE and be at least as long as the probe. Use brushes that are properly sized and shaped to remove accumulated material from the nozzle and probe liner if used.
6.2.4 Filter Storage Container. Use a sealed filter holder, wide-mouth amber glass jar with PTFE-lined cap, or glass petri dish sealed with PTFE tape. Purchase precleaned amber glass jars and petri dishes, or clean according to the glassware cleaning procedures listed in Section 8.1.1.1 of this method.
6.2.5 Field Balance. Use a weighing device capable of measurements to an accuracy of 0.5 g.
6.2.6 Aluminum Foil. Use heavy duty aluminum foil cleaned by rinsing three times with hexane or toluene and stored in a pre-cleaned glass petri dish or glass jar. Do not use aluminum foil to wrap or contact filter samples due to the possibility of reaction between the sample and the aluminum.
6.2.7 Silica Adsorbent Storage Container. Use an air-tight container to store silica gel.
6.2.8 Glass Sample Storage Container. Recover samples in amber glass bottles, 500- or 1000-milliliters (mL) with leak-free PTFE-lined caps. Either purchase precleaned bottles or clean containers according to glassware cleaning procedures listed in Section 8.1.1.1 of this method.
6.3 Sample Extraction Equipment.
6.3.1 Sample Container. Use 125- and 250-mL amber glass bottles with PTFE-lined caps.
6.3.2 Test Tubes. Use glass test tubes or small ( e.g., 5 to 10 mL) amber vials.
6.3.3 Soxhlet/Dean-Stark Extraction Apparatus.
6.3.3.1 Soxhlet Apparatus. Use 200-mL capacity thimble holder capable of holding 43 × 123-millimeter (mm) extraction thimbles, with receiving flask (typically round-bottom).
6.3.3.2 Moisture Trap. Use Dean-Stark or Barret with fluoropolymer stopcock trap to fit between the Soxhlet extractor body and the condenser as shown in Figure 23-3 of this method.
Note: Dean-Stark or Barret traps are used to remove water with extraction solvents that are less dense and insoluble in water.
6.3.3.3 Extraction Thimble. Use quartz, glass, or glass fiber thimble, typically 43 × 123 mm to fit Soxhlet apparatus. The use of cellulose thimbles for sample extraction in this method is prohibited.
6.3.3.4 Heating Mantle. Use a hemispherical shaped heating mantle to fit round-bottom flask.
6.3.4 Kuderna-Danish (KD) Concentrator. Use an apparatus consisting of a three-ball Snyder column, a flask with leak-free joint to accept the three-ball Snyder column at the top, a leak-free joint to receive a graduated concentration tube at the bottom and a heating mantle.
Note: Rotary evaporation has only been demonstrated when analyzing PCDD/PCDF. The KD with Snyder column is recommended when analyzing for PAH and/or PCB to avoid evaporation loss resulting in failed performance criteria for pre-extraction spike recovery.
6.3.5 Nitrogen Evaporative Concentrator. Use a nitrogen evaporative concentrator equipped with a water bath with the temperature controlled in the range of 30 to 60 °C (86 to 140 °F) (N-Evap Organomation Associates, Inc., South Berlin, MA, or equivalent).
6.3.6 Separatory Funnels. Use glass or PTFE 2-liter separatory funnels.
6.4 Glass Liquid Chromatography Columns.
6.4.1 Pasteur Pipettes. Use disposable pipettes, or glass serological pipettes typically 150 mm long × 6 mm ID.
6.4.2 Liquid Chromatography Columns. 200 to 300 mm long × 20 mm ID with 250-mL reservoir.
6.5 Analytical Equipment.
6.5.1 Gas Chromatograph. Use a gas chromatograph consisting of the following components:
6.5.1.1 GC Oven. Use an oven capable of maintaining the separation column at the proper operating temperature ± 1.0 °C (1.8 °F) and performing programmed increases in temperature at rates of at least 40 °C/min with isothermal hold.
6.5.1.2 GC Temperature Monitor. Use a temperature monitor to measure column oven temperature to ± 1.0 °C (1.8 °F).
6.5.1.3 GC Flow System. Use an electronic pressure control or equivalent gas metering system to control carrier gas flow or pressure.
6.5.1.4 GC Injection Port. Use a split/splitless injection port in the splitless mode or on-column injection port for the capillary column.
6.5.2 Capillary GC Column. Use different columns for the analysis of the different target compound classes in this method, if needed. Perform the resolution checks in Sections 10.2.3.5 and 10.2.3.6 of this method to document the required resolution. Compound separation must meet the resolution specifications in Section 10.2.3.5 of this method and the identification specifications found in Section 11.4.3.4 of this method.
6.5.2.1 PCDD/PCDF Column. Gas chromatographic columns used to measure PCDD/PCDF should be capable of achieving separation of the 17 PCDD/PCDF target compounds from the nearest eluting target compound(s). The valley height resolution between 2,3,7,8-substituted TeCDD and TeCDF and the nearest eluting isomers must not exceed 25% of the taller of the two peaks. The valley height resolution between all other target PCDD/PCDF compounds and the nearest eluting targets (or interference) must not exceed 40% of the taller of the two peaks.
Note: Fishman, et al. (see Section 16.3 of this method) demonstrated that all TEF isomers can be fully differentiated from closely eluting isomers using either of two sets of non-polar and polar stationary phase combinations. One set consisted of 5% phenyl methylpolysiloxane (DB-5, HP-5MS, Rtx-5MS, Equity-5) and 50% cyanopropylmethyl, 50% phenylmethylsiloxane (DB-225, SP 2331) GC columns and the other set consisted of 5% phenyl, 94% methyl, 1% vinyl silicone bonded-phase (DB-5MS, ZB-5MS, VF-5MS, CP-Sil 8 CB LowBleed/MS) with 50% cyanopropylmethyl, 50% phenylmethylsiloxane (SP-2331).
6.5.2.2 PAH Column. Use column systems for measuring PAH that can achieve separation of anthracene and phenanthrene at m/z 178 such that the valley between the peaks does not exceed 50% of the taller of the two peaks, and benzo[ b ]fluoranthene and benzo[ k ]fluoranthene such that the valley between the peaks is less than 60% of the height of the taller peak. These requirements are achievable using a 30-m narrow bore (0.25 mm ID) 5% phenyl polysilphenylene-siloxane (BPX5 or equivalent) bonded-phase, fused-silica capillary column.
6.5.2.3 PCB Column. Use column systems for measuring PCB that can achieve unique resolution and identification of the toxics for determination of a TEQ PCB using toxic equivalency factors (TEF). Resolution is shown by a valley between the peaks not exceeding 40% of the taller of the two peaks. Isomers may be unresolved if they have the same TEF and RRF and if these unresolved isomers are uniquely resolved from all other congeners. These requirements are achievable using several 30-meter (m) narrow bore (0.25 mm ID) columns including 8% phenyl polycarborane-siloxane (HT8), DB-XLB, and poly (50% n-octyl/50% methyl siloxane) (SPB-Octyl). Quantification of unresolved isomers should use the nearest eluting target PCB pre-extraction standard in Appendix A of this method, unless otherwise specified in applicable rule, regulation, or permit.
Note: If all 209 PCB are analyzed the 17 toxic PCB congeners should be resolved and reported while the other PCB can be reported as totals by homolog, for example, total TrCB.
6.5.3 Mass Spectrometer. Instrument employing 28 to 70 electron volt ionization. The instrument and data system must be capable of repetitive monitoring of at least 12 exact m/z values with a mass resolution defined in Section 10.2.1 within the measurement mass range. The recommended lock-mass ions to be used for mass drift correction are presented in Tables 23-4, 23-5, and 23-6 of this method for PCDD/PCDF, PAH, and PCB, respectively, as applicable to target analytes. Mass drifts of 5 parts per million (ppm) or more can have serious effects on instrument performance.
6.5.4 Mass Spectrometer Data System. Use a data system compatible with the mass spectrometer and capable of sequencing and monitoring multiple groups of selected ions.
6.5.5 Analytical Balance. Use an analytical balance to measure within 0.1 milligram (mg).
7.0 Reagents, Media, and Standards
7.1 Filter. Glass fiber filters, without organic binder, exhibiting at least 99.95% efficiency (<0.05% penetration) on 0.3-micron dioctyl phthalate smoke particles.
7.1.1 Conduct a QC check on the filter lot prior to the field test to demonstrate that filters are free from contamination or interference by extracting and analyzing a minimum of three filters from each lot as follows. Spike with pre-extraction and pre-extraction filter recovery standards for target compounds to be measured and extract each filter separately with toluene as described in Section 11 of this method. After extraction, remove the filters and the solvent from the filters under clean conditions ( e.g., a clean nitrogen stream). Analyze the extracts according to the procedures in Section 11 of this method, including adding pre-analysis standard. This filter check analysis must meet the performance requirements in Section 13.1 of this method. Ongoing analysis of LMB can be used to fulfill this check. If criteria are not met for target compounds, repeat with additional filters from the lot or evaluate another lot.
7.2 Adsorbent Resin. Amberlite® XAD-2 resin. All adsorbent resin must meet the cleanliness criteria described for LMB in Section 13.1 of this method following the same extraction, concentration, cleanup, and analysis steps as field samples. This method recommends using the procedures provided in Appendix B to this method to clean the resin before use, if needed. However, this method allows alternative cleanup procedures that use automated extraction equipment if the adsorbent meets the required performance criteria described for LMB in Section 13.1 of this method.
7.2.1 Conduct a QC check on the cleaned adsorbent lot or batch following the extraction and analyses procedures in Section 11 of this method, including adding applicable labeled standards. The cleaned adsorbent must meet the criteria described for LMB in Section 13.1 of this method. An LMB conducted with an adsorbent lot or batch can serve this purpose.
7.2.2 Storage. Store adsorbent in a solvent-rinsed nonporous clean container and secure lid.
7.3 Glass Wool. Clean the glass wool to meet the specifications in Section 13.1 of this method. Glass wool is dried of the solvent and stored in a clean glass container with a PTFE-lined screw cap.
7.4 Water. Use deionized or distilled water meeting requirements in Section 13.1 of this method and store in its original container or in a clean glass container with a PTFE-lined screw cap.
7.5 Silica Gel. Indicating type for sampling, 6-16 mesh. If previously used, dry at 175 °C (347 °F) for two hours. Use new silica gel as received. As an alternative, use other types of desiccants (equivalent or better), subject to the approval of the Administrator.
7.6 Methylene Chloride. Pesticide grade or better.
7.7 Sample Recovery Reagents.
7.7.1 Acetone. Pesticide grade or better.
7.7.2 Toluene. Pesticide grade or better.
7.8 Sample Extraction and Cleanup.
7.8.1 Potassium Hydroxide. American Chemical Society (ACS) grade, 2% (weight/volume) in water.
7.8.2 Sodium Sulfate. Granulated or powdered, reagent grade. Evaluate for cleanliness prior to use with an LMB. The LMB must meet the requirements in Section 13.1 of this method for target compounds. Store in a clean glass container with a PTFE-lined screw cap.
7.8.3 Sulfuric Acid. Reagent grade.
7.8.4 Sodium Hydroxide. 1.0 N. Weigh 40 g of sodium hydroxide into a 1-liter volumetric flask. Dilute to 1 liter with water.
7.8.5 Hexane. Pesticide grade or better.
7.8.6 Methanol. Pesticide grade or better.
7.8.7 Toluene. Pesticide grade or better.
7.8.8 High-Boiling Alkanes Used as Keeper Solvents ( e.g., tetradecane, nonane, decane). Pesticide grade. Note: Lower homologous series alkanes (nonane or decane) are necessary for higher volatility targets such as MoCB and naphthalene to maintain retention during concentration procedures. However, do not take samples to dryness when using these lower alkane homologs.
7.8.9 Liquid Column Chromatography Packing Materials. Use the following column chromatography packing materials, as needed, to prepare sample extracts by fractionation and removal of interferences. Commercially prepacked cleaning columns may be available for this purpose. The liquid column chromatography packing materials must be adequate to clean the samples to be fit for purpose and meet the performance criteria of this method. All procedures for preparing column chromatography packing materials are recommendations shown to meet the performance specifications required for the recovery of labeled compounds described in Section 13 of this method.
7.8.9.1 Alumina. Use either acidic or basic alumina in the cleanup of sample extracts. Use the same type of alumina for all samples in an analytical sequence, including those used to demonstrate LMB performance.
7.8.9.1.1 Acidic Alumina (Sigma-Aldrich® 199966 or equivalent). Brockmann activity grade 1, 100-200 mesh. Prior to use, activate the alumina by heating for 12 hours at 130 °C (266 °F). Store in a desiccator. You may use pre-activated alumina purchased from a supplier as received.
7.8.9.1.2 Basic Alumina (Sigma-Aldrich® 19943 or equivalent). Brockmann activity grade 1. Activate by heating to 600 °C (1,112 °F) for a minimum of 24 hours. Do not heat to over 700 °C (1,292 °F) because this can lead to reduced capacity for retaining the target compounds. Store at 130 °C (266 °F) in a covered flask. Recommended storage time for acidic alumina is up to five days from baking. Use prepacked alumina columns immediately after opening the vacuum-sealed pouch or container.
7.8.9.2 Florisil®. Activated, 60-100 mesh recommended. Heat previously activated Florisil® in a glass container loosely covered with aluminum foil in an oven at 130 to 150 °C (266 to 302 °F) for a minimum of 24 hours. Allow to cool and store activated Florisil® silica in a desiccator.
7.8.9.3 Silica Gel. Use either activated, acid- or base-coated silica gel in the cleanup of sample extracts. Use the same type of silica gel for all samples in an analytical sequence, including those used to demonstrate LMB performance.
7.8.9.3.1 Activated Silica Gel. Supelco® 1-3651, Bio-Sil® A, 100-200 mesh (or equivalent). Prior to use, silica gel should be activated by solvent rinsing and heat activation. It is recommended to rinse with methylene chloride and activate the silica gel by heating for at least 1 hour at 180 °C (356 °F). After allowing to cool, rinse the silica gel sequentially with methanol and toluene. Heat the rinsed silica gel at 50 °C (122 °F) for 10 minutes, then increase the temperature gradually to 180 °C (356 °F) over 25 minutes and maintain the gel at this temperature for 90 minutes. Allow to cool in a desiccator to room temperature and store in a glass container with a PTFE-lined screw cap. Alternative conditioning procedure may be used if the performance criteria in Section 13.1 are met for target compounds.
7.8.9.3.2 Acidic Silica Gel (30% weight/weight). Combine 100 g of activated silica gel with 44 g of concentrated sulfuric acid in a clean screw-capped glass container and agitate thoroughly. Disperse the solids with a stirring rod until obtaining a uniform mixture of acid-coated silica gel. Store the mixture in a glass container with a PTFE-lined screw cap.
7.8.9.3.3 Basic Silica Gel. Combine 30 g of 1 N sodium hydroxide with 100 g of activated silica gel in a clean screw-capped glass container and agitate thoroughly. Disperse solids with a stirring rod until obtaining a uniform mixture of base-coated silica gel. Store the mixture in glass container with a PTFE-lined screw cap.
7.8.9.4 Carbon/Celite® 545 (or equivalent solid support). Use of a carbon-based column cleanup material ( e.g., one of the many including for example Carbopack® B or C) to further remove non-planar impurities from the samples prior to analysis may be necessary. You must evaluate alternative carbon-based sorbents for this purpose prior to their use. An 18% weight/weight mixture of Carbopack® C and Celite® 545 has been used for this purpose and should be activated at 130 °C (266 °F) for a minimum of 6 hours. Allow to cool and store this mixture in a desiccator.
7.8.10 Nitrogen. 99.999% (ultra-high) purity.
7.9 Sample Analysis.
7.9.1 Helium. 99.999% (ultra-high) purity.
7.9.2 Spiking Standards. Prepare spiking standards quantitatively at a convenient concentration ( e.g., 10 nanograms (ng)/mL) or use commercial standards if available, to enable accurate spiking of a labeled standard at various stages of the sample and extract preparation. You may adjust the sample fortification concentrations from those recommended in Tables 23-7, 23-8, and 23-9 of this method to accommodate the concentration of target compounds anticipated in samples if the performance criteria in Section 13 of this method are met.
Note: When adjusting the fortification concentrations in the final sample extract, consider variables such as the aliquot of extract used and injection volume of samples and calibration.
7.9.3 Pre-Sampling Adsorbent Standard. Prepare stock standard solutions in nonane to enable spiking so that the isotopically labeled compounds in the final sample extract are at the concentration shown under the heading “Pre-sampling Adsorbent Standard” in Tables 23-7, 23-8, and 23-9 of this method, for applicable target compound classes.
7.9.4 Pre-extraction Filter Recovery Standard. Prepare stock standard solutions in nonane to enable spiking so that the isotopically labeled compounds in the final sample extract are at the concentration shown under the heading “Pre-extraction Filter Recovery Standard” in Tables 23-7, 23-8, and 23-9 of this method, for applicable target compound classes.
7.9.5 Pre-extraction Standard. Prepare stock standard solutions in nonane to enable spiking so that the isotopically labeled compounds in the final sample extract are at the concentration shown under the heading “Pre-extraction Standard” in Tables 23-7, 23-8, and 23-9 of this method, for applicable target compound classes.
7.9.6 Pre-analysis Standard. Prepare stock standard solutions in nonane to enable spiking so that the isotopically labeled compounds in the final sample extract are at the concentration shown under the heading “Pre-analysis Standard” in Tables 23-7, 23-8, and 23-9 of this method, for applicable target compound classes.
8.0 Sample Collection, Preservation, and Storage
8.1 Sampling. This method involves collection and recovery of trace concentrations of target semivolatile organic compounds. Therefore, field sampling and recovery staff should be trained and experienced in the best practices for handling and using organic solvents in field environments to recover and protect samples from contamination.
8.1.1 Pretest Preparation.
8.1.1.1 Cleaning Glassware. Clean glassware thoroughly before using. This section provides a recommended procedure, but any protocol that consistently results in contamination-free glassware meeting the LMB criteria in Section 13.1 of this method is acceptable.
8.1.1.1.1 Soak all glassware in hot soapy water (Alconox® or equivalent).
8.1.1.1.2 Rinse with hot tap water.
8.1.1.1.3 Rinse with deionized/distilled water.
8.1.1.1.4 Rinse with methanol.
8.1.1.1.5 Rinse with toluene.
8.1.1.1.6 Baking glassware up to 400 °C (752 °F) for a minimum of 2 hours may be necessary to remove contaminants or interferents from particularly dirty samples. Allow glassware to cool after baking.
Note: Repeated baking of glassware may cause active sites on the glass surface that may irreversibly adsorb target compounds.
8.1.1.1.7 Cover glassware openings with clean glass fitting caps or cleaned aluminum foil (see Section 6.2.6 of this method).
8.1.1.1.8 Rinse glassware immediately before use with acetone and toluene.
Note: To prepare heavily soiled glassware, remove surface residuals from the glassware by soaking in hot soapy water, rinsing with hot water, then soaking with a non-chromic acid oxidizing cleaning reagent in a strong acid ( e.g., NOCHROMIX® prepared according to manufacturer's directions). After the acid soak, rinse with hot water and repeat the cleaning procedures in Section 8.1.1.1 of this method.
8.1.1.2 Adsorbent Module. Load the modules in a clean area to avoid contamination. Fill a module with 20 to 40 g of XAD-2. Spike modules before the sampling event, but do not spike the modules in the field. Add the pre-sampling adsorbent standard to the top quarter of the adsorbent bed rather than onto the top or bottom of the adsorbent bed. Add sufficient spike (picograms (pg)/module) to result in the final sample theoretical concentrations specified in Tables 23-7, 23-8, and 23-9 of this method for PCDD/PCDF, PAH, and PCB, respectively, and to be above the lowest calibration concentration to ensure the standard recovery is quantitative. For samples with known or anticipated target compound concentration significantly higher or lower than the specified concentration in these tables, adjust the pre-sampling adsorbent standard concentration to the expected native compound concentration, but no less than 10 times the method detection limit (MDL). Follow the XAD-2 with cleaned glass wool and tightly cap both ends of the module. For analysis that includes PAH, use spiked modules within 14 days of preparation. See Table 23-10 of this method for storage conditions.
8.1.1.3 Sampling Train. Figure 23-1 of this method shows the complete sampling train. Follow the best practices by maintaining all sampling train components according to the procedure described in APTD-0576 Maintenance, Calibration, and Operation of Isokinetic Source-sampling Equipment (U.S. EPA 1972).
8.1.1.4 Silica Gel. Weigh several 200 to 300 g portions of silica gel in an air-tight container to the nearest 0.5 g. Record the total weight of the silica gel plus container, on the outside of each container. As an alternative, directly weigh the silica gel in its impinger or sampling holder just prior to sampling.
8.1.1.5 Filter. Check each filter against light for irregularities and flaws or pinhole leaks. Pack the filters flat in a clean glass container. Do not mark filters with ink or any other contaminating substance.
8.1.2 Preliminary Determinations. Use the procedures specified in Section 8.2 of Method 5 of Appendix A-3 to 40 CFR part 60.
8.1.2.1 Sample Volume. Unless otherwise specified in an applicable rule, regulation, or permit, sample for a minimum of 2 minutes at each traverse point. This method recommends sampling a minimum of 2.5 dry standard cubic meters (dscm).
8.1.2.2 For continuously operating processes, use the same sampling time at each traverse point. To avoid timekeeping errors, use an integer, or an integer plus one-half minute, for each traverse point.
8.1.2.3 For batch processes, determine the minimum operating cycle duration, dividing the sampling time evenly between the required numbers of traverse points. After sampling all traverse points once, sample each point again for the same duration of time per sampling point in reverse order until the operating cycle is completed. Sample all traverse points at least once during each test run.
8.1.3 Preparation of Sampling Train.
8.1.3.1 During field preparation and assembly of the sampling train, keep all train openings where contamination can enter sealed until just prior to assembly or until sampling is about to begin. To protect the adsorbent module from radiant heat and sunlight, you must wrap the module with aluminum foil or other suitable material capable of shielding the module from light. The XAD-2 adsorbent resin temperature must never exceed 50 °C (122 °F) because thermal decomposition will occur. Clean and prepare a complete set of sampling train components that will contact the sample for each sampling run, including one complete set to be used as a field train proof blank as a tool to evaluate equipment preparation and potential contamination during sample recovery as described in Section 9.6 of this method.
8.1.3.2 Place approximately 100 mL of water in the second and third impingers but leave the first and fourth impingers empty. Transfer approximately 200 g or more of silica gel from its container to the fifth impinger. Weigh each impinger and the adsorbent module, including the fitting caps, to the nearest 0.5 g using the field balance and record the weight for moisture determination. Remove the aluminum foil from the adsorbent module before weighing. Keep the module out of direct sunlight and rewrap the module with foil immediately after recording the module weight.
8.1.3.3 Using tweezers or clean disposable surgical gloves, place a filter in the filter holder. Be sure that the filter is properly centered, and the gasket properly placed, to prevent the sample gas stream from circumventing the filter. Check the filter for tears after completing the assembly.
8.1.3.4 Prepare the inside of the sampling probe and nozzle by brushing each component while rinsing three times each with acetone and toluene. Install the selected nozzle, using the connecting systems described in Section 6.1.2 of this method. Mark the probe with heat resistant tape or by some other method to denote the proper distance into the stack or duct for each sampling point. Assemble the train as shown in Figure 23-1 of this method. Orient the adsorbent module vertically so condensed moisture drains into the first impinger. See APTD-0576 Maintenance, Calibration, and Operation of Isokinetic Source-sampling Equipment (U.S. EPA 1972) for details.
8.1.3.5 Turn on the recirculation pump to the adsorbent module and condenser coil and begin monitoring the temperature of the gas entering the adsorbent module. Ensure proper temperature of the gas entering the adsorbent module before proceeding.
8.1.4 Leak-Check Procedure. Same as Section 8.4 of Method 5 of Appendix A-3 to 40 CFR part 60.
8.1.5 Sampling Train Operation. Same as Sections 8.5.1 through 8.5.9 of Method 5 of Appendix A-3 to 40 CFR part 60.
8.1.5.1 Monitor the filter temperature with a sensor and record the filter temperature during sampling to ensure a sample gas temperature exiting the filter of 120 °C ± 14 °C (248 °F ± 25 °F), or such other temperature as specified by an applicable subpart of the standards or approved by the Administrator for an application of this method.
8.1.5.2 During testing, you must record the temperature of the gas entering the XAD-2 adsorbent module. The temperature of the gas must not exceed 20 °C (68 °F) for efficient capture of the target compounds.
8.2 Sample Recovery. Begin the cleanup procedure as soon as the probe is removed from the stack at the end of the sampling period. Seal the nozzle end of the sampling probe with PTFE tape or clean ( e.g., toluene rinsed) aluminum foil.
8.2.1 When the probe can be safely handled, wipe off all external particulate matter near the tip of the probe. Conduct a post-test leak check. Remove the probe from the train and close off both ends with PTFE tape or clean aluminum foil. Seal off the inlet to the train with PTFE tape, a ground glass cap, or clean aluminum foil.
8.2.2 Transfer the probe and impinger assembly to the cleanup area. This method recommends cleaning and enclosing this area to minimize the chances of losing or contaminating the sample. To avoid sample contamination and unnecessary exposure to toxic chemicals, smoking or eating in the sample recovery area shall not be allowed.
8.2.3 Inspect the train prior to and during disassembly. Note and record any abnormal conditions ( e.g., broken filters, colored impinger liquid). Recover and prepare samples for shipping as follows in Sections 8.2.4 through 8.2.12 of this method.
8.2.4 Container No. 1. Either seal the filter holder or carefully remove the filter from the filter holder and place it in its identified container. If it is necessary to remove the filter, use a pair of cleaned tweezers to handle the filter. If necessary, fold the filter such that the particulate cake is inside the fold. Carefully transfer to the container any particulate matter and filter fibers that adhere to the filter holder gasket by using a dry inert bristle brush and a sharp-edged blade. Seal the container and store cool (≤20 °C, 68 °F) for transport to the laboratory.
8.2.5 Adsorbent Module Sample. Remove the module from the train, tightly cover both ends with fitting caps and PTFE tape, remove the foil, drain the recirculating water from the module, weigh and record the module weight, and label the adsorbent module. Moisture measurement in the field using the Method 23 train requires weighing the adsorbent module before sampling and after sampling as part of the sample recovery.
8.2.6 Container No. 2. Quantitatively recover material deposited in the nozzle, the front half of the filter holder, and the cyclone, if used, by brushing while rinsing three times with acetone followed by three rinses with toluene. Collect all the rinses in Container No. 2.
8.2.7 Rinse the back half of the filter holder three times with acetone followed by three rinses with toluene. Rinse the sample transfer line between the filter and the condenser three times with acetone followed by three rinses with toluene. If using a separate condenser and adsorbent module, rinse the condenser three times with acetone followed by three rinses with toluene. Collect all the rinses in Container No. 2 and mark the level of the liquid on the container.
8.2.8 Moisture Weight. Weigh the adsorbent module, impingers, and silica gel impinger to within ±0.5 g using the field balance and record the weights. This information is required to calculate the moisture content of the effluent gas. For PCDD/PCDF-only measurements, discard the liquid after measuring and recording the weight.
8.2.9 Container No. 3. You must save and analyze impinger water samples if PAH and/or PCB are the target compounds. Quantitatively recover impinger water samples for analysis if PAH and/or PCB are the target compounds by rinsing three times with acetone followed by three rinses with toluene. Collect impinger water and rinses in Container No. 3 and mark the level of the liquid on the container.
8.2.10 Silica Gel. Note the color of the indicating silica gel to determine if it has been completely spent and report its condition on the field data sheet.
8.2.11 Field Sample Handling, Preservation, Storage, and Transport. Store all field samples temporarily in cool (≤20 °C, 68 °F) and dark conditions prior to transport to the laboratory. Ship samples cool (≤20 °C, 68 °F), shielded from ultraviolet light. In addition, follow the procedures in American Society for Testing and Materials (ASTM) D6911-15 (Guide for Packaging and Shipping Environmental Samples for Laboratory Analysis) for all samples, where appropriate. To avoid contamination of the samples, pay special attention to cleanliness during transport, field handling, sampling, recovery, and laboratory analysis, as well as during preparation of the adsorbent cartridges.
8.2.12 Sample Custody. Proper procedures and documentation for sample chain of custody are critical to ensuring data integrity. Follow the chain of custody procedures in ASTM D4840-99(2018)e1 (Standard Guide for Sample Chain-of-Custody Procedures) for all samples (including field samples and blanks).
8.3 Sample Storage Conditions and Laboratory Hold Times.
8.3.1 Table 23-10 of this method summarizes the sample storage conditions and laboratory hold times.
8.3.2 Store sampling train rinses and filter samples in the dark at the storage conditions in Table 23-10 from the time the laboratory receives the samples until analysis.
8.3.3 You may store adsorbent samples for PCDD/PCDF or PCB analysis prior to extraction in the dark at 6 °C (43 °F) or less for up to one year from the time the laboratory receives the samples.
Note: The hold times listed in this method for adsorbent samples for PCDD/PCDF and PCB are recommendations as these compounds are very stable under the conditions listed in this section.
8.3.4 Protect adsorbent samples destined for PAH analysis from ultraviolet light. You may store adsorbent samples for PAH analysis in the dark at 6 °C (43 °F) or less for up to 30 days from the time the laboratory receives the samples.
8.3.5 Analyze PAH extracts within 40 days of extraction.
8.3.6 You may store sample aliquots including archived extracts of PCDD/PCDF, PAH and/or PCB samples in the dark at −10 °C (14 °F) or less for up to one year. Sample extracts must not be stored with pierced septa.
Note: The hold times listed in this method for sample aliquots for PCDD/PCDF and PCB are recommendations as these compounds are very stable under the conditions listed in this section.
9.0 Quality Control
Note: In recognition of advances that are occurring in sampling and analytical technology, and to allow the test team to overcome analyte sensitivity and matrix interferences, this method allows certain options to increase sample collection volume and to improve separations and the quality of the analysis results for target analytes. It is the laboratory's responsibility to establish the conditions for optimum sample extraction, cleanup, and concentration to meet the performance criteria in this method. However, you may not change the fundamental sampling and analysis techniques, isokinetic sampling with an adsorbent collection media followed by sample extraction, and HRMS detection and isotopic dilution quantification procedures. Section 13 of this method specifies the performance criteria to ensure that options employed for a sample set and analytes of interest are equal to or better than the specificity of the techniques in this method. The minimum requirements of this method consist of the initial demonstration of capability (IDC) and ongoing QC requirements. The analysis team shall perform an IDC to demonstrate acceptable accuracy and precision with this method as described in Section 9.3. The ongoing QC includes performing CCVs and LMBs to evaluate an individual laboratory's performance against the criteria in this method. The method includes analysis of samples spiked with labeled compounds to evaluate and document data quality. Laboratory performance is compared to established performance criteria to determine if the results of analyses meet the performance characteristics and requirements of the method.
9.1 Record and report data and information that will allow an independent reviewer to validate the determination of each target compound concentration. Record and report the data as described in Sections 9.1.1 through 9.1.7 of this method and performance criteria results required in Section 13 of this method.
9.1.1 Sample numbers and other sample identifiers. Each sample must have a unique identifier.
9.1.2 Field sample volume.
9.1.3 Field sampling date.
9.1.4 Extraction dates.
9.1.5 Analysis dates and times.
9.1.6 Analysis sequence/run chronology.
9.1.7 Quantitation Reports.
9.1.7.1 This method does not consider EMPC-flagged data to be zero concentrations. Calculate and report the EMPC concentrations.
9.1.7.2 In determining compliance with any PCDD and PCDF standard developed using zero for values that are below the EDL of the method, including federal emission standards using Method 23 promulgated under 40 CFR parts 60 and 63 prior to March 20, 2023, use zero for the determination of total and weighted concentrations when the target compound is not detected. For all other circumstances, unless otherwise specified in applicable regulations, permits, or other requirements, when a target compound is measured at or below EDL, use EDL as the concentration for calculating compliance.
9.1.7.3 For each sample you must report EDLs, MDLs, LMBs and Field Train Proof Blank results and target compound analysis results.
9.2 Isotopically Labeled Standard Recovery.
9.2.1 Pre-sampling Adsorbent Standard and Pre-extraction Filter Recovery Standard Recoveries. Pre-sampling adsorbent standard and pre-extraction filter recovery standard recoveries must demonstrate on a per sample basis that recovery of the labeled standard achieved the requirements in Section 13 of this method. Recoveries below the acceptable range for the pre-sampling adsorbent standard may be an indication of breakthrough in the sampling train.
9.2.1.1 If the pre-sampling adsorbent standard average percent recovery is below 70%, the sampling run is not valid, and the stack test must be repeated. As an alternative, you do not have to repeat the stack test for invalid analyses if the pre-sampling adsorbent standard average percent recovery is 25% or more and you divide the final results by the fraction of the pre-sampling adsorbent standard average percent recovery.
9.2.1.2 If the percent recovery of all the pre-extraction filter recovery standard compounds is below 70%, you may reanalyze the sample. If the recovery is still below the limit, the filter sampling extraction is not valid, and you must repeat the stack or vent sampling and subsequent analysis.
9.2.2 Pre-extraction Standard Recoveries. Pre-extraction standard recoveries must demonstrate on a per sample basis that recovery of the labeled standard achieved the requirements in Section 13.15 of this method. If the recovery criteria are not met, you may reanalyze the sample. If the recovery criteria are still not met, the sampling run is not valid, and the stack test must be repeated. Recoveries outside the acceptable range for pre-extraction standard are an indication that sample preparation procedures did not adequately address sample and or sample matrix processing to recover native target compounds.
9.2.3 Pre-analysis Standard Response. Pre-analysis standard recoveries must demonstrate on a per sample basis that adequate labeled standard signal meets the requirements in Section 13.16 of this method. Add pre-analysis standard to every sample (including blanks, QC samples, and calibration solutions) in a known concentration. If the prepared samples do not meet the pre-analysis standard response criteria, you may reanalyze and/or prepare and analyze archive samples to attempt meeting requirements for the compounds that do not meet the pre-analysis standard response criteria. Poor sensitivity compared to initial calibration response may indicate injection errors or instrument drift.
9.3 Initial Demonstration of Capability (IDC). The IDC must be successfully performed prior to analyzing field samples by meeting the QC requirements in Table 23-18. The IDC must be repeated if changes are made to analytical parameters not previously validated during the IDC. This may include, for example, changing the sample volume, selecting alternate quantitation ions, extending the calibration range, adding additional pre-analysis standard, or adding additional pre-extraction standard. The same calibration range used during the IDC must be used for the analysis of field samples.
9.3.1 Perform initial calibration following the procedures in Section 10. The lowest calibration standard used to establish the initial calibration must not be less than three times the MDL. The initial calibration must meet performance criteria in Section 13.9.
9.3.2 Lowest Calibration Concentration Confirmation. Establish a target concentration for the lowest calibration standard based on the intended use of the method. The lowest calibration concentration may be established by a laboratory or programmatic lowest quantitative reporting requirement. The laboratory calibration curve must be set at or below this level. Perform seven replicate analyses of a calibration sample prepared at proposed lowest calibration concentration. The replicate analyses of the lowest calibration concentrations standards must meet the criteria in Sections 13.9 and 13.17.1.
Note: Consider that establishing the lowest calibration concentration too low may cause repeated failure of ongoing QC requirements.
9.3.3 Calculate Lowest Calibration Statistics. Calculate the mean and standard deviation for each analyte in these replicates (those used in Section 9.3.2). Determine the Half Range for the Prediction Interval of Results (HRPIR) using Equation 23-13. Calculate the Upper and Lower Limits for the Prediction Interval of Results (PIR) with Equations 23-14 and 23-15.
9.3.4 Lowest Calibration Point Acceptance Criteria. The laboratory's ability to measure analyte concentrations down to the lowest calibration point is confirmed if the criteria presented in Section 13.17.1 are met. If these criteria are not met, the lowest calibration point as been set too low and must be confirmed at a higher concentration.
9.3.5 Demonstration of Low System Background. Analyze an LMB after the highest standard in the calibration range. If an automated extraction system is used, an LMB must be extracted on each port. Performance criteria are presented in Section 13.1. Note: When using automated systems, the same systems must be used for samples and QC samples, such as blanks and resin checks.
9.3.6 Initial Calibration Verification. A QCS must be analyzed during the IDC, and then following each initial calibration thereafter (at a minimum quarterly). A QCS is a mid-level standard prepared from a second source standard or prepared from a source of standards different from the source of calibration standards. The purpose of the QCS is to verify the integrity of the primary calibration standards. The acceptance criterion is presented in Section 13.11.
9.3.7 MDL. Perform an MDL determination using a minimum of seven spiked combined filter/sorbent media samples, spiked within 2 to 10 times of the expected MDL, and seven LMBs (combined filter/sorbent media) through all the steps of the method following the requirements in 40 CFR part 136 Appendix B. Confirm target compounds meet the qualitative identification criteria in Sections 13.12 and 13.13. The criteria for the MDL determination are presented in Section 13.6.1 of this method.
9.3.8 MDL Confirmation. Confirm newly determined MDLs by preparing a low-level spiked combined filter/sorbent media sample by spiking the sorbent with native target compounds at 1 to 5 times the MDL and pre-extraction standard at the concentration used to analyze field samples and analyze. The criterion for the MDL confirmation is presented in Section 13.6.1 of this method.
9.3.9 Demonstration of Precision. Prepare, extract, and analyze seven replicate spiked samples in a valid Extraction Batch. Fortify the spiked samples near the midpoint of the initial calibration curve. The criterion is presented in Section 13.17.2 and Table 23-18. Demonstration is repeated for failed compounds only.
9.3.10 Demonstration of Accuracy. Using the same set of replicate data generated for Section 9.3.9 of this method, calculate the average % recovery. The criterion is presented in Section 13.17.3 and Table 23-18. Demonstration is repeated for failed compounds only.
9.4 LMBs. Evaluate background contamination from glassware, equipment, solvents, standards, and media used for sample batches using an LMB prepared and analyzed identically to the field samples, including the same labeled standards, media, sodium sulfate, glass wool, glassware, solvents, etc. An LMB must be extracted with every batch of samples. Analyze an LMB at least once during each analytical sequence or every 12 hours, whichever period is shorter. If multiple LMB are required for an analytical sequence, report the initial LMB associated with each 12 hour analysis period.
9.5 EDL. Calculate the EDL using Equation 23-11 of this method.
Note: If the applicable compliance limit is total dioxin or total furan, report the sum of the EDLs for all the target compounds. If the applicable rule limit is a TEQ value, report the sum of the EDLs for all target compounds multiplied by their corresponding compound specific TEF.
9.6 Field Train Proof Blank Assessment. Conduct at least one field train proof blank for each test series at a single facility. A field train proof blank is used to evaluate equipment preparation and potential contamination during sample recovery and consists of a fully assembled train at the sampling site. Prepare and assemble the field train proof blank train in a manner identical to that described in Sections 8.1.3 and 8.1.4 of this method using glassware from the same preparation batch as the field samples. The field train proof blank train must remain assembled for the same average amount of time samples are collected. Recover the field train proof blank train as described in Section 8.2 of this method. Follow all subsequent steps for field train proof blank train sample preparation and analysis used for field samples including data reporting. Section 13.1 of this method describes the criteria for the field train proof blank.
10.0 Calibration and Standardization
10.1 Sampling System. Same as Sections 6.1 and 10.1 through 10.7 of Method 5 of Appendix A-3 to 40 CFR part 60.
10.2 HRGC/HRMS System.
10.2.1 Mass Resolution. Tune the HRMS instrument to a resolving power of at least 10,000 at 10% percent of the peak height or 25,000 at 50% percent of the peak height. The resolving power for PAH and PCB analysis may be 8,000 at 10% of the peak height or 15,000 at 50% of the peak height. Assess the resolution at three exact m/z's representing the low-, mid-, and high-m/z range of the masses used to measure the target compound class. You may use peak matching and the chosen perfluoro-kerosene (PFK) or perfluorotributylamine (FC43) reference peak to verify that the exact mass is within 5 ppm of the required value.
10.2.2 Initial Calibration. Calibrate the HRGC/HRMS system using a minimum of five concentrations over a range that brackets expected field sample concentrations and the concentration of isotopically labeled standards in spiked samples. Tables 23-11, 23-12, and/or 23-13 of this method show the calibration concentrations recommended by this method, as applicable to the target compound classes. Determine the initial relative response factors for the target compounds and isotopically labeled standards using the initial calibration. Criteria for the initial calibration is in Section 13.9 of this method.
10.2.2.1 Lock-Mass Ions. Tables 23-4, 23-5, and 23-6 of this method present the recommended mass spectrometer lock-mass ions for PCDD/PCDF, PAH, and PCB, respectively. The reference compounds PFK or FC43 have ions that may be selected as your lock-mass and QC check ions. Monitor the QC check ions specified in these tables to verify instrument stability during the analysis (see Section 13.8 for performance criteria). Additional cleanup of the sample extract (or archive extract) and reanalysis is necessary for failure to maintain the lock-mass during analysis.
10.2.2.2 The relative standard deviation (RSD) for the mean calibration relative response factor from each of the unlabeled analytes and isotopically labeled compounds used in an analysis must be less than or equal to the values in Table 23-14 of this method.
10.2.2.3 The signal-to-noise (S/N) ratio for the GC/MS signal present in every selected ion current profile must be greater than or equal to 10 in all concentrations of calibration standards for unlabeled targets and isotopically labeled standards. The ion abundance ratios must be within the control limits in Table 23-15 of this method.
Note: An interference with PFK m/z 223.9872 may preclude meeting 10:1 S/N for the DiCB congeners at the optional Cal 1 level (Table 23-11). If this interference occurs, 10:1 S/N must be met at the Cal 2 level.
10.2.3 Continuing Calibration Verification.
10.2.3.1 Prepare the CCV standard at the same time as the batch of field samples using the same labeled standards. Prepare CCV standards at mid-level of the calibration (C3 level from Tables 23-11, 23-12, or 23-13 of this method). Inject a CCV standard, for the target compound class, at least once every 12 hours during an analysis sequence. Calculate the RRF for each compound and compare each RRF to the corresponding mean RRF obtained during the initial calibration. The RRF for each native compound measured in a CCV must not deviate from the initial calibration RRF by more than the limits shown in Table 23-14.
10.2.3.2 The ion abundance ratios must be within the allowable control limits shown in Table 23-15 of this method.
10.2.3.3 The S/N ratio for the GC/MS signal present in every selected ion current profile must be greater than or equal to 10.
10.2.3.4 Repeat the initial calibration when there is a failure to meet the requirements for acceptable CCV standard analysis.
10.2.3.5 Column Separation Check. Use the results from a CCV to verify and document the resolution required in Section 13.2, 13.3, or 13.4 of this method for the target compound classes analyzed with this method. If target compounds are not sufficiently resolved to meet the requirement, an analysis on a confirmation column is recommended (see Section 13.5 of this method).
10.2.3.6 If you use a confirmation column, perform the resolution check in Section 10.2.3.5 of this method to document the required resolution on the confirmation column. See Section 13.5 of this method on confirmation columns, if needed.
11.0 Analysis Procedure
11.1 Sample Extraction and Concentration. The sample extraction procedures in this method are the same for PCDD, PCDF, PCB and PAH targets. Figure 23-4 provides a flow chart showing sample container combination and extraction steps. Do not allow samples and extracts destined for PAH or PCB analysis to concentrate to dryness because the lower molecular weight PAH and the mono- through tri-chlorobiphenyls may be totally or partially lost. Note: Rotary evaporation is applicable when analyzing for PCDD/PCDF only. Snyder column apparatus is recommended when analyzing for PAH and PCB.
11.1.1 Optional Soxhlet Precleaning. Place an extraction thimble (see Section 6.3.3.3 of this method) and a plug of glass wool into the Soxhlet apparatus equipped with a Dean-Stark trap, charge the apparatus with toluene, and reflux for a minimum of 3 hours. Remove the toluene and discard it. Remove the extraction thimble from the extraction system and place it in a glass beaker to catch the solvent rinses from sample transfer to the extraction thimble. Retain the clean glass wool plug. Alternatively, confirm that the LMB for associated reagents, materials, and media meets the performance requirements in Section 13.1 of this method.
11.1.2 Container No. 1 (Filter) Preparation. Spike the filter with the appropriate pre-extraction filter recovery standard to result in the final sample extract concentrations shown in Tables 23-7, 23-8, and 23-9 of this method taking care that all spike liquid is distributed on the filter. Allow the filter to dry enough to prevent overspill, then transfer the filter and the contents of Container No. 1 directly to the glass extraction thimble in the glass solvent rinse catch beaker so that the filter will be completely immersed in the solvent during extraction.
11.1.3 Adsorbent Module. Spike the adsorbent with the appropriate pre-extraction standard to result in the final sample extract concentrations shown in Tables 23-7, 23-8, and 23-9 of this method, as applicable, spiked into the adsorbent, not on top of the adsorbent. Transfer the adsorbent material to the glass extraction thimble in the glass solvent rinse catch beaker. Rinse the module into the thimble in the beaker with the contents of Container No. 1. Alternatively, suspend the adsorbent module directly over the extraction thimble in a beaker, then, using a wash bottle containing methanol, flush the XAD-2 into the thimble onto the filter. Thoroughly rinse the interior of the glass module that contained the XAD-2 with toluene.
11.1.4 Container No. 2 (Acetone and Toluene Rinses). Concentrate the sample to a volume of no less than 5 mL. Concentrate samples containing toluene using a heating mantle and three-ball Snyder column or a rotary evaporator. Rinse sample Container No. 2 three times with small portions of toluene and add these to the concentrated solution and concentrate further to no less than 5 mL. This residue contains particulate matter removed in the rinse of the train probe and nozzle. Rinse the concentrated material from Container No. 2 into the glass extraction thimble containing the filter and the XAD-2 resin.
11.1.5 Transfer the solvent contained in the glass solvent rinse catch beaker to the extraction apparatus solvent reservoir. Rinse the beaker into the Soxhlet extraction apparatus solvent reservoir three times with small portions of toluene.
11.1.6 Container No. 3 (Impinger Water and Rinses). For PAH and PCB analysis, transfer the contents of Container No. 3 to a separatory funnel. Adjust to pH 2 with 6 N sulfuric acid, if necessary. Rinse the sample container with three successive 10-mL aliquots of the toluene and add these rinses to the separatory funnel. Extract the sample by vigorously shaking the separatory funnel for 5 minutes. After complete separation of the phases, remove the solvent and filter it through a bed of precleaned, dry sodium sulfate into the Soxhlet extraction apparatus solvent reservoir. Repeat the extraction step two additional times. Adjust the pH to 11 with 6 N sodium hydroxide, re-extract the impinger water and rinses, and filter it through a bed of precleaned, dry sodium sulfate into the Soxhlet extraction apparatus solvent reservoir. Rinse the sodium sulfate into the extraction apparatus solvent reservoir with fresh solvent and discard the sodium sulfate.
11.1.7 Add the appropriate pre-extraction standard for the target compound classes (to result in the final sample extract concentrations shown in Tables 23-7, 23-8, and 23-9 of this method) to the extraction thimble containing the combined filter and adsorbent sample fractions. Cover the contents of the extraction thimble with the cleaned glass wool plug to prevent the XAD-2 resin from splashing into the solvent reservoir of the extractor. Place the extraction thimble into the Soxhlet extraction apparatus.
11.1.8 Pour additional toluene to fill the solvent reservoir to approximately two-thirds capacity. Add PTFE boiling chips and assemble the apparatus.
11.1.9 Adjust the heat source to cause the extractor to cycle approximately three times per hour. Extract the sample for sufficient time to meet the pre-extraction standard recovery performance criteria in Section 13.15 of this method. The solvent should cycle completely through the system a minimum of 48 times.
11.2 Sample Aliquots for Cleanup and Analysis.
11.2.1 After extraction, allow the Soxhlet apparatus to cool.
11.2.2 Initial Extract Concentration. You may perform an initial concentration of the sample extract using the techniques ( e.g., Kuderna Danish, rotary evaporation, nitrogen blowdown) found to recover the pre-extraction standard sufficient to meet the performance criteria in Section 13.15 of this method. Concentrate initial extracts in toluene using a heating mantle and three-ball Snyder column or a rotary evaporator. Concentrate the field train proof blank and LMB samples in the same manner as samples.
Note: To meet isotopically labeled standard recoveries for low molecular weight PCB and PAH, do not evaporate samples to dryness and do not use a rotary evaporator to concentrate extracts.
11.2.3 Allow the sample extract to cool. You should use a minimum of one half of the sample extract for PCDD/PCDF analysis. You may archive the remaining sample extract or further split the sample extract for PCB and/or PAH analysis and archive.
Note: If using amount other than half the sample extract, adjust the spiking amount of the labeled standards accordingly.
11.2.4 If necessary, further concentrate the sample extract for cleanup and analysis using concentration techniques ( e.g., Kuderna Danish, rotary evaporation, nitrogen blowdown) found to recover the pre-extraction standard sufficient to meet the performance criteria in Section 13 of this method.
11.3 Sample Cleanup and Fractionation. You may process a separate aliquot/split of the sample extract for each of the compound classes analyzed by this method. Sample cleanup for each compound class may include techniques in addition to column chromatography such as acid/base back-extraction, Gel Permeation Chromatography, or high-performance liquid chromatography (HPLC) to isolate target compounds from interferences. This section includes a description of column chromatography shown to meet the performance criteria in Sections 9.2 and 13 of this method. The following sample cleanup and fractionation procedures are recommended but not required. You may modify cleanup column dimensions to meet manual or automated cleanup procedures as technology changes and improves. You must evaluate the cleanup and fractionation procedures used to confirm acceptable recovery of isotopically labeled standards. The alternative procedures must provide sufficient cleanup to meet method identification criteria (Section 11.4.3.4 of this method) and recovery criteria (Section 9.2 of this method). Section 13 of this method summarizes the method performance requirements.
Note: Recommendations in this section provide a cleanup approach that may allow multiple compound class measurement from a single aliquot of the original sample extract. Typically, Florisil® and alumina are used to separate PAH and PCDPE from PCDD and PCDF target compounds. Use acid, neutral, and basic silica gel and cleanup procedures to remove nonpolar and polar interferences from samples destined for PCB and PCDD/PCDF analysis. Use Carbopack®/Celite® (or other equivalent carbon-based column material) to remove other nonpolar interferences.
11.3.1 PAH and PCDPE Fractionation and Cleanup. You may use a Florisil® column to remove PAH and PCDPE from the sample extract. You may also fractionate sample extracts using Florisil® as the first cleanup step to separate PAH for analysis.
Note: High concentrations of PAH may interfere, leading to failure of performance criteria for PCDD/PCDF or PCB analysis.
11.3.1.1 Pack a 6-mm ID chromatographic column or equivalent diameter glass pipet with a glass wool plug followed by approximately 1.5 g (approximately 2 mL) of activated Florisil®. Add approximately 1 cm (approximately 1 mL) of anhydrous sodium sulfate followed by a glass wool plug to the head of the column. Pre-elute the column with 10 mL of methylene chloride followed by 10 mL of hexane and discard the eluate.
11.3.1.2 When the solvent is within 1 mm of the packing, transfer the concentrated extract (up to 5 mL) to the top of the Florisil® column, rinse the sample container twice with 1 to 2 mL of hexane, adding each rinse to the column, and elute the column with 35 mL of 5% dichloromethane in hexane. This fraction (Fraction 1) should contain target PCB, and selected hydrocarbons and chlorinated monoaromatic compounds.
11.3.1.3 Elute the column with 35 mL of 15% of dichloromethane in hexane and collect the eluate. This fraction (Fraction 2) should contain target PCDD/PCDF compounds.
11.3.1.4 Elute the column with 50 mL of 50% dichloromethane in hexane. The fraction (Fraction 3) should contain target PAH.
11.3.1.5 If necessary to remove any remaining polar organic compounds, elute the column with 70 mL of 15% acetone in hexane.
11.3.2 PCDD/PCDF and PCB Fractionation and Cleanup. You may remove PAH from the original aliquot of sample extract used for PCDD/PCDF analysis as described in Section 11.3.1 of this method. Design the column cleanup chromatography for PCDD/PCDF and PCB such that two consecutive fractions are collected (one with PCDD/PCDF and one with PCB) without impacting the detection limits. Depending on the source and sample matrix of the original sample, one or more of the following column cleanup approaches may be necessary to further remove polyhalogenated diphenyl ethers. You may use any number of permutations found in the referenced literature for this cleanup if the pre-extraction standard recoveries from field and LMB samples meet the associated performance criteria in Section 13 of this method. Alternatively, you may use an automated cleanup approach that meets the labeled spike recovery requirements in Section 13 of this method.
11.3.2.1 Silica Gel Column Chromatography. Pack one end of a glass column, approximately 20 mm ID × 230 mm long, with glass wool. Add in sequence to the glass column, 1 g of silica gel, 2 g of sodium hydroxide impregnated silica gel, 1 g of silica gel, 4 g of acid-modified silica gel, 1 g of silica gel, and 1 cm layer of anhydrous sodium sulfate. Pre-elute the column with 30 to 50 mL of hexane leaving a small quantity of hexane above the sodium sulfate layer. Discard the pre-elution hexane. Add the sample extract, dissolved in 5 mL of hexane to the head of the column. Allow the sample to flow into the column leaving a small quantity of hexane above the sodium sulfate layer. Rinse the extract container with two additional 5-mL rinses of hexane and apply each rinse to the column separately as the previous addition elutes. Elute the column with an additional 90 mL of hexane and retain the entire eluate. Concentrate this solution to a volume of about 1 mL using the nitrogen evaporative concentrator (see Section 6.3.5 of this method).
11.3.2.2 Silver Nitrate Silica Gel Column Chromatography. Pack a column (6 mm ID, 150 mm in length) sequentially with 1 g of silica gel and 1 g of 10% silver nitrate silica gel followed by a layer of about 10 mm of sodium sulfate (anhydrous). Wash the column sufficiently with hexane, elute until the liquid level reaches to the upper end of the column, and then transfer the concentrated sample (about 5 mL). Rinse the container several times with a small amount of hexane, elute with 200 mL of hexane at a flow rate about 2.5 mL/min (approximately one drop per second) to elute PCDD/PCDF.
11.3.2.3 Multi-layer Silica Gel Column Chromatography. You may use a multi-layer silica gel column in place of separate silica columns. Pack a column of 20 mm ID and 300 mm in length sequentially by the dry pack method with 0.9 g of silica gel, 3.0 g of 2% potassium hydroxide silica gel, 0.9 g of silica gel, 4.5 g of 44% sulfuric acid silica gel, 6.0 g of 22% sulfuric acid silica gel, 0.9 g of silica gel, 3.0 g of 10% silver nitrate silica gel, 2.0 g of silica gel and 6.0 g of sodium sulfate (anhydrous). Wash the column sufficiently with hexane, elute until the liquid level reaches to the upper end of the column, and then load the sample solution. Rinse the container several times with a small amount of hexane, elute with 150-200 mL of hexane at a flow rate about 2.5 mL/min (approximately one drop per second) to elute PCDD/PCDF.
11.3.2.4 Basic Alumina Column Chromatography. Pack a column (20 mm ID, 300 mm in length) with approximately 6 to 12 g of basic alumina. Pre-elute the column with 50 to 100 mL of hexane. Transfer the concentrated extract from the previous column cleanup to the top of the basic alumina column. Allow the sample to flow into the column leaving a small quantity of solvent above the top of the bed. Rinse the extract container with two additional 1-mL rinses of hexane and apply each rinse to the column separately as the previous addition elutes. Elute the column with 100 mL hexane to remove the interferences. Elute the PCDD/PCDF from the column with 20 to 40 mL of 50% methylene chloride in hexane. The ratio of methylene chloride to hexane may vary depending on the activity of the alumina used in the column preparation. Do not let the head of the column go without solvent. The first 100 mL hexane eluate is not used for subsequent PCDD/PCDF analysis. The eluate is concentrated to approximately 0.5 mL using the nitrogen evaporative concentrator.
11.3.2.5 Carbopack® C/Celite® 545 Column or Equivalent. Cut both ends from a 10 mL disposable Pasteur pipette (see Section 6.4.1 of this method) to produce a 10 cm column. Fire-polish both ends and flare both ends if desired. Insert a glass wool plug at one end and pack the column with 0.55 g of Carbopack®/Celite® (see Section 7.8.9.4 of this method) to form an adsorbent bed approximately 2 cm long. Insert a glass wool plug on top of the bed to hold the adsorbent in place. Pre-elute the column with 5 mL of toluene followed by 2 mL of methylene chloride:methanol:toluene (15:4:1 volume/volume (v/v)), 1 mL of methylene chloride:cyclohexane (1:1 v/v), and 5 mL of hexane. If the flow rate of eluate exceeds 0.5 mL/minute, discard the column. Do not let the head of the column go without solvent. Add the sample extract to the column. Rinse the sample container twice with 1 mL portions of hexane and apply separately to the column. Apply 2 mL of hexane to the head of the column to complete the transfer. Elute the interfering compounds with two 3 mL portions of hexane, 2 mL of methylene chloride:cyclohexane (1:1 v/v), and 2 mL of methylene chloride:methanol:toluene (15:4:1 v/v). Discard the eluate. Invert the column and elute the PCDD/PCDF with 20 mL of toluene. If carbon particles are present in the eluate, filter through glass-fiber filter paper. Concentrate the eluate to approximately 0.5 mL using the nitrogen evaporative concentrator for further cleanup or analysis by HRGC/HRMS.
11.4 PCDD, PCDF, PCB and PAH Analysis.
11.4.1 Analyze the sample extract with an HRGC/HRMS using the instrumental parameters in Sections 11.4.2 and 11.4.3 of this method.
11.4.1.1 Immediately prior to analysis, add an aliquot (typically 20 microliters (µl)) of the pre-analysis standard to result in the final sample extract concentrations in Tables 23-7, 23-8, and 23-9 of this method to each sample as appropriate for the compounds you are measuring by this method.
11.4.1.2 Inject an aliquot of the sample extract into the GC, typically 1 µl. You may perform separate analyses using different GC columns for each of the target compound classes. Perform calibration and sample analysis for each target compound class using the same instrument operating conditions including injection volume.
11.4.1.2.1 If target compounds are not resolved sufficiently from other target compounds or interferences in the sample to meet the requirements in Section 10.2.3.5 or 10.2.3.6 of this method, as applicable to the compound class being analyzed, or as otherwise specified in an applicable regulation, permit, or other requirement, analyze sample (or another aliquot of the sample) using an alternative column that provides elution order to uniquely quantify the target compounds subject to interference on the first GC column.
11.4.1.2.2 You may use column systems other than those recommended in this method provided the analyst is able to demonstrate, using calibration and CCVs, that the alternative column system is able to meet the applicable specifications of Section 10.2.3.5 or 10.2.3.6 of this method.
11.4.2 Example Gas Chromatograph Operating Conditions.
11.4.2.1 Injector. Configured for capillary column, splitless, 250 °C (482 °F).
11.4.2.2 Carrier Gas. Helium, 1 to 2 mL/min.
11.4.2.3 Oven. Optimize the GC temperature program to achieve the required separation and target compound recovery for the GC column in use. Table 23-16 of this method presents the typical conditions for a DB5-MS column.
11.4.3 High-Resolution Mass Spectrometer.
11.4.3.1 Ionization Mode. Electron ionization.
11.4.3.2 Source Temperature. Maintain the source temperature in the range of 250 to 300 °C (482 to 572 °F).
11.4.3.3 Ion Monitoring Mode. Tables 23-4, 23-5, and 23-6 of this method summarize the various ions to be monitored for PCDD/PCDF, PAH, and PCB, respectively.
11.4.3.4 Identification Criteria for Target Compounds. Use the following identification criteria for the characterization of target compounds in this method. The available native and isotopically labeled standards allow the unique identification of all PCDD/PCDF, PAH, and selected PCB congeners analyzed in this method. Also see Sections 13.12 and 13.13 of this method for identification criteria for PCDD/PCDF/PCB and PAH target compounds, respectively.
11.4.3.4.1 For PCDD/PCDF and PCB, Table 23-15 of this method provides acceptance limits for the integrated ion abundance ratio of primary and secondary target compound ions. When the ion abundance ratio for a target analyte is outside the performance criteria, you may reanalyze samples on an alternative GC column to resolve chemical interferences, tune the mass spectrometer to operate at a higher mass resolution to discriminate against the interference(s), and/or further cleanup an archived sample to remove the interference(s). Report analysis results as an EMPC when a response meets identification criteria except the ion abundance ratio criteria or when a peak representing a PCDPE has been detected at the retention time. This method does not consider EMPC-flagged data to be zero concentrations.
Note: Some EMPCs may be caused by poor ion statistics when the concentration of the target compound is at or near the DL.
11.4.3.4.2 The retention time for the analytes must be within 3 seconds of the corresponding labeled pre-extraction standard.
11.4.3.4.3 The signals for the two exact masses in Tables 23-4 and 23-6 of this method for PCDD/PCDF and PCB, respectively, must be present and must reach their maximum response within two seconds of each other.
11.4.3.4.4 Identify and quantify specific target compounds or isomers that do not have corresponding pre-extraction standard compounds by comparing to the pre-extraction standard of the same compound class with the nearest retention time to target compound.
11.4.3.4.5 For the identification of specific PCB congeners, the retention time of the native congener must be within 0.006 relative retention time (RRT) units of the pre-extraction standard.
11.4.3.4.6 For qualitative identification, the S/N ratio for the GC signal present in every selected ion current profile for native compound response must be greater than or equal to 2.5.
11.4.3.4.7 The separation of target compounds, including 2,3,7,8-TeCDD and 2,3,7,8-TeCDF, must satisfy the separation criteria in Section 10.2.3.5 of this method and all the identification criteria specified in Sections 11.4.3.4.1 through 11.4.3.4.6 of this method. See Section 13.5 of this method on confirmation columns, if needed.
11.4.3.4.8 Chlorodiphenyl Ether Interference. If chromatographic peaks are detected at the retention time of any PCDF in any of the m/z channels used to monitor PCDPE, there is evidence of a positive interference and you may opt to flag data noting the interference and keep the value to calculate PCDF concentration as EMPC or reanalyze to remove or shift the interference. This method recommends alumina (see Section 11.3.2.4 of this method) and Florisil® (see Section 11.3.1 of this method) liquid column chromatography packing materials for removal of PCDPE during sample cleanup.
11.4.3.4.9 The recommended MS lock-mass ions are specified in Tables 23-4, 23-5, and 23-6 of this method for PCDD/PCDF, PAH, and PCB, respectively. Monitor the QC check ions to verify instrument stability during the analysis. If the QC check ion signal varies by more than 25% from the average response across the run, flag results for all isomers at corresponding retention time as the lock-mass ions or QC check ions. You have the option to reanalyze after additional cleanup on the sample (or an archived portion of the sample if the archive is available), or after dilution of the sample. Alternately, determine through additional quality review whether the target analyte and its corresponding isotopically labeled standard are equally affected by the change in lock-mass ions and/or QC check ions. When you reanalyze a sample, ensure all concentration calculations are reported from the reanalyzed sample.
11.4.3.4.10 For the identification of PAH, the RRT of each native to its labeled compound must be within 0.006 RRT units compared to the corresponding RRTs in the continuing calibration. The signals for the characteristic ion listed in Table 23-5 of this method must be present.
11.4.3.5 Quantitation. Measure the response of each native target compound and the corresponding pre-extraction standard. Using the CCV RRF, calculate the mass of each target compound, using equations in Section 12.7 of this method. Use the pre-extraction standard to correct the native target compounds result for variations in performance of the extraction, cleanup, and concentration steps of the analysis. Recovery of pre-extraction standard must meet the minimum specifications in Section 9.2. of this method to ensure that the method performance and reliability have not been compromised by unacceptable losses during sample processing. Table 23-17 of this method shows the assignments for pre-extraction standard compounds for use in calculating the response factor and the concentrations of PCB. Recoveries of all labeled standard compounds must meet the minimum recovery specifications in Section 13 of this method. Note: Unacceptably low recoveries can be an indication of a sample processing step that caused the low recoveries, such as spiking errors.
11.4.3.5.1 Use Equation 23-7 to calculate the amount of each target compound or group in the sample.
11.4.3.5.2 Use Equation 23-8 to calculate the concentration per dscm of each target compound or group in the gas.
11.4.3.5.3 Quantify native PCDD and PCDF in its homologous series using the corresponding native and pre-extraction standard response in its homologous series. For example, use 13 C 12 -2,3,7,8-TeCDD to calculate the concentrations of all other tetra chlorinated isomers.
11.4.3.5.4 As an option or as required or specified in applicable regulations, permits, or other requirements, you may quantify any or all other PCB congeners as resolved or coeluting combinations using the RRF of the nearest eluting native target PCB in the same homolog group and the pre-extraction standard assigned in Appendix A to this method.
11.4.3.5.5 As an option or as required or specified in applicable regulations, permits, or other requirements, report the total concentration of congeners at a given level of chlorination (homolog; i.e., total TrCB, total PeCB, total HxCB, etc.) by summing the concentrations of all congeners identified in the retention time window for the homologs as assigned in Appendix A to this method.
11.4.3.5.6 As an option or if required in an applicable regulation, permit or other requirement, total PCB may be reported by summing all congeners identified at all window-defining congeners (WDCs) as assigned in Appendix A to this method.
12.0 Data Analysis and Calculations
Note: Same as Section 12 of Method 5 of Appendix A-3 to 40 CFR part 60, with the following additions.
12.1 Nomenclature.
A1 n = Integrated ion current of the primary m/z values for the target native compound.
A1 pe = Integrated ion current of the primary m/z values for the pre-extraction standard compound (assigned in Tables 23-4, 23-5, and 23-6 of this method).
A1 pa = Integrated ion current of the primary m/z values for the pre-analysis standard compound.
A2 n = Integrated ion current of the secondary m/z values for the target native compound. For PAH A2 n = 0.
A2 pe = Integrated ion current of the secondary m/z's for the pre-extraction standard compound. For PAH A2 l = 0.
A2 pa = Integrated ion current of the secondary m/z values for the pre-analysis standard compound.
C i = Mass of compound i in the sample, pg.
C idscm = Concentration of target native compound i in the emission gas, pg/dscm.
C T = Total mass of target compounds in the sample, pg/sample.
dscm = Dry standard cubic meters of gas volume sample measured by the dry gas meter, corrected to standard conditions.
H ai = Summed heights of the noise for each quantitation ion for native target compounds.
H ci = Summed heights of the noise at the primary and secondary m/z's of the pre-extraction standard i.
L PIR = Lower limit for the prediction interval of results.
n = Number of values.
PD = Percent Difference in the RRF of the continuing calibration verification compared to the average RRF of the initial calibration, %.
Q n = Quantity of the target native compound, pg.
Q pe = Quantity of the pre-extraction standard, pg.
Q pa = Quantity of the pre-analysis standard, pg.
R = Recovery of pre-sampling adsorbent standard and pre-extraction filter recovery standard, %.
R pe = Recovery of pre-extraction standard, %.
RRF i = Relative response factor of a native target compound or pre-sampling adsorbent standard and pre-extraction filter recovery standard at calibration level i.
RRF pe = Relative response factor of a pre-extraction standard compound.
RRF ccv = Relative response factor of a native target compound or pre-sampling adsorbent standard and pre-extraction filter recovery standard in the continuing calibration verification.
RSD = Relative standard deviation, in this case, of RRFs over the calibration levels, %.
SD = Standard deviation.
SD RRF = Standard deviation of initial calibration RRFs.
U PIR = Upper limit for the prediction interval of results.
WDC = Window-defining congener representing an isotopically labeled compound that defines the beginning or end of a retention time window bracketing a target homolog.
12.2 Individual Compound RRF for Each Calibration Level i. Equation 23-1 for the response factor of each target native compound relative to its labeled pre-extraction standard analog includes the integrated ion current of both the primary and secondary m/z values for each compound in the calibration standard, excluding PAH, which use only primary m/z values. Use Equation 23-2 to calculate the RRF for pre-extraction standard.

Note: the units for Q pe and Q n in Eq. 23-1 and the units for Q pa and Q pe in Equation 23-2 must be the same.
12.3 Average RRF for Each Compound Over the Minimum of Five Calibration Levels.

12.4 Percent RSD of the RRFs for a Compound Over the Calibration Levels. The requirement for the initial calibration RSD is in Section 13.9 and Table 23-14 of this method.

12.5 Standard Deviation of the RRFs for a Compound Over the Calibration Levels.

12.6 Percent Difference of the RRF of the Continuing Calibration Verification Compared to the Average RRF from the Initial Calibration for Each Target Compound. Use Equation 23-1 to calculate the RRF for the continuing calibration verification for comparison to the average RRF from the initial calibration. The requirement for the continuing calibration verification % difference is in Section 13.10 and Table 23-14 of this method.

12.7 Amount of Individual Target Compound i in the Sample by Isotope Dilution (pg). This equation corrects for the target native compound recovery based on its labeled pre-extraction standard analog. This equation is also used to calculate the amount of pre-sampling adsorbent standard and pre-extraction filter recovery standard recovered.

Note: For the quantitation of the pre-sampling adsorbent standard and the pre-extraction filter recovery standard, use a corresponding pre-extraction isomer (or homolog) with the closest retention time.
12.8 Concentration of the Individual Target Compound or Group i in the Emission Gas (pg/dscm). The total concentration of a target compound group in the sample can be calculated by substituting C T from Eq. 23-12 for C i in Equation 23-8.

12.9 Recovery of Labeled Compound Standards. Use Equation 23-9 to determine the recovery of pre-sampling adsorbent standard and the pre-extraction filter recovery standard. Use Equation 23-10 to determine the recovery of the pre-extraction standard. The recovery performance criteria for these standards are in Sections 13.14, 13.15, and 13.16 of this method.

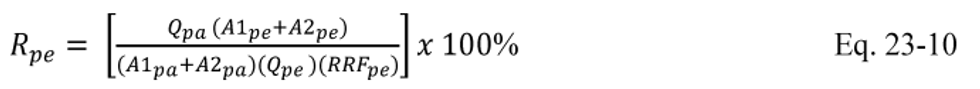
Note: Recovery may be calculated based on mass instead of concentration, as needed.
Note: R pe must be corrected for the fraction of the original sample extract used for analysis. ( e.g., if half of the extract is used for analysis of the target class, R pe must be multiplied by a factor of 2).
12.10 Estimated Detection Limit (EDL).

12.11 Total Target Compound Mass.

Note: Unless otherwise specified in applicable regulations, permits or other requirements, count any target compounds reported as non-detected as EDL when calculating the concentration of target compounds in the sample.
12.12 Upper and Lower Limits for the Prediction Interval of Results (PIR)
Half Range (HR) for the Predication Interval of Results

Note: 3.963 is a constant value for seven replicates.
Upper and Lower Limits for the Prediction Interval of Results

13.0 Method Performance
Data generated with this method must be fit for purpose. Applicable results of method performance criteria in this section must be reported. Consequences of failed quality criteria are provided with the criteria in this section.
13.1 Background Assessment—Field Train Proof Blank, LMB and Materials. Determine the contribution to target compound concentration from reagents, media and glassware used to make target compound measurements. Conduct at least one field train proof blank for each test series at a single facility. Analyze at least one LMB sample during an analytical sequence or every 12 hours, whichever is shorter. Native target compound concentrations in the field train proof blank, LMB and materials assessment must be less than or equal to three times the EDL of the method or 10 times lower than the quantitation limit required by the end use of the data ( e.g., compliance limit or other limits set by consent decree or permit), whichever is higher. The field train proof blank, LMB and materials assessment must also meet the performance specifications in Tables 23-7, 23-8, and 23-9, as applicable to the compound target list.
13.2 GC column or column systems used to measure PCDD/PCDF must meet the column separation requirements in Section 6.5.2.1 of this method and the applicable requirements in Sections 10.2.3.5 and 11.4.3.4 of this method using the continuing calibration verification. Failure to meet this chromatographic resolution criterion requires data from this analysis to be flagged explaining the potential bias of the results.
13.3 GC column or column systems used to measure PAH must meet the column separation requirements in Section 6.5.2.2 of this method and the applicable requirements in Sections 10.2.3.5 and 11.4.3.4 of this method using the continuing calibration check. Failure to meet this chromatographic resolution criterion requires data from this analysis to be flagged explaining the potential bias of the results.
13.4 GC column or column systems used to measure PCB must meet the column separation requirements in Section 6.5.2.3 of this method and the applicable requirements in Sections 10.2.3.5 and 11.4.3.4 of this method using the continuing calibration check and be able to achieve unique resolution and identification of the toxics for determination of a TEQ PCB. The rule requiring the use of this method will establish which WHO TEF to use. Failure to meet this chromatographic resolution criterion requires data from this analysis to be flagged explaining the potential bias of the results.
13.5 Confirmation Column. If target compounds are not sufficiently resolved from other target compounds or interferences in the sample to meet the requirements for target compounds in Sections 13.2, 13.3, and/or 13.4 of this method, analyze sample (or another aliquot of the sample) using an alternative column that provides elution order to uniquely quantify the target compounds subject to interference on the first GC column. When using a confirmation column, document the required resolution.
13.6 Detection Limits.
13.6.1 MDL. The MDLs are determined following the procedures in Section 9.3.7 of this method. MDLs are confirmed by preparing and analyzing a spiked sample (spiked at 1 to 5 times the determined MDL, see Section 9.3.8), then confirm that the target compounds meet the qualitative identification criteria in Section 11.4.3.4 of this method. If the MDL confirmation criteria are not met, the MDL determination is repeated with a higher spike concentration until criteria are met.
13.6.2 EDL. If the sample specific EDLs are less than 50% of the emission standard, the EDLs are acceptable.
13.7 Tune. The groups of monitored ions are listed in Tables 23-4, 23-5, and 23-6 of this method, as applicable for the target compound class. Tune the instrument to meet the required resolving power in Section 10.2.1 for the desired target compound class. Assess the resolution at three exact m/z's representing the low-, mid-, and high-m/z range of the masses used to measure the target compound class. You may use peak matching and the chosen PFK (or FC43) reference peak to verify that the exact mass is within 5 ppm of the required value.
13.8 Lock-Mass Ions. The MS lock-mass and QC check ions in Tables 23-4, 23-5, and 23-6 of this method are recommended for PCDD/PCDF, PCB, or PAH, respectively. The reference compounds PFK or FC43 have ions that may be selected as your lock-mass and QC check ions. Monitor the QC check ions specified in these tables to verify instrument stability during the analysis; these must not vary >25% from the average response. Additional cleanup on sample extract (or archive extract) and reanalysis is necessary for failure to maintain lock-mass during analysis.
13.9 Initial Calibration.
13.9.1 The RSD for mean RRF from each of the target analytes and labeled standards in the calibration samples must not exceed the values in Table 23-14 of this method.
13.9.2 The S/N in every selected ion current profile must be ≥10 for all unlabeled targets and labeled standards in the calibration samples.
13.9.3 The ion abundance ratios must be within the control limits in Table 23-15 of this method.
13.10 Continuing Calibration Verification.
13.10.1 The RRF for each unlabeled and labeled compound measured in a CCV must not deviate from the initial calibration RRF by more than the limits shown in Table 23-14 of this method.
13.10.2 The ion abundance ratios must be within the control limits in Table 23-15 of this method.
13.10.3 The S/N ratio for the GC/MS signal present in every selected ion current profile must be greater than or equal to 10.
13.10.4 Repeat the initial calibration when there is a failure to meet the requirements for an acceptable CCV analysis.
13.10.5 Column Separation Check. Use the results from a CCV to verify and document the resolution required in Sections 13.2, 13.3, or 13.4 of this method for the target compound classes analyzed with this method. The separation criteria are applicable to all the compounds in a target class whether analyzed by a single or multiple GC columns. If a confirmation column is used, document required resolution (see Section 13.5).
13.11 QCS. A QCS must be analyzed during the IDC and after initial calibrations (at a minimum quarterly). The acceptance criterion for the QCS is 70-130% of the true value. If the accuracy for any analyte fails the recovery criterion, prepare a fresh standard dilution and repeat. If the freshly prepared QCS fails, determine the cause, recalibrate the instrument if necessary and reanalyze the QCS.
13.12 Compound Identification for PCDD/PCDF and PCB.
13.12.1 Target compounds must have ion abundance ratios within the control limits in Table 23-15 of this method. PAH target compounds have single ion identifiers with no ion abundance ratio requirement. Report analysis results as an EMPC when a response meets identification criteria but fails the ion abundance ratio criteria or when a peak representing a PCDPE has been detected at the target compound retention time.
13.12.2 The retention time for the analytes must be within 3 seconds of the corresponding pre-extraction standard.
13.12.3 The monitored ions, shown in Table 23-4 of this method for a given PCDD/PCDF, must reach their maximum response within 2 seconds of each other.
13.12.4 The monitored ions, shown in Table 23-6 of this method for a given PCB, must reach their maximum response within 2 seconds of each other.
13.12.5 For the identification of specific PCB, the RRT of the native congener must be within 0.006 RRT units of the pre-extraction standard RRT.
13.12.6 The S/N ratio for the monitored ions for native compounds must be greater than or equal to 2.5.
13.12.7 Identify and quantify isomers that do not have corresponding pre-extraction standard compounds by comparing to the pre-extraction standard of the same compound class with the nearest retention time to the target compound.
13.12.8 If chromatographic peaks are detected at the retention time of any PCDD/PCDF in any of the m/z channels used to monitor PCDPE, there is evidence of interference and positive bias. Data must be flagged to indicate an interference. You may report the total with bias for the affected target. To reduce the bias, you may use a confirmatory column or perform additional clean up on an archived sample followed by reanalysis.
13.13 Compound Identification for PAH.
13.13.1 The signals for the characteristic ion listed in Table 23-5 of this method must be present.
13.13.2 The RRT between each native and labeled compound must be within 0.006 RRT units.
13.14 Pre-sampling Adsorbent Standard and Pre-extraction Filter Recovery Standard Recovery. Recoveries of pre-sampling adsorbent standard added to the sample and pre-extraction filter recovery standard added to the filter must be between 70 and 130% (see Tables 23-7, 23-8, and 23-9 of this method).
13.14.1 If the recovery of all the pre-sampling adsorbent standard compounds is below 70%, the sampling runs are not valid, and you must repeat the stack or vent sampling. As an alternative, you do not have to repeat the test if the average pre-sampling adsorbent standard recovery is 25% or more and you divide the final results by the average fraction of pre-sampling adsorbent standard recovery.
13.14.2 If the recovery of all the pre-extraction filter recovery standard compounds is below 70%, you may reanalyze the sample. If the recovery criteria are still not met, the sampling recovery is not valid, and you must repeat the stack or vent sampling.
13.15 Pre-extraction Standard Recovery. Recoveries of all pre-extraction standard compounds added to the sample must be between 20 to 130% for PCDD/PCDF and PAH (see Tables 23-7 and 23-8 of this method) and between 20 to 145% for PCB (see Table 23-9 of this method). If the recovery criteria are not met, you may reanalyze the sample and/or prepare and analyze the archive sample. If the recovery criteria are still not met, the sampling run is not valid, and the stack test must be repeated.
13.16 Pre-analysis Standard Response. Response of all pre-analysis standard compounds must show a S/N for every selected ion current profile of ≥ 10. If the minimum response is not met, you must reanalyze the sample. Poor sensitivity compared to initial calibration response may indicate injection errors or instrument drift.
13.17 IDC—Lowest calibration concentration, Demonstration of precision, Demonstration of accuracy .
13.17.1 Lowest calibration concentration. The Upper PIR Limit must be less than, or equal, to 150%; and the Lower PIR Limit must be greater than, or equal to, 50%. If these criteria are not met, the lowest calibration point has been set too low and must be confirmed at a higher concentration.
13.17.2 Demonstration of precision. The percent relative standard deviation (%RSD) of the concentrations from the replicate analyses must be less than 20% for all target analytes. Demonstration would be repeated for failed compounds only.
13.17.3 Demonstration of accuracy. The average % recovery for each target analyte must be within 70 to 130%. Demonstration would be repeated for failed compounds only.
13.18 Requirements for Equivalency. The Administrator considers any modification of this method, beyond those expressly permitted in this method as options, to be a major modification subject to application and approval of alternative test procedures following EPA Guidance Document 22 currently found at: https://www.epa.gov/emc/emc-guideline-documents.
13.19 Records. As part of the laboratory's quality system, the laboratory must maintain records of modifications to this method.
14.0 Pollution Prevention
The target compounds used as standards in this method are prepared in extremely small amounts and pose little threat to the environment when managed properly. Prepare standards in volumes consistent with laboratory use to minimize the disposal of excess volumes of expired standards.
15.0 Waste Management
15.1 The laboratory is responsible for complying with all federal, state, and local regulations governing waste management, particularly the hazardous waste identification rules and land disposal restrictions, and for protecting the air, water, and land by minimizing and controlling all releases from fume hoods and bench operations. The laboratory must also comply with any sewage discharge permits and regulations. The EPA's Environmental Management Guide for Small Laboratories (EPA 233-B-98-001) provides an overview of requirements.
15.2 Samples containing hydrogen chloride or sulfuric acid to pH <2 are hazardous and must be handled and disposed in accordance with federal, state, and local regulations.
15.3 For further information on waste management, consult The Waste Management Manual for Laboratory Personnel and Less is Better-Laboratory Chemical Management for Waste Reduction, available from the American Chemical Society's Department of Government Relations and Science Policy, 1155 16th Street NW, Washington, DC 20036.
16.0 Bibliography
1. American Society of Mechanical Engineers. Analytical Procedures to Assay Stack Effluent Samples and Residual Combustion Products for Polychlorinated Dibenzo-p-Dioxins (PCDD) and Polychlorinated Dibenzofurans (PCDF). Prepared for the U.S. Department of Energy and U.S. Environmental Protection Agency. Washington, DC. December 1984. 23 p.
2. American Society of Mechanical Engineers. Sampling for the Determination of Chlorinated Organic Compounds in Stack Emissions. Prepared for U.S. Department of Energy and U.S. Environmental Protection Agency. Washington DC. December 1984. 25 p.
3. Fishman, V.N., Martin, G.D. and Lamparski, L.L., Comparison of a variety of gas chromatographic columns with different polarities for the separation of chlorinated dibenzo-p-dioxins and dibenzofurans by high-resolution mass spectrometry, Journal of Chromatography A 1139 (2007) 285-300.
4. International Agency for Research on Cancer. Environmental Carcinogens Methods of Analysis and Exposure Measurement, Volume 11—Polychlorinated Dioxins and Dibenzofurans. IARC Scientific Publications No. 108, 1991.
5. Stieglitz, L., Zwick, G., Roth, W. Investigation of different treatment techniques for PCDD/PCDF in fly ash. Chemosphere 15: 1135-1140; 1986.
6. Triangle Laboratories. Case Study: Analysis of Samples for the Presence of Tetra Through Octachloro-p-Dibenzodioxins and Dibenzofurans. Research Triangle Park, NC. 1988. 26 p.
7. U.S. Environmental Protection Agency. Method 8290A—Polychlorinated Dibenzo-p-dioxins (PCDDs) and Polychlorinated Dibenzofurans (PCDFs) by High-Resolution Gas Chromatography/High-Resolution Mass Spectrometry (HRGC/HRMS), Revision 1. February 2007. In: Test Methods for Evaluating Solid Waste, Physical/Chemical Methods, EPA publication SW-846. Washington, DC.
8. U.S. Environmental Protection Agency. Office of Air Programs Publication No. APTD-0576: Maintenance, Calibration, and Operation of Isokinetic Source Sampling Equipment. Research Triangle Park, NC. March 1972.
9. U.S. Environmental Protection Agency. Method 1625C-Semivolatile Organic Compounds by Isotope Dilution GCMS. June 1989.
10. U.S. Environmental Protection Agency. Method 1613B-Tetra- through Octa-Chlorinated Dioxins and Furans by Isotope Dilution HRGC/HRMS. October 1994.
11. U.S. Environmental Protection Agency. Method 1668C-Chlorinated Biphenyl Congeners in Water, Soil, Sediment, Biosolids, and Tissue by HRGC/HRMS. April 2010.
12. Tondeur, Y., Nestrick, T., Silva, Héctor A., Vining, B., Hart, J. Analytical procedures for the determination of polychlorinated-p-dioxins, polychlorinated dibenzofurans, and hexachlorobenzene in pentachlorophenol, Chemosphere Volume 80, Issue 2, June 2010 pages 157-164.
13. U.S. Environmental Protection Agency. Definition and Procedure for the Determination of the Method Detection Limit, Revision 2. EPA 821-R-16-006. December 2016.
14. Tondeur Y, Niederhut WJ, Missler SR. A hybrid HRGC/MS/MS Method for the Characterization of Tetrachlorodibenzo-p-Dioxins in Environmental Samples; Bio. Med. and Environ. Mass Spectr. 14, pages 449-456, 1987.
15. Gianluca R., Mosca S., Guerriero E., Rotatori M. Development of a new automated clean-up system for the simultaneous analysis of polychlorinated dibenzo- p -dioxins (PCDDs), dibenzofurans (PCDFs) and `dioxin-like' polychlorinated biphenyls (dl-PCB) in flue gas emissions by GPC-SPE. J. Environ. Monit. 14, pages 1082-1090, 2012.
16. U.S. Environmental Protection Agency. The National Dioxin Air Monitoring Network (NDAMN) Report of the Results of Atmospheric Measurements of Polychlorinated Dibenzo- p -Dioxins (PCDDs), Polychlorinated Dibenzofurans (PCDFs), and Dioxin-like Polychlorinated Biphenyl (PCBs) in Rural and Remote Areas of the United States from June 1998 through November 2004. EPA/600/R-13/183F. August 2013.
17. Guo, Y., Kannan, K. Analytical Methods for the Measurement of Legacy and Emerging Persistent Organic Pollutants in Complex Sample Matrices. Comprehensive Analytical Chemistry. Vol. 67. January 2015.
18. U.S. Environmental Protection Agency. USEPA Contract Laboratory Program (CLP) National Functional Guidelines for Chlorinated Dibenzo-p-Dioxins (CDDs) and Chlorinated Dibenzofurans (CDFs) Data Review. EPA-540-R-11-016. September 2011.
17.0 Tables, Diagrams, Flowcharts, and Validation Data
| Polychlorinated dibenzo- p -dioxins | CAS a Registry No. | Polychlorinated dibenzofurans | CAS a Registry No. |
|---|---|---|---|
| a Chemical Abstract Service. | |||
| 2,3,7,8-TeCDD | 1746-01-6 | 2,3,7,8-TeCDF | 51207-31-9 |
| 1,2,3,7,8-PeCDD | 40321-76-4 | 1,2,3,7,8-PeCDF | 57117-41-6 |
| 1,2,3,4,7,8-HxCDD | 39227-28-6 | 2,3,4,7,8-PeCDF | 57117-31-4 |
| 1,2,3,6,7,8-HxCDD | 57653-85-7 | 1,2,3,4,7,8-HxCDF | 70648-26-9 |
| 1,2,3,7,8,9-HxCDD | 19408-74-3 | 1,2,3,6,7,8-HxCDF | 57117-44-9 |
| 1,2,3,4,6,7,8-HpCDD | 35822-46-9 | 1,2,3,7,8,9-HxCDF | 72918-21-9 |
| Total TeCDD | 41903-57-5 | 2,3,4,6,7,8-HxCDF | 60851-34-5 |
| Total PeCDD | 36088-22-9 | 1,2,3,4,6,7,8-HpCDF | 67562-39-4 |
| Total HxCDD | 34465-46-8 | 1,2,3,4,7,8,9-HpCDF | 55673-89-7 |
| Total HpCDD | 37871-00-4 | Total TeCDF | 55722-27-5 |
| OCDD | 3268-87-9 | Total PeCDF | 30402-15-4 |
| Total HxCDF | 55684-94-1 | ||
| Total HpCDF | 38998-75-3 | ||
| OCDF | 39001-02-0 | ||
| Polycyclic aromatic hydrocarbons | CAS a Registry No. | Polycyclic aromatic hydrocarbons | CAS a Registry No. |
|---|---|---|---|
| a Chemical Abstract Service. | |||
| Naphthalene | 91-20-3 | Chrysene | 218-01-9 |
| 2-Methylnaphthalene | 91-57-6 | Benzo[ b ]fluoranthene | 205-99-2 |
| Acenaphthylene | 208-96-8 | Benzo[ k ]fluoranthene | 207-08-9 |
| Acenaphthene | 83-32-9 | Perylene | 198-55-8 |
| Fluorene | 86-73-7 | Benzo[ a ]pyrene | 50-32-8 |
| Anthracene | 120-12-7 | Benzo[ e ]pyrene | 192-97-2 |
| Phenanthrene | 85-01-8 | Benzo[ g,h,i ]perylene | 191-24-2 |
| Fluoranthene | 206-44-0 | Indeno[ 1,2,3-cd ]pyrene | 193-39-5 |
| Pyrene | 129-00-0 | Dibenz[ a,h ]anthracene | 53-70-3 |
| Benz[ a ]anthracene | 56-55-3 | ||
| PCB congener | BZ No. a | CAS b Registry No. | PCB congener | BZ No. a | CAS b Registry No. |
|---|---|---|---|---|---|
| a BZ No.: Ballschmiter and Zell 1980, or International Union of Pure and Applied Chemistry (IUPAC) number. | |||||
| b Chemical Abstract Service. | |||||
| 2,4′-DiCB | 8 | 34883-43-7 | 2,2′,3,3′,4,4′-HxCB | 128 | 38380-07-3 |
| 2,2′,5-TrCB | 18 | 37680-65-2 | 2,2′,3,4,4′,5′-HxCB | 138 | 35065-28-2 |
| 2,4,4′-TrCB | 28 | 7012-37-5 | 2,2′,4,4′,5,5′-HxCB | 153 | 35065-27-1 |
| 2,2′,3,5′-TeCB | 44 | 41464-39-5 | 2,3,3′,4,4′,5-HxCB | 156 | 38380-08-4 |
| 2,2′,5,5′-TeCB | 52 | 35693-99-3 | 2,3,3′,4,4′,5′-HxCB | 157 | 69782-90-7 |
| 2,3′,4,4′-TeCB | 66 | 32598-10-0 | 2,3′,4,4′,5,5′-HxCB | 167 | 52663-72-6 |
| 3,3′,4,4′-TeCB | 77 | 32598-13-3 | 3,3′,4,4′,5,5′-HxCB | 169 | 32774-16-6 |
| 3,4,4′,5-TeCB | 81 | 70362-50-4 | 2,2′,3,3′,4,4′,5-HpCB | 170 | 35065-30-6 |
| 2,2′,4,5,5′-PeCB | 101 | 37680-73-2 | 2,2′,3,4,4′,5,5′-HpCB | 180 | 35065-29-3 |
| 2,3,3′,4,4′-PeCB | 105 | 32598-14-4 | 2,2′,3,4′,5,5′,6-HpCB | 187 | 52663-68-0 |
| 2,3,4,4′,5-PeCB | 114 | 74472-37-0 | 2,3,3′,4,4′,5,5′-HpCB | 189 | 39635-31-9 |
| 2,3′,4,4′,5-PeCB | 118 | 31508-00-6 | 2,2′,3,3′,4,4′,5,6-OcCB | 195 | 52663-78-2 |
| 2′,3,4,4′,5-PeCB | 123 | 65510-44-3 | 2,2′,3,3′,4,4′,5,5′,6-NoCB | 206 | 40186-72-9 |
| 3,3′,4,4′,5-PeCB | 126 | 57465-28-8 | 2,2′,3,3′,4,4′,5,5′,6,6′-DeCB | 209 | 2051-24-3 |
| Mass a | Ion type b | Elemental composition | Target analyte b | Mass a | Ion type b | Elemental composition | Target analyte b |
|---|---|---|---|---|---|---|---|
| a The following nuclidic masses were used to calculate exact masses: H = 1.007825, C = 12.000000, 13 C = 13.003355, F = 18.9984, O = 15.994915, 35 C l= 34.968853, 37 Cl = 36.965903. | |||||||
| b (S) = Labeled Standard. LOCK = Lock-Mass Ion PFK or FC43. QC = Quality Control Check Ion. Note: Consider monitoring 328 m/z if a high level of PCB is expected. | |||||||
| 263.9871 | LOCK | C 5 F 10 N | FC43 | 383.8639 | M | 13 C 12 H 2 35 Cl 6 O | HxCDF (S). |
| 292.9825 | LOCK | C 7 F 11 | PFK | 385.8610 | M 2 | 13 C 12 H 2 35 Cl 5 37 ClO | HxCDF (S). |
| 303.9016 | M | C 12 H 4 35 Cl 4 O | TeCDF | 389.8157 | M 2 | C 12 H 2 35 Cl 5 37 ClO 2 | HxCDD. |
| 305.8987 | M 2 | C 12 H 4 35 Cl 37 ClO | TeCDF | 391.8127 | M 4 | C 12 H 2 35 Cl 4 37 Cl 2 O 2 | HxCDD. |
| 313.9839 | QC | C 6 F 12 N | FC43 | 392.9760 | LOCK | C 9 F 15 | PFK. |
| 315.9419 | M | 13 C 12 H 4 35 Cl 4 O | TeCDF (S) | 401.8559 | M 2 | 13 C 12 H 2 35 Cl 5 37 ClO 2 | HxCDD (S). |
| 316.9745 | M 2 | 13 C 12 H 4 35 Cl 3 37 ClO | TeCDF (S) | 403.8529 | M 4 | 13 C 12 H 2 35 Cl 4 37 Cl 2 O | HxCDD (S). |
| 317.9389 | M 2 | 13 C 12 H 4 35 Cl 2 37 ClO | TeCDF (S) | 425.9775 | QC | C 9 F 16 N | FC43. |
| 319.8965 | M | C 12 H 4 35 Cl 4 O 2 | TeCDD | 445.7555 | M 4 | C 12 H 2 35 Cl 6 37 Cl 2 O | OCDPE. |
| 321.8936 | M 2 | C 12 H 4 35 Cl 3 37 ClO 2 | TeCDD | 407.7818 | M 2 | C 12 H 35 Cl 6 37 ClO | HpCDF. |
| 325.9839 | QC | C 7 F 12 N | FC43 | 409.7789 | M 4 | C 12 H 35 Cl 5 37 Cl 2 O | HpCDF. |
| 330.9792 | QC | C 7 F 13 | PFK | 417.8253 | M | 13 C 12 H 35 Cl 7 O | HpCDF (S). |
| 331.9368 | M | 13 C 12 H 4 35 Cl 4 O 2 | TeCDD (S) | 419.8220 | M 2 | 13 C 12 H 35 Cl 6 37 ClO | HpCDF (S). |
| 333.9339 | M 2 | 13 C 12 H 4 35 Cl 3 37 ClO 2 | TeCDD (S) | 423.7766 | M 2 | C 12 H 35 Cl 6 37 ClO 2 | HpCDD. |
| 339.8597 | M 2 | C 12 H 3 35 Cl 4 37 ClO | PeCDF | 425.7737 | M 4 | C 12 H 35 Cl 5 37 Cl 2 O 2 | HpCDD. |
| 341.8567 | M 4 | C 12 H 3 35 Cl 3 37 Cl 2 O | PeCDF | 430.9729 | QC | C 9 F 17 | PFK. |
| 354.9792 | LOCK | C 9 F 13 | PFK | 435.8169 | M 2 | 13 C 12 H 35 Cl 6 37 ClO 2 | HpCDD (S). |
| 351.9000 | M 2 | 13 C 12 H 3 35 Cl 4 37 ClO | PeCDF (S) | 437.8140 | M 4 | 13 C 12 H 35 Cl 5 37 Cl 2 O 2 | HpCDD (S). |
| 353.8970 | M 4 | 13 C 12 H 3 35 Cl 3 37 Cl 2 O | PeCDF (S) | 442.9728 | LOCK | C 10 F 17 | PFK. |
| 355.8546 | M 2 | C 12 H 3 35 Cl 4 37 ClO 2 | PeCDD | 479.7165 | M 4 | C 12 H 35 Cl 7 37 Cl 2 O | NCPDE. |
| 357.8516 | M 4 | C 12 H 3 35 Cl 3 37 Cl 2 O 2 | PeCDD | 430.9729 | LOCK | C 9 F 17 | PFK. |
| 367.8949 | M 2 | 13 C 12 H 3 35 Cl 4 37 ClO 2 | PeCDD (S) | 441.7428 | M 2 | C 12 35 Cl 7 37 ClO | OCDF. |
| 369.8919 | M 4 | 13 C 12 H 3 35 Cl 3 37 Cl 2 O 2 | PeCDD (S) | 443.7399 | M 4 | C 12 35 Cl 6 37 Cl 2 O | OCDF. |
| 375.9807 | QC | C 8 F 14 N | FC43 | 457.7377 | M 2 | C 12 35 Cl 7 37 ClO 2 | OCDD. |
| 375.8364 | M 2 | C 12 H 4 35 Cl 5 37 ClO | HxCDPE | 459.7348 | M 4 | C 12 35 Cl 6 37 Cl 2 O 2 | OCDD. |
| 409.7974 | M 2 | C 12 H 3 35 Cl 6 37 ClO | HpCPDE | 463.9743 | QC | C 9 F 18 N | FC43. |
| 373.8208 | M 2 | C 12 H 2 35Cl 5 37 ClO | HxCDF | 469.7779 | M 2 | 13 C 12 35 Cl 7 37 ClO 2 | OCDD (S). |
| 375.8178 | M 4 | C 12 H 2 35 Cl 4 37 Cl 2 O | HxCDF | 471.7750 | M 4 | 13 C 12 35 Cl 6 37 Cl 2 O 2 | OCDD (S). |
| 375.9807 | QC | C 8 F 14 N | FC43 | 513.6775 | M 4 | C 12 35 Cl 8 37 Cl 2 O 2 | DCDPE. |
| 442.9728 | QC | C 10 F 17 | PFK. | ||||
| Aromatic ring No. | Mass a | Ion type b | Elemental composition | Target analyte |
|---|---|---|---|---|
| a Isotopic masses used for accurate mass calculation: 1 H = 1.0078, 12 C = 12.0000, 13 C = 13.0034, 2 H = 2.0141. | ||||
| b LOCK = Lock-Mass Ion PFK or FC43. QC = Quality Control Check Ion. | ||||
| 2 | 128.0624 | M | C 10 H 8 | Naphthalene. |
| 130.9920 | LOCK | PFK/FC43. | ||
| 2 | 134.0828 | M | 13 C 6 12 C 4 H 8 | 13 C 6 -Naphthalene. |
| 2 | 142.078 | M | C 11 H 10 | 2-Methylnaphthalene. |
| 2 | 148.0984 | M | 13 C 6 12 C 5 H 10 | 13 C 6 -2-Methylnaphthalene. |
| 2 | 152.0624 | M | C 12 H 8 | Acenaphthylene. |
| 2 | 158.0828 | M | 13 C 6 12 C 6 H 8 | 13 C 6 -Acenaphthylene. |
| 2 | 154.078 | M | C 12 H 10 | Acenaphthene. |
| 2 | 160.078 | M | 13 C 6 12 C 6 H 10 | 13 C 6 -Acenaphthene. |
| 2 | 166.078 | M | C 13 H 10 | Fluorene. |
| 169.988 | QC | PFK/FC43. | ||
| 2 | 172.0984 | M | 13 C 6 12 C 7 H 10 | 13 C 6 -Fluorene. |
| 3 | 178.078 | M | C 14 H 10 | Phenanthrene. |
| 3 | 184.0984 | M | 13 C 6 12 C 8 H 10 | 13 C 6 -Phenanthrene. |
| 3 | 178.078 | M | C 14 H 10 | Anthracene. |
| 3 | 184.078 | M | 13 C 6 12 C 8 H 10 | 13 C 6 -Anthracene. |
| 3 | 202.078 | M | C 16 H 10 | Fluoranthene. |
| 204.9888 | QC | PFK. | ||
| 3 | 208.0984 | M | 13 C 6 12 C 10 H 10 | 13 C 6 -Fluoranthene. |
| 4 | 202.078 | M | C 16 H 10 | Pyrene. |
| 4 | 205.078 | M | 13 C 3 12 C 13 H 10 | 13 C 3 -Pyrene. |
| 213.9898 | QC | FC43. | ||
| 218.9856 | LOCK | FC43. | ||
| 4 | 228.0936 | M | C 18 H 12 | Benz[ a ]anthracene. |
| 230.9856 | LOCK | PFK. | ||
| 4 | 234.114 | M | 13 C 6 C 12 H 12 | 13 C 6 -Benz[ a ]anthracene. |
| 4 | 228.0936 | M | C 18 H 12 | Chrysene. |
| 4 | 234.114 | M | 13 C 6 12 C 12 H 12 | 13 C 6 -Chrysene. |
| 4 | 252.0936 | M | C 20 H 12 | Benzo[ b ]fluoranthene. |
| 4 | 258.114 | M | 13 C 6 12 C 14 H 12 | 13 C 6 -Benzo[ b ]fluoranthene. |
| 4 | 252.32 | M | C 20 H 12 | Benzo[ k ]fluoranthene. |
| 4 | 258.114 | M | 13 C 6 12 C 14 H 12 | 13 C 6 -Benzo[ k ]fluoranthene. |
| 5 | 252.0936 | M | C 20 H 12 | Benzo[ e ]pyrene. |
| 5 | 256.1072 | M | 13 C 4 12 C 16 H 12 | 13 C 4 -Benzo[ e ]pyrene. |
| 5 | 256.1072 | M | 13 C 4 12 C 16 H 12 | 13 C 4 -Benzo[ a ]pyrene. |
| 5 | 252.0936 | M | C 20 H 12 | Benzo[ a ]pyrene. |
| 5 | 252.0936 | M | C 20 H 12 | Perylene. |
| 5 | 264.1692 | M | C 20 D 12 | d 12 -Perylene. |
| 268.9824 | QC | PFK. | ||
| 263.9871 | LOCK | FC43. | ||
| 6 | 276.0936 | M | C 22 H 12 | Indeno[ 1,2,3-cd ]pyrene. |
| 6 | 282.114 | M | 13 C 6 12 C 16 H 12 | 13 C 6 -Indeno[ 1,2,3,cd ]pyrene. |
| 5 | 278.1092 | M | C 22 H 14 | Dibenz[ a,h ]anthracene. |
| 280.9824 | LOCK | PFK. | ||
| 5 | 284.1296 | M | 13 C 6 12 C 16 H 14 | 13 C 6 -Dibenz[ a,h ]anthracene. |
| 6 | 276.0936 | M | C 22 H 12 | Benzo[ g,h,i ]perylene. |
| 6 | 288.1344 | M | 13 C 12 12 C 10 H 12 | 13 C 12 -Benzo[ g,h,i ]perylene. |
| 313.9839 | QC | FC43. | ||
| Chlorine substitution | Mass a | Ion type b | Elemental composition | Target analyte | Chlorine substitution | Mass a | Ion type b | Elemental composition | Target analyte |
|---|---|---|---|---|---|---|---|---|---|
| a Isotopic masses used for accurate mass calculation: 1 H = 1.0078, 12 C = 12.0000, 13 C = 13.0034, 35 Cl = 34.9689, 37 Cl = 36.9659, 19 F = 18.9984. An interference with PFK m/z 223.9872 may preclude meeting 10:1 S/N for the DiCB congeners at optional Cal 1 level (Table 23-11). If this interference occurs, 10:1 S/N must be met at the Cal 2 level. | |||||||||
| b LOCK = Lock-Mass Ion PFK or FC43. QC = Quality Control Check Ion. | |||||||||
| Fn-1; Cl-1 | 188.0393 | M | 12 C 12 H 9 35 Cl | Cl-1 PCB | Fn-5; Cl-5,6,7 | 323.8834 | M | 12 C 12 H 5 35 Cl 5 | Cl-5 PCB. |
| 190.0363 | M 2 | 12 C 12 H 9 37 Cl | Cl-1 PCB | 325.8804 | M 2 | 12 C 12 H 5 35 Cl 4 37 Cl | Cl-5 PCB. | ||
| 200.0795 | M | 13 C 12 H 9 35 Cl | 13 C 12 Cl-1 PCB | 327.8775 | M 4 | 12 C 12 H 5 35 Cl 3 37 Cl 2 | Cl-5 PCB. | ||
| 202.0766 | M 2 | 12 C 12 H 9 37 Cl | 13 C 12 Cl-1 PCB | 337.9207 | M 2 | 13 C 12 H 5 35 Cl 4 37 Cl | 13 C 12 Cl-5 PCB. | ||
| 218.9856 | LOCK | C 4 F 9 | PFK | 339.9178 | M 4 | 13 C 12 H 5 35 Cl 3 37 Cl 2 | 13 C 12 Cl-5 PCB. | ||
| Fn-2; Cl-2,3 | 222.0003 | M | 12 C 12 H 8 35 Cl 2 | Cl-2 PCB | 354.9792 | LOCK | C 9 F 13 | PFK. | |
| 223.9974 | M 2 | 12 C 12 H 8 35 Cl 37 Cl | Cl-2 PCB | 359.8415 | M 2 | 12 C 12 H 4 35 Cl 5 37 Cl | Cl-6 PCB. | ||
| 225.9944 | M 4 | 12 C 12 H 8 37 Cl 2 | Cl-2 PCB | 361.8385 | M 4 | 12 C 12 H 4 35 Cl 4 37 Cl 2 | Cl-6 PCB. | ||
| 234.0406 | M | 13 C 12 H 8 35 Cl 2 | 13 C 12 Cl-2 PCB | 363.8356 | M 6 | 12 C 12 H 4 35 Cl 3 37 Cl 3 | Cl-6 PCB. | ||
| 236.0376 | M 2 | 13 C 12 H 8 35 Cl 37 Cl | 13 C 12 Cl-2 PCB | 371.8817 | M 2 | 13 C 12 H 4 35 Cl 5 37 Cl | 13 C 12 Cl-6 PCB. | ||
| 242.9856 | LOCK | C 4 F 9 | PFK | 373.8788 | M 4 | 13 C 12 H 4 35 Cl 4 37 Cl 2 | 13 C 12 Cl-6 PCB. | ||
| 255.9613 | M | 12 C 12 H 7 35 Cl 3 | Cl-3 PCB | 393.8025 | M 2 | 12 C 12 H 3 35 Cl 6 37 Cl | Cl-7 PCB. | ||
| 257.9584 | M 2 | 12 C 12 H 7 35 Cl 2 37 Cl | Cl-3 PCB | 395.7995 | M 4 | 12 C 12 H 3 35 Cl 5 37 Cl 2 | Cl-7 PCB. | ||
| 268.0016 | M | 13 C 12 H 7 35 Cl 3 | 13 C 12 Cl-3 PCB | 397.7966 | M 6 | 12 C 12 H 3 35 Cl 4 37 Cl 3 | 37 Cl 3 Cl-7 PCB. | ||
| 269.9986 | M 2 | 13 C 12 H 7 35 Cl 2 37 Cl | 13 C 12 Cl-3 PCB | 405.8428 | M 2 | 13 C 12 H 3 35 Cl 6 37 Cl | 13 C 12 Cl-7 PCB. | ||
| Fn-3; Cl-3,4,5 | 255.9613 | M | 12 C 12 H 7 35 Cl 3 | Cl-3 PCB | 407.8398 | M 4 | 13 C 12 H 3 35 Cl 5 37 Cl 2 | 13 C 12 Cl-7 PCB. | |
| 257.9584 | M 2 | 12 C 12 H 7 35 Cl 2 37 Cl | Cl-3 PCB | 454.9728 | QC | C 11 F 17 | PFK. | ||
| 259.9554 | M 4 | 12 C 12 H 7 35 Cl 37 Cl 2 | Cl-3 PCB | Fn-6; Cl-7,8,9,10 | 393.8025 | M 2 | 12 C 12 H 3 35 Cl 6 37 Cl | Cl-7 PCB. | |
| 268.0016 | M | 13 C 12 H 7 35 Cl 3 | 13 C 12 Cl-3 PCB | 395.7995 | M 4 | 12 C 12 H 3 35 Cl 5 37 Cl 2 | Cl-7 PCB. | ||
| 269.9986 | M 2 | 13 C 12 H 7 35 Cl 2 37 Cl | 13 C 12 Cl-3 PCB | 397.7966 | M 6 | 12 C 12 H 3 35 Cl 4 37 Cl 3 | Cl-7 PCB. | ||
| 280.9825 | LOCK | C 6 F 11 | PFK | 405.8428 | M 2 | 13 C 12 H 3 35 Cl 6 37 Cl | 13 C 12 Cl-7 PCB. | ||
| 289.9224 | M | 12 C 12 H 6 35 Cl 4 | Cl-4 PCB | 407.8398 | M 4 | 13 C 12 H 3 35 Cl 5 37 Cl 2 | 13 C 12 Cl-7 PCB. | ||
| 291.9194 | M 2 | 12 C 12 H 6 35 Cl 3 37 Cl | Cl-4 PCB | 427.7635 | M 2 | 12 C 12 H 2 35 Cl 7 37 Cl | Cl-8 PCB. | ||
| 293.9165 | M 4 | 12 C 12 H 6 35 Cl 2 37 Cl 2 | Cl-4 PCB | 429.7606 | M 4 | 12 C 12 H 2 35 Cl 6 37 Cl 2 | Cl-8 PCB. | ||
| 301.9626 | M | 13 C 12 H 6 35 Cl 4 | 13 C 12 Cl-4 PCB | 431.7576 | M 6 | 12 C 12 H 2 35 Cl 5 37 Cl 3 | Cl-8 PCB. | ||
| 303.9597 | M 2 | 13 C 12 H 6 35 Cl 3 37 Cl | 13 C 12 Cl-4 PCB | 439.8038 | M 2 | 13 C 12 H 2 35 Cl 7 37 Cl | 13 C 12 Cl-8 PCB. | ||
| 323.8834 | M | 12 C 12 H 5 35 Cl 5 | Cl-5 PCB | 441.8008 | M 4 | 13 C 12 H 2 35 Cl 6 37 Cl 2 | 13 C 12 Cl-8 PCB. | ||
| 325.8804 | M 2 | 12 C 12 H 5 35 Cl 4 37 Cl | Cl-5 PCB | 454.9728 | QC | C 11 F 17 | PFK. | ||
| 327.8775 | M 4 | 12 C 12 H 5 35 Cl 3 37 Cl 2 | Cl-5 PCB | 427.7635 | M 2 | 12 C 12 H 2 35 Cl 7 37 Cl | Cl-8 PCB. | ||
| 337.9207 | M 2 | 13 C 12 H 5 35 Cl 4 37 Cl | 13 C 12 Cl-5 PCB | 429.7606 | M 4 | 12 C 12 H 2 35 Cl 6 37 Cl 2 | Cl-8 PCB. | ||
| 339.9178 | M 4 | 13 C 12 H 5 35 Cl 3 37 Cl 2 | 13 C 12 Cl-5 PCB | 431.7576 | M 6 | 12 C 12 H 2 35 Cl 5 37 Cl 3 | Cl-8 PCB. | ||
| Fn-4; Cl-4,5,6 | 289.9224 | M | 12 C 12 H 6 35 Cl 4 | Cl-4 PCB | 439.8038 | M 2 | 13 C 12 H 2 35 Cl 7 37 Cl | 13 C 12 Cl-8 PCB. | |
| 291.9194 | M 2 | 12 C 12 H 6 35 Cl 3 37 Cl | Cl-4 PCB | 441.8008 | M 4 | 13 C 12 H 2 35 Cl 6 37 Cl 2 | 13 C 12 Cl-8 PCB. | ||
| 293.9165 | M 4 | 12 C 12 H 6 35 Cl 2 37 Cl 2 | Cl-4 PCB | 442.9728 | QC | C 10 F 17 | PFK. | ||
| 301.9626 | M 2 | 13 C 12 H 6 35 Cl 3 37 Cl | 13 C 12 Cl-4 PCB | 454.9728 | LOCK | C 11 F 17 | PFK. | ||
| 303.9597 | M 4 | 13 C 12 H 6 35 Cl 2 37 Cl 2 | 13 C 12 Cl-4 PCB | 461.7246 | M 2 | 12 C 12 H 1 35 Cl 8 37 Cl | Cl-9 PCB. | ||
| 323.8834 | M | 12 C 12 H 5 35 Cl 5 | Cl-5 PCB | 463.7216 | M 4 | 12 C 12 H 1 35 Cl 7 37 Cl 2 | Cl-9 PCB. | ||
| 325.8804 | M 2 | 12 C 12 H 5 35 Cl 4 37 Cl | Cl-5 PCB | 465.7187 | M 6 | 12 C 12 H 1 35 Cl 6 37 Cl 3 | Cl-9 PCB. | ||
| 327.8775 | M 4 | 12 C 12 H 5 35 Cl 3 37 Cl 2 | Cl-5 PCB | 473.7648 | M 2 | 13 C 12 H 1 35 Cl 8 37 Cl | 13 C 12 Cl-9 PCB. | ||
| 330.9792 | LOCK | C 7 F 15 | PFK | 475.7619 | M 4 | 13 C 12 H 1 35 Cl 7 37 Cl 2 | 13 C 12 Cl-9 PCB. | ||
| 337.9207 | M 2 | 13 C 12 H 5 35 Cl 4 37 Cl | 13 C 12 Cl-5 PCB | 495.6856 | M 2 | 13 C 12 H 4 5 Cl 9 37 Cl | Cl-10 PCB. | ||
| 339.9178 | M 4 | 13 C 12 H 5 35 Cl 3 37 Cl 2 | 13 C 12 Cl-5 PCB | 499.6797 | M 6 | 12 C 12 H 4 35 Cl 8 37 Cl 2 | Cl-10 PCB. | ||
| 359.8415 | M 2 | 13 C 12 H 4 35 Cl 5 37 Cl | Cl-6 PCB | 501.6767 | M 8 | 12 C 12 H 4 35 Cl 7 37 Cl 3 | Cl-10 PCB. | ||
| 361.8385 | M 4 | 13 C 12 H 4 35 Cl 4 37 Cl 2 | Cl-6 PCB | 507.7258 | M 2 | 13 C 12 H 4 35 Cl 9 37 Cl | 13 C 12 Cl-10 PCB. | ||
| 363.8356 | M 6 | 12 C 12 H 4 35 Cl 3 37 Cl 3 | Cl-6 PCB | 509.7229 | M 4 | 13 C 12 H 4 35 Cl 8 37 Cl 2 | 13 C 12 Cl-10 PCB. | ||
| 371.8817 | M 2 | 13 C 12 H 4 35 Cl 5 37 Cl | 13 C 12 Cl-6 PCB | 511.7199 | M 6 | 13 C 12 H 4 35 Cl 7 37 Cl 3 | 13 C 12 Cl-10 PCB. | ||
| 373.8788 | M 4 | 13 C 12 H 4 35 Cl 4 37 Cl 2 | 13 C 12 Cl-6 PCB | ||||||
| Compound | pg/μL in final extract b | Spike recovery |
|---|---|---|
| a Changes in the amounts of labeled standards added to the sample or its representative extract will necessitate an adjustment of the calibration solutions to prevent the introduction of inconsistencies. Spike concentration assumes 1 µL sample injection volume for analysis or the injection volume for calibration standards and samples is the same. | ||
| b Labeled standard concentrations are recommendations (equivalent mass per sample of 25 pg pre-extraction standard, as an example, based on a 200 µL extract volume split in half before cleanup with a 20 µL aliquot of a 500 pg/µL spiking solution). Recommendations are based on assumption that half of the extract will be archived before cleanup. Spike levels may be adjusted for different split levels. | ||
| Note: all standards used should be reported. | ||
| Pre-sampling Adsorbent Standard | ||
| 13 C 12 -1,2,3,4-TeCDD | 50 | 70-130% |
| 13 C 12 -1,2,3,4,7-PeCDD | 50 | 70-130% |
| 13 C 12 -1,2,3,4,6-PeCDF | 50 | 70-130% |
| 13 C 12 -1,2,3,4,6,9-HxCDF | 50 | 70-130% |
| 13 C 12 -1,2,3,4,6,8,9-HpCDF | 50 | 70-130% |
| Pre-extraction Filter Recovery Standard | ||
| 13 C 12 -1,2,7,8-TeCDF | 50 | 70-130% |
| 13 C 12 -1,2,3,4,6,8-HxCDD | 50 | 70-130% |
| Pre-extraction Standard | ||
| 13 C 12 -2,3,7,8-TeCDD | 50 | 20-130% |
| 13 C 12 -2,3,7,8-TeCDF | 50 | 20-130% |
| 13 C 12 -1,2,3,7,8-PeCDD | 50 | 20-130% |
| 13 C 12 -1,2,3,7,8-PeCDF | 50 | 20-130% |
| 13 C 12 -2,3,4,7,8-PeCDF | 50 | 20-130% |
| 13 C 12 -1,2,3,4,7,8-HxCDD | 50 | 20-130% |
| 13 C 12 -1,2,3,6,7,8-HxCDD | 50 | 20-130% |
| 13 C 12 -1,2,3,7,8,9-HxCDD | 50 | 20-130% |
| 13 C 12 -1,2,3,4,7,8-HxCDF | 50 | 20-130% |
| 13 C 12 -1,2,3,6,7,8-HxCDF | 50 | 20-130% |
| 13 C 12 -2,3,4,6,7,8-HxCDF | 50 | 20-130% |
| 13 C 12 -1,2,3,7,8,9-HxCDF | 50 | 20-130% |
| 13 C 12 -1,2,3,4,6,7,8-HpCDD | 50 | 20-130% |
| 13 C 12 -1,2,3,4,6,7,8-HpCDF | 50 | 20-130% |
| 13 C 12 -1,2,3,4,7,8,9-HpCDF | 50 | 20-130% |
| 13 C 12 -OCDD | 100 | 20-130% |
| 13 C 12 -OCDF | 100 | 20-130% |
| Pre-analysis Standard | ||
| 13 C 12 -1,3,6,8-TeCDD | 50 | S/N≥10 |
| 13 C 12 -1,2,3,4-TeCDF | 50 | S/N≥10 |
| 13 C 12 -1,2,3,4,6,7-HxCDD | 50 | S/N≥10 |
| 13 C 12 -1,2,3,4,6,7,9-HpCDD | 50 | S/N≥10 |
| Alternate Recovery Standard | ||
| 13 C 12 -1,3,7,8-TeCDD | 50 | 20-130% |
| 13 C 12 -1,2,4,7,8-PeCDD | 50 | 20-130% |
| Compound | pg/µL in final extract b | Spike recovery |
|---|---|---|
| a Changes in the amounts of labeled standards added to the sample or its representative extract will necessitate an adjustment of the calibration solutions to prevent the introduction of inconsistencies. | ||
| b Labeled standard concentrations are recommendations (equivalent mass per sample of 25 pg pre-extraction standard, as an example, based on a 200 µL extract volume split in half before cleanup with a 20 µL aliquot of a 1000 pg/µL spiking solution). Recommendations are based on assumption that half of the extract will be archived before cleanup. Spike levels may be adjusted for different split levels. | ||
| Note: all standards used should be reported. | ||
| Pre-sampling Adsorbent Standard | ||
| 13 C 6 -Benzo[ c ]fluorene | 100 | 70-130% |
| 13 C 12 -Benzo[ j ]fluoranthene | 100 | 70-130% |
| Pre-extraction Filter Recovery Standard | ||
| d 10 -Anthracene | 100 | 70-130% |
| Pre-extraction Standard | ||
| 13 C 6 -Naphthalene | 100 | 20-130% |
| 13 C 6 -2-Methylnaphthalene | 100 | 20-130% |
| 13 C 6 -Acenaphthylene | 100 | 20-130% |
| 13 C 6 -Acenaphthene | 100 | 20-130% |
| 13 C 6 -Fluorene | 100 | 20-130% |
| 13 C 6 -Phenanthrene | 100 | 20-130% |
| 13 C 6 -Anthracene | 100 | 20-130% |
| 13 C 6 -Fluoranthene | 100 | 20-130% |
| 13 C 3 -Pyrene | 100 | 20-130% |
| 13 C 6 -Benz[ a ]anthracene | 100 | 20-130% |
| 13 C 6 -Chrysene | 100 | 20-130% |
| 13 C 6 -Benzo[ b ]fluoranthene | 100 | 20-130% |
| 13 C 6 -Benzo[ k ]fluoranthene | 100 | 20-130% |
| 13 C 4 -Benzo[ e ]pyrene | 100 | 20-130% |
| 13 C 4 -Benzo[ a ]pyrene | 100 | 20-130% |
| d 12 -Perylene | 100 | 20-130% |
| 13 C 6 -Indeno[ 1,2,3-cd ]pyrene | 100 | 20-130% |
| 13 C 6 -Dibenz[ a,h ]anthracene | 100 | 20-130% |
| 13 C 12 -Benzo[ g,h,i ]perylene | 100 | 20-130% |
| Pre-analysis Standard | ||
| d 10 -Acenaphthene | 100 | S/N≥10 |
| d 10 -Pyrene | 100 | S/N≥10 |
| d 12 -Benzo[ e ]pyrene | 100 | S/N≥10 |
| Compound | BZ No. b | pg/µL in final extract c | Spike recovery |
|---|---|---|---|
| a Changes in the amounts of spike standards added to the sample or its representative extract will necessitate an adjustment of the calibration solutions to prevent the introduction of inconsistencies. | |||
| b BZ No.: Ballschmiter and Zell 1980, or IUPAC number. | |||
| c Labeled standard concentrations are recommendations (equivalent mass per sample of 25 pg pre-extraction standard, as an example, based on a 200 µL extract volume split in half before cleanup with a 20 µL aliquot of a 1000 pg/µL spiking solution). Recommendations are based on assumption that half of the extract will be archived before cleanup. Spike levels may be adjusted for different split levels. | |||
| NOAAT = PCB considered toxic by the National Oceanic and Atmospheric Administration. | |||
| WHOT = PCB considered toxic by the World Health Organization. | |||
| Note: all standards used should be reported. | |||
| Pre-sampling Adsorbent Standard | |||
| 13 C 12 -3,3′-DiCB | 11L | 100 | 70-130% |
| 13 C 12 -2,4′,5-TrCB | 31L | 100 | 70-130% |
| 13 C 12 -2,2′,3,5′,6-PeCB | 95L | 100 | 70-130% |
| 13 C 12 -2,2′,4,4′,5,5′-HxCB | 153L | 100 | 70-130% |
| Pre-extraction Filter Recovery Standard | |||
| 13 C 12 -2,3,3′,4,5,5′-HxCB | 159L | 100 | 70-130% |
| Pre-extraction Standard | |||
| 13 C 12 -2-MoCB (WDC) | 1L | 100 | 20-145% |
| 13 C 12 -4-MoCB (WDC) | 3L | 100 | 20-145% |
| 13 C 12 -2,2′-DiCB (WDC) | 4L | 100 | 20-145% |
| 13 C 12 -4,4′-DiCB (WDC) | 15L | 100 | 20-145% |
| 13 C 12 -2,2′,6-TrCB (WDC) | 19L | 100 | 20-145% |
| 13 C 12 -3,4′,4′-TrCB (WDC) | 37L | 100 | 20-145% |
| 13 C 12 -2,2′,6,6′-TeCB (WDC) | 54L | 100 | 20-145% |
| 13 C 12 -3,3′,4,4′-TeCB (WDC) (WHOT) (NOAAT) | 77L | 100 | 20-145% |
| 13 C 12 -3,4,4′,5-TeCB (WHOT) | 81L | 100 | 20-145% |
| 13 C 12 -2,2′,4,6,6′-PeCB (WDC) | 104L | 100 | 20-145% |
| 13 C 12 -2,3,3′,4,4′-PeCB (WHOT) | 105L | 100 | 20-145% |
| 13 C 12 -2,3,4,4′,5-PeCB (WHO) | 114L | 100 | 20-145% |
| 13 C 12 -2,3′,4,4′,5-PeCB (WHOT) | 118L | 100 | 20-145% |
| 13 C 12 -2′,3,4,4′,5-PeCB (WHOT) | 123L | 100 | 20-145% |
| 13 C 12 -3,3′,4,4′,5-PeCB (WDC) (WHOT) | 126L | 100 | 20-145% |
| 13 C 12 -2,2′,4,4′,6,6′-HxCB (WDC) | 155L | 100 | 20-145% |
| 13 C 12 -2,3,3′,4,4′,5-HxCB (WHOT) | 156L | 100 | 20-145% |
| 13 C 12 -2,3,3′,4,4′,5′-HxCB (WHOT) | 157L | 100 | 20-145% |
| 13 C 12 -2,3′,4,4′,5,5′-HxCB (WHOT) | 167L | 100 | 20-145% |
| 13 C 12 -3,3′,4,4′,5,5′-HxCB (WDC) (WHOT) (NOAAT) | 169L | 100 | 20-145% |
| 13 C 12 -2,2′,3,3′,4,4′,5′-HpCB (NOAAT) | 170L | 100 | 20-145% |
| 13 C 12 -2,2′,3,4,4′,5,5′-HpCB (NOAAT) | 180L | 100 | 20-145% |
| 13 C 12 -2,2′,3,4′,5,6,6′-HpCB (WDC) | 188L | 100 | 20-145% |
| 13 C 12 -2,3,3′,4,4′,5,5′-HpCB (WDC) (WHOT) | 189L | 100 | 20-145% |
| 13 C 12 -2,2′,3′,3′,5,5′,6,6′-OcCB (WDC) | 202L | 100 | 20-145% |
| 13 C 12 -2,3′,3′,4,4′,5,5′,6-OcCB (WDC) | 205L | 100 | 20-145% |
| 13 C 12 -2,2′,3,3′,4,4′,5,5′,6-NoCB (WDC) | 206L | 100 | 20-145% |
| 13 C 12 -2,2′,3,3′,4,5,5′,6,6′-NoCB (WDC) | 208L | 100 | 20-145% |
| 13 C 12 -DeCB (WDC) | 209L | 100 | 20-145% |
| Pre-analysis Standard | |||
| 13 C 12 -2,5-DiCB | 9L | 100 | S/N≥10 |
| 13 C 12 -2,2′,5,5′-TeCB (NOAAT) | 52L | 100 | S/N≥10 |
| 13 C 12 -2,2′,4,5,5′-PeCB (NOAAT) | 101L | 100 | S/N≥10 |
| 13 C 12 -2,2′,3,4,4′,5′-HxCB (NOAAT) | 138L | 100 | S/N≥10 |
| 13 C 12 -2,2′,3,3′,4,4′,5,5′-OcCB | 194L | 100 | S/N≥10 |
| Optional Cleanup Standard | |||
| 13 C 12 -2-MoCB (NOAAT) | 28L | 100 | 20-130% |
| 13 C 12 -2,2′,4,5,5′-PeCB | 111L | 100 | 20-130% |
| 13 C 12 -2,2′,3,3′,5,5′,6,6′-OcCB | 178L | 100 | 20-130% |
| Alternate Recovery Standard | |||
| 13 C 12 -2,3′,4′,5-TeCB | 70L | 100 | 20-130% |
| 13 C 12 -2,3,4,4′-TeCB | 60L | 100 | 20-130% |
| 13 C 12 -3,3′,4,5,5′-PeCB | 127L | 100 | 20-130% |
| Sample type | PCDD/PCDF | PAH | PCB |
|---|---|---|---|
| a Samples and extracts must be stored in the dark. | |||
| b Hold times begin from the time the laboratory receives the sample. | |||
| c Room temperature is acceptable if PCDD/PCDF are the only target compounds. | |||
| Note: Hold times for PCDD/PCDF and PCB are recommendations. | |||
| Field Storage and Shipping Conditions | |||
| All Field Samples | ≤20 °C, (68 °F) | ≤20 °C, (68 °F) | ≤20 °C, (68 °F). |
| Laboratory Storage Conditions | |||
| Sampling Train Rinses and Filters | ≤6 °C (43 °F) | ≤6 °C (43 °F) | ≤6 °C (43 °F). |
| Adsorbent | ≤6 °C (43 °F) | ≤6 °C (43 °F) | ≤6 °C (43 °F). |
| Extract and Archive | <26 °C (79 °F) c | <−10 °C (14 °F) | <−10 °C (14 °F). |
| Laboratory Hold Times | |||
| Extract and Archive | One year | 45 Days | One year. |
| Standard compound | Cal 1 (optional) | Cal 2 | Cal 3 | Cal 4 | Cal 5 | Cal 6 | Cal 7 (optional) |
|---|---|---|---|---|---|---|---|
| a Assumes 1 μL injection volume or the injection volume for standards and samples is the same. | |||||||
| Target (Unlabeled) Analytes | 0.50 | 1.0 | 5.0 | 10.0 | 25 | 50 | 100 |
| Pre-sampling Adsorbent Standard | 50 | 50 | 50 | 50 | 50 | 50 | 50 |
| Pre-extraction Filter Recovery Standard | 50 | 50 | 50 | 50 | 50 | 50 | 50 |
| Pre-extraction Standard ( 13 C 12 -OCDD, 13 C 12 -OCDF−100 pg/μL) | 50 | 50 | 50 | 50 | 50 | 50 | 50 |
| Pre-analysis Standard | 50 | 50 | 50 | 50 | 50 | 50 | 50 |
| Alternate Recovery Standard | 50 | 50 | 50 | 50 | 50 | 50 | 50 |
| Standard compound | Cal 1 (optional) | Cal 2 | Cal 3 | Cal 4 | Cal 5 | Cal 6 | Cal 7 (optional) |
|---|---|---|---|---|---|---|---|
| a Assumes 1 μL injection volume. | |||||||
| Target (Unlabeled) Analytes | 1 | 2 | 4 | 20 | 80 | 400 | 1,000 |
| Pre-sampling Adsorbent Standard | 100 | 100 | 100 | 100 | 100 | 100 | 100 |
| Pre-extraction Filter Recovery Standard | 100 | 100 | 100 | 100 | 100 | 100 | 100 |
| Pre-extraction Standard | 100 | 100 | 100 | 100 | 100 | 100 | 100 |
| Pre-analysis Standard | 100 | 100 | 100 | 100 | 100 | 100 | 100 |
| Standard compound | Cal 1 (optional) | Cal 2 | Cal 3 | Cal 4 | Cal 5 | Cal 6 | Cal 7 (optional) |
|---|---|---|---|---|---|---|---|
| a Assumes 1 μL injection volume. | |||||||
| Target (Unlabeled) Analytes | 0.50 | 1 | 5 | 10 | 50 | 400 | 2,000 |
| Pre-sampling Adsorbent Standard | 100 | 100 | 100 | 100 | 100 | 100 | 100 |
| Pre-extraction Filter Recovery Standard | 100 | 100 | 100 | 100 | 100 | 100 | 100 |
| Pre-extraction Standard | 100 | 100 | 100 | 100 | 100 | 100 | 100 |
| Pre-analysis Standard | 100 | 100 | 100 | 100 | 100 | 100 | 100 |
| Alternate Standard | 100 | 100 | 100 | 100 | 100 | 100 | 100 |
| Analyte group | Initial calibration RRF RSD | Continuing calibration RRF compared to ICAL RRF (PD) |
|---|---|---|
| Native (Unlabeled) Analytes | 10 | 25 |
| Pre-sampling Adsorbent Standard | 20 | 25 |
| Pre-extraction Filter Recovery Standard | 20 | 25 |
| Pre-extraction Standard | 20 | 30 |
| Alternative Recovery Standard | 20 | 30 |
| Number of chlorine atoms | Ion type | Theoretical ratio | Lower control limit | Upper control limit |
|---|---|---|---|---|
| a Used only for 13 C-HxCDF. | ||||
| b Used only for 13 C-HpCDF. | ||||
| 1 | M/M 2 | 3.13 | 2.66 | 3.60 |
| 2 | M/M 2 | 1.56 | 1.33 | 1.79 |
| 3 | M/M 2 | 1.04 | 0.88 | 1.20 |
| 4 | M/M 2 | 0.77 | 0.65 | 0.89 |
| 5 | M 2/M 4 | 1.55 | 1.32 | 1.78 |
| 6 | M 2/M 4 | 1.24 | 1.05 | 1.43 |
| 6 a | M/M 2 | 0.51 | 0.43 | 0.59 |
| 7 | M 2/M 4 | 1.05 | 0.89 | 1.21 |
| 7 b | M/M 2 | 0.44 | 0.37 | 0.51 |
| 8 | M 2/M 4 | 0.89 | 0.76 | 1.02 |
| 9 | M 2/M 4 | 0.77 | 0.65 | 0.89 |
| 10 | M 4/M 6 | 1.16 | 0.99 | 1.33 |
| Column parameter | PCDD/PCDF | PAH | PCB |
|---|---|---|---|
| Injector temperature | 250 °C | 320 °C | 270 °C. |
| Initial oven temperature | 100 °C | 100 °C | 100 °C. |
| Initial hold time (minutes) | 2 | 2 | 2. |
| Temperature program | 100 to 190 °C at 40 °C/min, then 190 to 300 °C at 3°C/min | 100 to 300 °C at 8°C/min | 100 to 150 °C at 15 °C/min, then 150 to 290 °C at 2.5 °C/min. |
| PCB Congener | BZ No. a | Labeled analog | BZ No. |
|---|---|---|---|
| a BZ No.: Ballschmiter and Zell 1980, or IUPAC number. | |||
| b Assignments assume the use of the SPB-Octyl column. In the event you choose another column, you may select the labeled standard having the same number of chlorine substituents and the closest retention time to the target analyte in question as the labeled standard to use for quantitation. | |||
| NOAAT = PCB considered toxic by the National Oceanic and Atmospheric Administration. | |||
| WHOT = PCB considered toxic by the World Health Organization. | |||
| 2,4′-DiCB (NOAAT) | 8 | 13 C 12 -2,2′-DiCB | 4L |
| 2,2′,5-TrCB (NOAAT) | 18 | 13 C 12 -2,2′,6-TrCB | 19L |
| 2,4,4′-TrCB (NOAAT) | 28 | 13 C 12 -2,2′,6-TrCB | 19L |
| 2,2′,3,5′-TeCB (NOAAT) | 52 | 13 C 12 -2,2′,6,6′-TeCB | 54L |
| 2,2′,5,5′-TeCB (NOAAT) | 52 | 13 C 12 -2,2′,6,6′-TeCB | 54L |
| 2,3′,4,4′-TeCB (NOAAT) | 66 | 13 C 12 -2,2′,6,6′-TeCB | 54L |
| 3,3′,4,4′-TeCB (NOAAT) (WHOT) | 77 | 13 C 12 -3,3′,4,4′-TeCB | 77L |
| 3,4,4′,5-TeCB (WHOT) | 81 | 13 C 12 -3,4,4″,5-TeCB | 81L |
| 2,2′,4,5,5′-PeCB (NOAAT) | 101 | 13 C 12 -2,2′,4,5,5′-PeCB | 104L |
| 2,3,3′,4,4′-PeCB (NOAAT) (WHOT) | 105 | 13 C 12 -2,3,3′,4,4′-PeCB | 105L |
| 2,3,4,4′,5-PeCB (WHOT) | 114 | 13 C 12 -2,3,4,4′,5-PeCB | 114L |
| 2,3′,4,4′,5-PeCB (WHOT) | 118 | 13 C 12 -2,3′,4,4′,5-PeCB | 118L |
| 2′,3,4,4′,5-PeCB (WHOT) | 123 | 13 C 12 -2′,3,4,4′,5-PeCB | 123L |
| 3,3′,4,4′,5-PeCB (NOAAT) (WHOT) | 126 | 13 C 12 -3,3′,4,4′,5-PeCB | 126L |
| 2,2′,3,3′,4,4′-HxCB (NOAAT) | 128 | 13 C 12 -2,2′,4,4′,6,6′-HxCB | 155L |
| 2,2′,3,4,4′,5′-HxCB (NOAAT) | 138 | 13 C 12 -2,2′,4,4′,6,6′-HxCB | 155L |
| 2,2′,4,4′,5,5′-HxCB (NOAAT) | 153 | 13 C 12 -2,2′,4,4′,6,6′-HxCB | 155L |
| 2,3,3′,4,4′,5-HxCB (WHOT) | 156 | 13 C 12 -2,3,3′,4,4′,5-HxCB | 156L |
| 2,3,3′,4,4′,5′-HxCB (WHOT) | 157 | 13 C 12 -2,3,3′,4,4′,5′-HxCB | 157L |
| 2,3′,4,4′,5,5′-HxCB (WHOT) | 167 | 13 C 12 -2,3′,4,4′,5,5′-HxCB | 167L |
| 3,3′,4,4′,5,5′-HxCB (NOAAT) (WHOT) | 169 | 13 C 12 -3,3′,4,4′,5,5′-HxCB | 169L |
| 2,2′,3,3′,4,4′,5-HpCB (NOAAT) | 170 | 13 C 12 -2,2′,3,3′,4,4′,5′-HpCB | 170L |
| 2,2′,3,4,4′,5,5′-HpCB (NOAAT) | 180 | 13 C 12 -2,2′,3,4,4′,5,5′-HpCB | 180L |
| 2,2′,3,4′,5,5′,6-HpCB (NOAAT) | 187 | 13 C 12 -2,2′,3,4′,5,6,6′-HpCB | 188L |
| 2,3,3′,4,4′,5,5′-HpCB (WHOT) | 189 | 13 C 12 -2,3,3′,4,4′,5,5′-HpCB | 189L |
| 2,2′,3,3′,4,4′,5,6-OcCB (NOAAT) | 195 | 13 C 12 -2,2′ 3,3′,5,5′,6,6′-OcCB | 202L |
| 2,2′,3,3′,4,4′,5,5′,6-NoCB (NOAAT) | 206 | 13 C 12 -2,2′,3,3′,4,4′,5,5′,6-NoCB | 206L |
| 2,2′,3,3′,4,4′,5,5′,6,6′-DeCB (NOAAT) | 209 | 13 C 12 -DeCB | 209L |
| Section | Requirement | Specification and frequency | Acceptance criteria |
|---|---|---|---|
| 9.3.5 | Demonstration of low system background | Analyze an LMB after the highest calibration standard Note: If an automated extraction system is used, an LMB must be extracted on each port | Confirm that the LMB is free from contamination as defined in Section 13.1. |
| 9.3.7 | Determination of MDL | Prepare, extract, and analyze 7 replicate spiked samples (spiked within 2 to 10 times of the expected MDL) and 7 LMBs See 40 CFR Part 136 Appendix B | See MDL confirmation. |
| 9.3.8 | MDL confirmation | Prepare, extract, and analyze a spiked sample (spiked at the MDL) | Confirm that the target compounds meet the qualitative identification criteria in Section 11.4.3.4 of this method. |
| 9.3.9 | Demonstration of precision | Prepare, extract, and analyze 7 replicate spiked samples (spiked near mid-range) | Percent relative standard deviation must be ≤20%. |
| 9.3.10 | Demonstration of accuracy | Calculate mean recovery for replicate spiked samples in Section 9.3.9 | Mean recovery within 70-130% of true value. |
| 9.3.2 | Lowest Calibration Concentration Confirmation | Establish a target concentration for the lowest calibration based on the intended use of the method | Upper PIR ≤150%. Lower PIR ≥50%. |
| 9.3.6 | Calibration Verification | Analyze a mid-level QCS | Within limits in Section 13.11. |
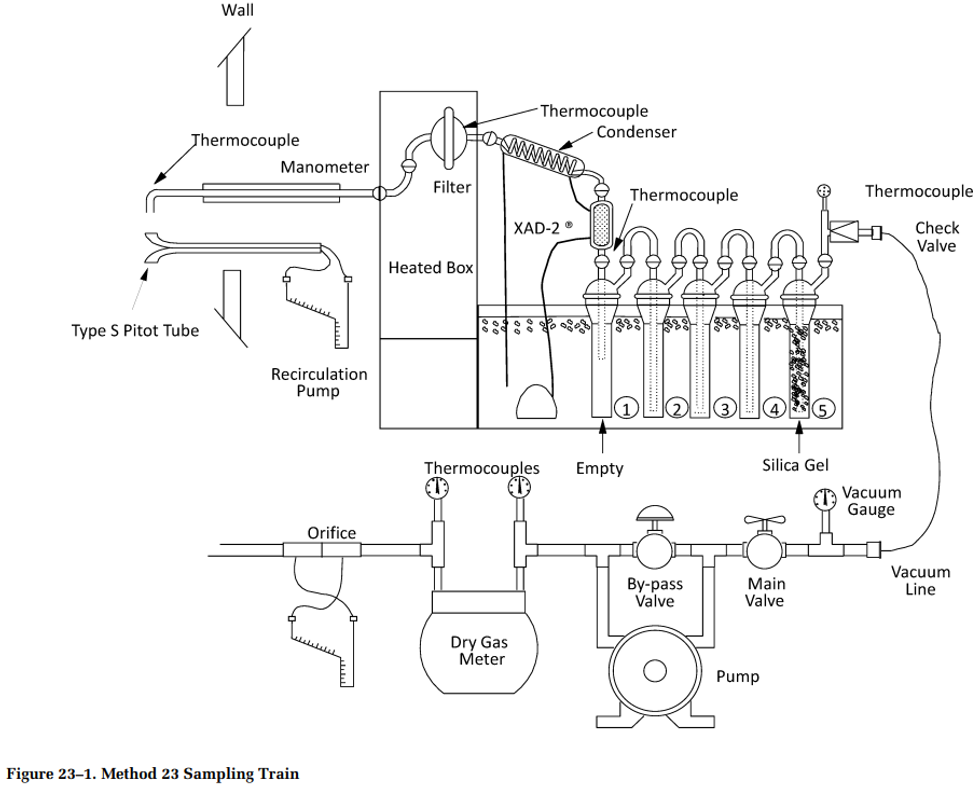
Figure 23-1. Method 23 Sampling Train
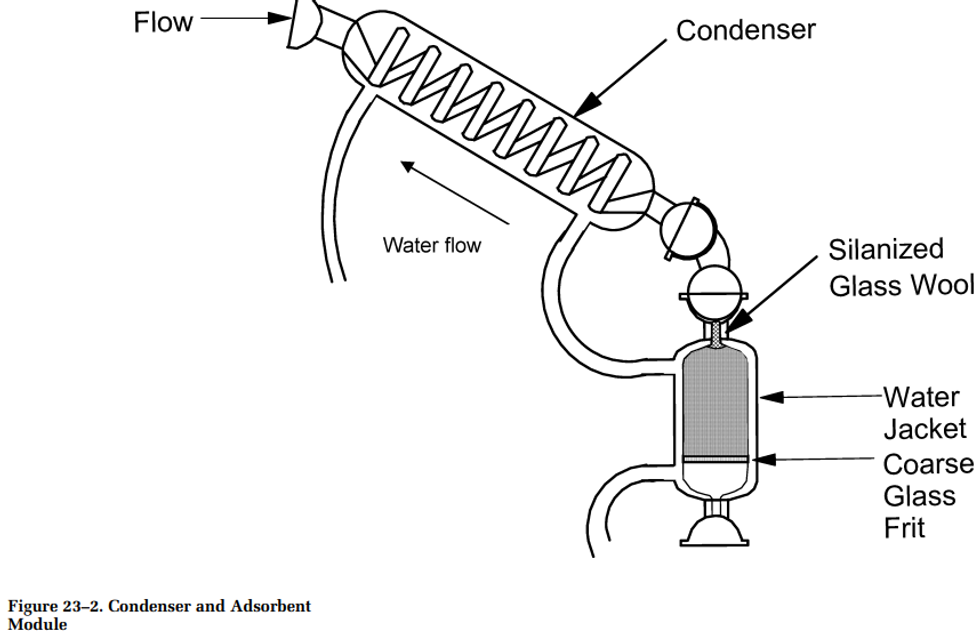
Figure 23-2. Condenser and Adsorbent Module
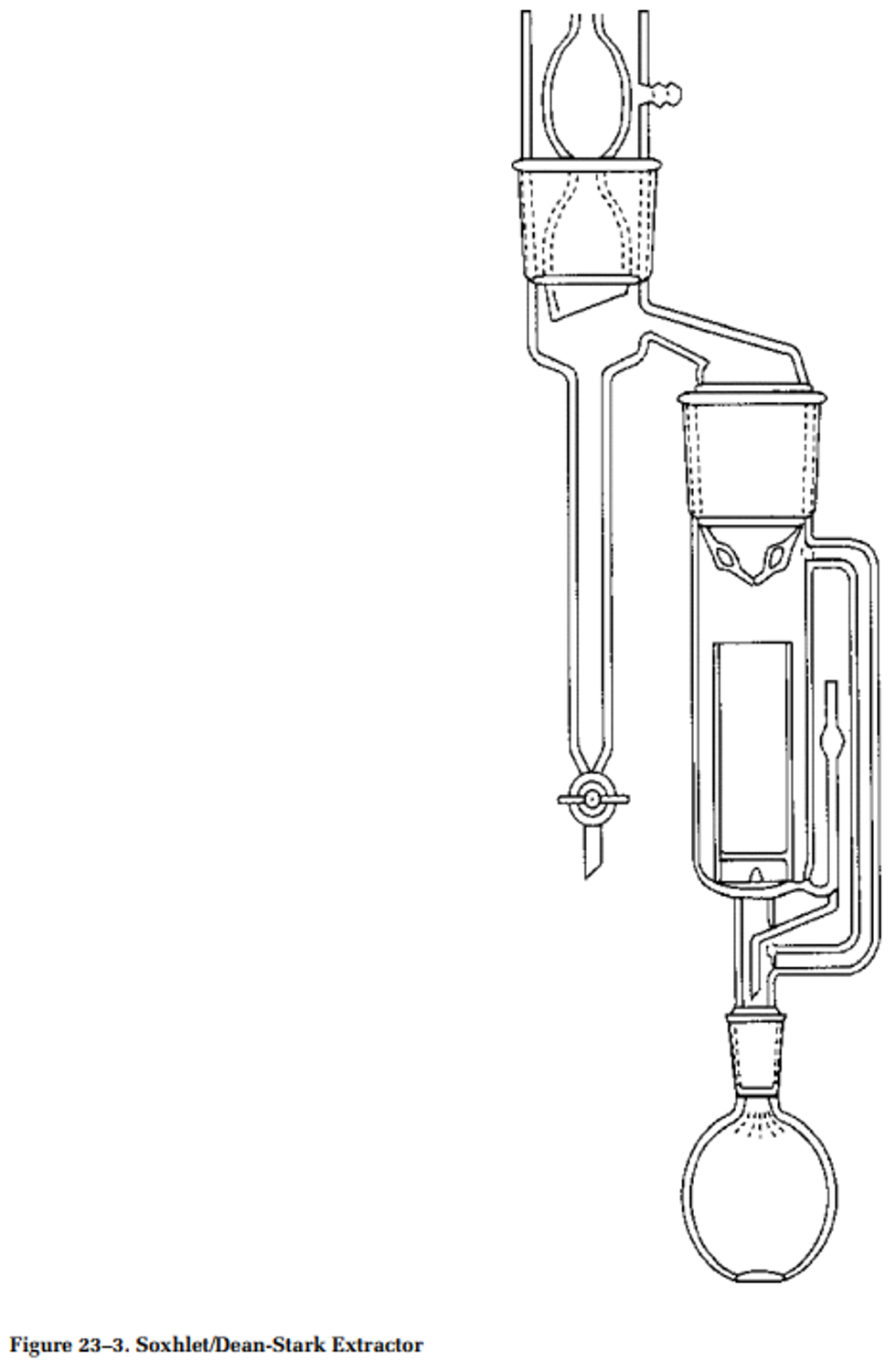
Figure 23-3. Soxhlet/Dean-Stark Extractor
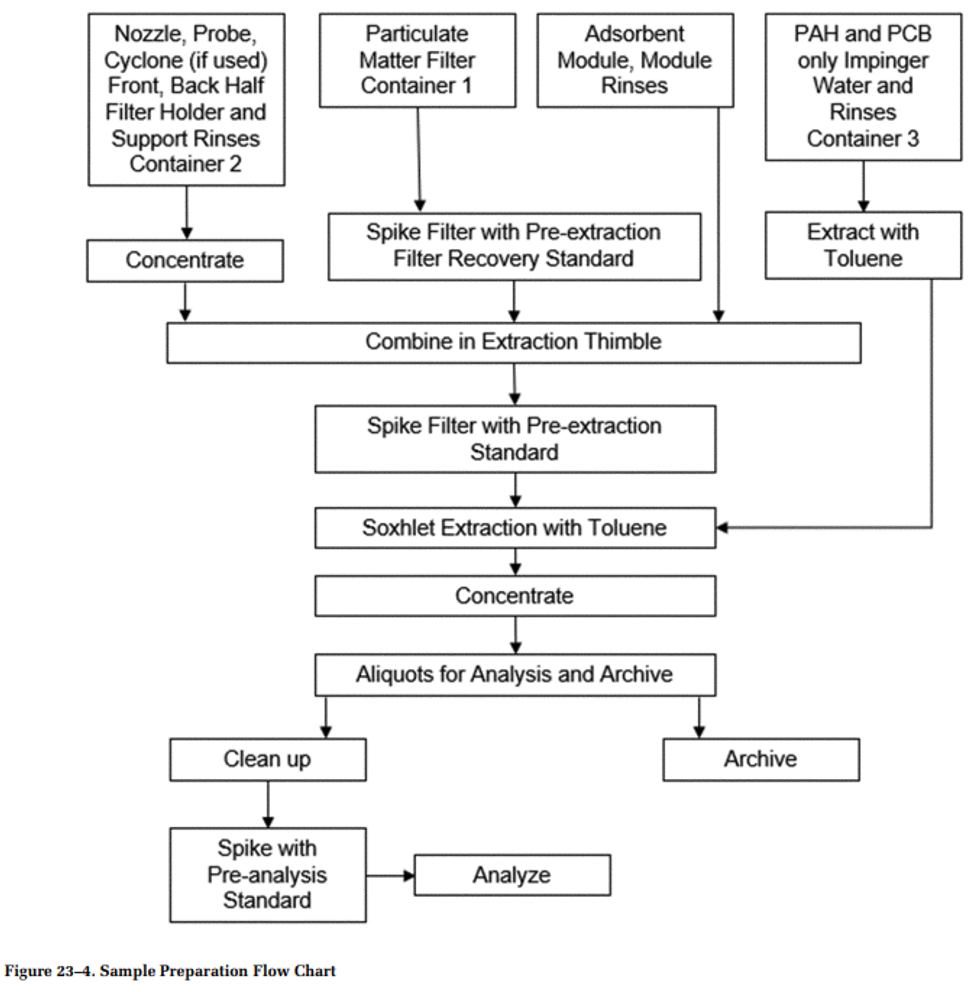
Figure 23-4. Sample Preparation Flow Chart
Appendix A to Method 23
| Pre-extraction standard | BZ b No. | Unlabeled target analyte | BZ b No. | Pre-extraction standard | BZ b No. | Unlabeled target analyte | BZ b No. |
|---|---|---|---|---|---|---|---|
| a Assignments assume the use of the SPB-Octyl column. In the event you choose another column, you may select the labeled standard having the same number of chlorine substituents and the closest retention time to the target analyte in question as the labeled standard to use for quantitation. | |||||||
| b BZ No.: Ballschmiter and Zell 1980, also referred to as IUPAC number. | |||||||
| MoCB | DiCB | ||||||
| 13 C 12 -2-MoCB | 1L | 2-MoCB | 1 | 13 C 12 -2,2′-DiCB | 4L | 2,2′-DiCB | 4 |
| 13 C 12 -2-MoCB | 1L | 3-MoCB | 2 | 13 C 12 -2,2′-DiCB | 4L | 2,3-DiCB | 5 |
| 13 C 12 -4-MoCB | 3L | 4-MoCB | 3 | 13 C 12 -2,2′-DiCB | 4L | 2,3′-DiCB | 6 |
| 13 C 12 -2,2′-DiCB | 4L | 2,4-DiCB | 7 | ||||
| 13 C 12 -2,2′-DiCB | 4L | 2,4′-DiCB | 8 | ||||
| 13 C 12 -2,2′-DiCB | 4L | 2,5-DiCB | 9 | ||||
| 13 C 12 -2,2′-DiCB | 4L | 2,6-DiCB | 10 | ||||
| 13 C 12 -2,2′-DiCB | 4L | 3,3′-DiCB | 11 | ||||
| 13 C 12 -2,2′-DiCB | 4L | 3,4-DiCB | 12 | ||||
| 13 C 12 -2,2′-DiCB | 4L | 3,4′-DiCB | 13 | ||||
| 13 C 12 -2,2′-DiCB | 4L | 3,5-DiCB | 14 | ||||
| 13 C 12 -4,4′-DiCB | 15L | 4,4′-DiCB | 15 | ||||
| TrCB | |||||||
| 13 C 12 -2,2′,6-TrCB | 19L | 2,2′,3-TrCB | 16 | 13 C 12 -3,4,4′-TrCB | 19L | 2,4,4′-TrCB | 28 |
| 13 C 12 -2,2′,6-TrCB | 19L | 2,2′,4-TrCB | 17 | 13 C 12 -3,4,4′-TrCB | 19L | 2,4,5-TrCB | 29 |
| 13 C 12 -2,2′,6-TrCB | 19L | 2,2′,5-TrCB | 18 | 13 C 12 -3,4,4′-TrCB | 19L | 2,4,6-TrCB | 30 |
| 13 C 12 -2,2′,6-TrCB | 19L | 2,2′,6-TrCB | 19 | 13 C 12 -3,4,4′-TrCB | 19L | 2,4′,5-TrCB | 31 |
| 13 C 12 -2,2′,6-TrCB | 19L | 2,3,3′-TrCB | 20 | 13 C 12 -3,4,4′-TrCB | 19L | 2,4′,6-TrCB | 32 |
| 13 C 12 -2,2′,6-TrCB | 19L | 2,3,4-TrCB | 21 | 13 C 12 -3,4,4′-TrCB | 19L | 2′,3,4-TrCB | 33 |
| 13 C 12 -2,2′,6-TrCB | 19L | 2,3,4′-TrCB | 22 | 13 C 12 -3,4,4′-TrCB | 19L | 2′,3,5-TrCB | 34 |
| 13 C 12 -2,2′,6-TrCB | 19L | 2,3,5- TrCB | 23 | 13 C 12 -3,4,4′-TrCB | 19L | 3,3′,4-TrCB | 35 |
| 13 C 12 -2,2′,6-TrCB | 19L | 2,3,6- TrCB | 23 | 13 C 12 -3,4,4′-TrCB | 19L | 3,3′,5-TrCB | 36 |
| 13 C 12 -2,2′,6-TrCB | 19L | 2,3′,4-TrCB | 25 | 13 C 12 -3,4′,4′-TrCB | 37L | 3,4,4′-TrCB | 37 |
| 13 C 12 -2,2′,6-TrCB | 19L | 2,3′,5-TrCB | 26 | 13 C 12 -3,4′,4′-TrCB | 37L | 3,4,5-TrCB | 38 |
| 13 C 12 -2,2′,6-TrCB | 19L | 2,3′,6-TrCB | 27 | 13 C 12 -3,4′,4′-TrCB | 37L | 3,4′,5-TrCB | 39 |
| TeCB | |||||||
| 13 C 12 -2,2′,6,6′-TeCB | 54L | 2,2′,3,3′-TeCB | 40 | 13 C 12 -2,2′,6,6′-TeCB | 54L | 2,3,4,5-TeCB | 61 |
| 13 C 12 -2,2′,6,6′-TeCB | 54L | 2,2′,3,4-TeCB | 41 | 13 C 12 -2,2′,6,6′-TeCB | 54L | 2,3,4,6-TeCB | 62 |
| 13 C 12 -2,2′,6,6′-TeCB | 54L | 2,2′,3,4′-TeCB | 42 | 13 C 12 -2,2′,6,6′-TeCB | 54L | 2,3,4′,5-TeCB | 63 |
| 13 C 12 -2,2′,6,6′-TeCB | 54L | 2,2′,3,5-TeCB | 43 | 13 C 12 -2,2′,6,6′-TeCB | 54L | 2,3,4′,6-TeCB | 64 |
| 13 C 12 -2,2′,6,6′-TeCB | 54L | 2,2′,3,5′-TeCB | 44 | 13 C 12 -2,2′,6,6′-TeCB | 54L | 2,3,5,6-TeCB | 65 |
| 13 C 12 -2,2′,6,6′-TeCB | 54L | 2,2′,3,6-TeCB | 45 | 13 C 12 -2,2′,6,6′-TeCB | 54L | 2,3′,4,4′-TeCB | 66 |
| 13 C 12 -2,2′,6,6′-TeCB | 54L | 2,2′,3,6′-TeCB | 46 | 13 C 12 -2,2′,6,6′-TeCB | 54L | 2,3′,4,5-TeCB | 67 |
| 13 C 12 -2,2′,6,6′-TeCB | 54L | 2,2′,4,4′-TeCB | 47 | 13 C 12 -2,2′,6,6′-TeCB | 54L | 2,3′,4,5′-TeCB | 68 |
| 13 C 12 -2,2′,6,6′-TeCB | 54L | 2,2′,4,5-TeCB | 48 | 13 C 12 -2,2′,6,6′-TeCB | 54L | 2,3′,4,6-TeCB | 69 |
| 13 C 12 -2,2′,6,6′-TeCB | 54L | 2,2′,4,5′-TeCB | 49 | 13 C 12 -2,2′,6,6′-TeCB | 54L | 2,3′,4′,5-TeCB | 70 |
| 13 C 12 -2,2′,6,6′-TeCB | 54L | 2,2′,4,6-TeCB | 50 | 13 C 12 -2,2′,6,6′-TeCB | 54L | 2,3′,4′,6-TeCB | 71 |
| 13 C 12 -2,2′,6,6′-TeCB | 54L | 2,2′,4,6′-TeCB | 51 | 13 C 12 -2,2′,6,6′-TeCB | 54L | 2,3′,5,5′-TeCB | 72 |
| 13 C 12 -2,2′,6,6′-TeCB | 54L | 2,2′,5,5′-TeCB | 52 | 13 C 12 -2,2′,6,6′-TeCB | 54L | 2,3′,5′,6-TeCB | 73 |
| 13 C 12 -2,2′,6,6′-TeCB | 54L | 2,2′,5,6′-TeCB | 53 | 13 C 12 -2,2′,6,6′-TeCB | 54L | 2,4,4′,5-TeCB | 74 |
| 13 C 12 -2,2′,6,6′-TeCB | 54L | 2,2′,6,6′-TeCB | 54 | 13 C 12 -2,2′,6,6′-TeCB | 54L | 2,4,4′,6-TeCB | 75 |
| 13 C 12 -2,2′,6,6′-TeCB | 54L | 2,3,3′,4′-TeCB | 55 | 13 C 12 -2,2′,6,6′-TeCB | 54L | 2′,3,4,5-TeCB | 76 |
| 13 C 12 -2,2′,6,6′-TeCB | 54L | 2,3,3′,4′-TeCB | 56 | 13 C 12 -3,3′,4,4′-TeCB | 77L | 3,3′,4,4′-TeCB | 77 |
| 13 C 12 -2,2′,6,6′-TeCB | 54L | 2,3,3′,5-TeCB | 57 | 13 C 12 -3,3′,4,4′-TeCB | 77L | 3,3′,4,5-TeCB | 78 |
| 13 C 12 -2,2′,6,6′-TeCB | 54L | 2,3,3′,5′-TeCB | 58 | 13 C 12 -3,3′,4,4′-TeCB | 77L | 3,3′,4,5′-TeCB | 79 |
| 13 C 12 -2,2′,6,6′-TeCB | 54L | 2,3,3′,6-TeCB | 59 | 13 C 12 -3,3′,4,4′-TeCB | 77L | 3,3′,5,5′-TeCB | 80 |
| 13 C 12 -2,2′,6,6′-TeCB | 54L | 2,3,4,4′-TeCB | 60 | 13 C 12 -3,4,4′,5-TeCB | 81L | 3,4,4′,5-TeCB | 81 |
| PeCB | |||||||
| 13 C 12 -2,2′,4,6,6′-PeCB | 104L | 2,2′,3,3′,4-PeCB | 82 | 13 C 12 -2,3,3′,4,4′-PeCB | 105L | 2,3,3′,4,4′-PeCB | 105 |
| 13 C 12 -2,2′,4,6,6′-PeCB | 104L | 2,2′,3,3′,5-PeCB | 83 | 13 C 12 -2,3,3′,4,4′-PeCB | 105L | 2,3,3′,4,5-PeCB | 106 |
| 13 C 12 -2,2′,4,6,6′-PeCB | 104L | 2,2′,3,3′,6-PeCB | 84 | 13 C 12 -2,3,3′,4,4′-PeCB | 105L | 2,3,3′,4′,5-PeCB | 107 |
| 13 C 12 -2,2′,4,6,6′-PeCB | 104L | 2,2′,3,4,4′-PeCB | 85 | 13 C 12 -2,3,3′,4,4′-PeCB | 105L | 2,3,3′,4,5′-PeCB | 108 |
| 13 C 12 -2,2′,4,6,6′-PeCB | 104L | 2,2′,3,4,5-PeCB | 86 | 13 C 12 -2,3,3′,4,4′-PeCB | 105L | 2,3,3′,4,6-PeCB | 109 |
| 13 C 12 -2,2′,4,6,6′-PeCB | 104L | 2,2′,3,4,5′-PeCB | 87 | 13 C 12 -2,3,3′,4,4′-PeCB | 105L | 2,3,3′,4′,6-PeCB | 110 |
| 13 C 12 -2,2′,4,6,6′-PeCB | 104L | 2,2′,3,4,6-PeCB | 88 | 13 C 12 -2,3,3′,4,4′-PeCB | 105L | 2,3,3′,5,5′-PeCB | 111 |
| 13 C 12 -2,2′,4,6,6′-PeCB | 104L | 2,2′,3,4,6′-PeCB | 89 | 13 C 12 -2,3,3′,4,4′-PeCB | 105L | 2,3,3′,5,6-PeCB | 112 |
| 13 C 12 -2,2′,4,6,6′-PeCB | 104L | 2,2′,3,4′,5-PeCB | 90 | 13 C 12 -2,3,3′,4,4′-PeCB | 105L | 2,3,3′,5′,6-PeCB | 113 |
| 13 C 12 -2,2′,4,6,6′-PeCB | 104L | 2,2′,3,4′,6-PeCB | 91 | 13 C 12 -2,3,4,4′,5-PeCB | 114L | 2,3,4,4′,5-PeCB | 114 |
| 13 C 12 -2,2′,4,6,6′-PeCB | 104L | 2,2′,3,5,5′-PeCB | 92 | 13 C 12 -2,3,4,4′,5-PeCB | 114L | 2,3,4,4′,6-PeCB | 115 |
| 13 C 12 -2,2′,4,6,6′-PeCB | 104L | 2,2′,3,5,6-PeCB | 93 | 13 C 12 -2,3,4,4′,5-PeCB | 114L | 2,3,4,5,6-PeCB | 116 |
| 13 C 12 -2,2′,4,6,6′-PeCB | 104L | 2,2′,3,5,6′-PeCB | 94 | 13 C 12 -2,3,4,4′,5-PeCB | 114L | 2,3,4′,5,6-PeCB | 117 |
| 13 C 12 -2,2′,4,6,6′-PeCB | 104L | 2,2′,3,5′,6-PeCB | 95 | 13 C 12 -2,3′,4,4′,5-PeCB | 118L | 2,3′,4,4′,5-PeCB | 118 |
| 13 C 12 -2,2′,4,6,6′-PeCB | 104L | 2,2′,3,6,6′-PeCB | 96 | 13 C 12 -2,3′,4,4′,5-PeCB | 118L | 2,3′,4,4′,6-PeCB | 119 |
| 13 C 12 -2,2′,4,6,6′-PeCB | 104L | 2,2′,3′,4,5-PeCB | 97 | 13 C 12 -2,3′,4,4′,5-PeCB | 118L | 2,3′,4,5,5′-PeCB | 120 |
| 13 C 12 -2,2′,4,6,6′-PeCB | 104L | 2,2′,3′,4,6-PeCB | 98 | 13 C 12 -2,3′,4,4′,5-PeCB | 118L | 2,3′,4,5,′6-PeCB | 121 |
| 13 C 12 -2,2′,4,6,6′-PeCB | 104L | 2,2′,4,4′,5-PeCB | 99 | 13 C 12 -2,3′,4,4′,5-PeCB | 118L | 2′,3,3′,4,5-PeCB | 122 |
| 13 C 12 -2,2′,4,6,6′-PeCB | 104L | 2,2′,4,4′,6-PeCB | 100 | 13 C 12 -2′,3,4,4′,5-PeCB | 123L | 2′,3,4,4′,5-PeCB | 123 |
| 13 C 12 -2,2′,4,6,6′-PeCB | 104L | 2,2′,4,5,5′-PeCB | 101 | 13 C 12 -2′,3,4,4′,5-PeCB | 123L | 2′,3,4,5,5′-PeCB | 124 |
| 13 C 12 -2,2′,4,6,6′-PeCB | 104L | 2,2′,4,5,6′-PeCB | 102 | 13 C 12 -2′,3,4,4′,5-PeCB | 123L | 2′,3,4,5,6′-PeCB | 125 |
| 13 C 12 -2,2′,4,6,6′-PeCB | 104L | 2,2′,4,5,′6-PeCB | 103 | 13 C 12 -3,3′,4,4′,5-PeCB | 126L | 3,3′,4,4′,5-PeCB | 126 |
| 13 C 12 -2,2′,4,6,6′-PeCB | 104L | 2,2′,4,6,6′-PeCB | 104 | 13 C 12 -3,3′,4,4′,5-PeCB | 126L | 3,3′,4,5,5′-PeCB | 127 |
| HxCB | |||||||
| 13 C 12 -2,2′,4,4′,6,6′-HxCB | 155L | 2,2′,3,3′,4,4′-HxCB | 128 | 13 C 12 -2,2′,4,4′,6,6′-HxCB | 155L | 2,2′,3,4′,5′,6-HxCB | 149 |
| 13 C 12 -2,2′,4,4′,6,6′-HxCB | 155L | 2,2′,3,3′,4,5-HxCB | 129 | 13 C 12 -2,2′,4,4′,6,6′-HxCB | 155L | 2,2′,3,4′,6,6′-HxCB | 150 |
| 13 C 12 -2,2′,4,4′,6,6′-HxCB | 155L | 2,2′,3,3′,4,5′-HxCB | 130 | 13 C 12 -2,2′,4,4′,6,6′-HxCB | 155L | 2,2′,3,5,5′,6-HxCB | 151 |
| 13 C 12 -2,2′,4,4′,6,6′-HxCB | 155L | 2,2′,3,3′,4,6-HxCB | 131 | 13 C 12 -2,2′,4,4′,6,6′-HxCB | 155L | 2,2′,3,5,6,6′-HxCB | 152 |
| 13 C 12 -2,2′,4,4′,6,6′-HxCB | 155L | 2,2′,3,3′,4,6′-HxCB | 132 | 13 C 12 -2,2′,4,4′,6,6′-HxCB | 155L | 2,2′,4,4′,5,5′-HxCB | 153 |
| 13 C 12 -2,2′,4,4′,6,6′-HxCB | 155L | 2,2′,3,3′,5,5′-HxCB | 133 | 13 C 12 -2,2′,4,4′,6,6′-HxCB | 155L | 2,2′,4,4′,5′,6-HxCB | 154 |
| 13 C 12 -2,2′,4,4′,6,6′-HxCB | 155L | 2,2′,3,3′,5,6-HxCB | 134 | 13 C 12 -2,2′,4,4′,6,6′-HxCB | 155L | 2,2′,4,4′,6,6′-HxCB | 155 |
| 13 C 12 -2,2′,4,4′,6,6′-HxCB | 155L | 2,2′,3,3′,5,6′-HxCB | 135 | 13 C 12 -2,3,3′,4,4′,5- HxCB | 156L | 2,3,3′,4,4′,5-HxCB | 156 |
| 13 C 12 -2,2′,4,4′,6,6′-HxCB | 155L | 2,2′,3,3′,6,6′-HxCB | 136 | 13 C 12 -2,3,3′,4,4′,5′-HxCB | 157L | 2,3,3′,4,4′,5′-HxCB | 157 |
| 13 C 12 -2,2′,4,4′,6,6′-HxCB | 155L | 2,2′,3,4,4′,5-HxCB | 137 | 13 C 12 -2,3,3′,4,4′,5′-HxCB | 157L | 2,3,3′,4,4′,6-HxCB | 158 |
| 13 C 12 -2,2′,4,4′,6,6′-HxCB | 155L | 2,2′,3,4,4′,5′-HxCB | 138 | 13 C 12 -2,3,3′,4,4′,5′-HxCB | 157L | 2,3,3′,4,5,5′-HxCB | 158 |
| 13 C 12 -2,2′,4,4′,6,6′-HxCB | 155L | 2,2′,3,4,4′,6-HxCB | 139 | 13 C 12 -2,3,3′,4,4′,5′-HxCB | 157L | 2,3,3′,4,5,6-HxCB | 160 |
| 13 C 12 -2,2′,4,4′,6,6′-HxCB | 155L | 2,2′,3,4,4′,6′-HxCB | 140 | 13 C 12 -2,3,3′,4,4′,5′-HxCB | 157L | 2,3,3′,4,5′,6-HxCB | 161 |
| 13 C 12 -2,2′,4,4′,6,6′-HxCB | 155L | 2,2′,3,4,5,5′-HxCB | 141 | 13 C 12 -2,3,3′,4,4′,5′-HxCB | 157L | 2,3,3′,4′,5,5′-HxCB | 162 |
| 13 C 12 -2,2′,4,4′,6,6′-HxCB | 155L | 2,2′,3,4,5,6-HxCB | 142 | 13 C 12 -2,3,3′,4,4′,5′-HxCB | 157L | 2,3,3′,4′,5,6-HxCB | 163 |
| 13 C 12 -2,2′,4,4′,6,6′-HxCB | 155L | 2,2′,3,4,5,6′-HxCB | 143 | 13 C 12 -2,3,3′,4,4′,5′-HxCB | 157L | 2,3,3′,4′,5′,6-HxCB | 164 |
| 13 C 12 -2,2′,4,4′,6,6′-HxCB | 155L | 2,2′,3,4,5′,6-HxCB | 144 | 13 C 12 -2,3,3′,4,4′,5′-HxCB | 157L | 2,3,3′,5,5′,6-HxCB | 165 |
| 13 C 12 -2,2′,4,4′,6,6′-HxCB | 155L | 2,2′,3,4,6,6′-HxCB | 145 | 13 C 12 -2,3,3′,4,4′,5′-HxCB | 157L | 2,3,4,4′,5,6-HxCB | 166 |
| 13 C 12 -2,2′,4,4′,6,6′-HxCB | 155L | 2,2′,3,4′,5,5′-HxCB | 146 | 13 C 12 -2,3′,4,4′,5,5′-HxCB | 167L | 2,3′,4,4′,5,5′-HxCB | 167 |
| 13 C 12 -2,2′,4,4′,6,6′-HxCB | 155L | 2,2′,3,4′,5,6-HxCB | 147 | 13 C 12 -2,3′,4,4′,5,5′-HxCB | 167L | 2,3′,4,4′,5′,6-HxCB | 168 |
| 13 C 12 -2,2′,4,4′,6,6′-HxCB | 155L | 2,2′,3,4′,5,6′-HxCB | 148 | 13 C 12 -3,3′,4,4′,5,5′-HxCB | 169L | 3,3′,4,4′,5,5′-HxCB | 169 |
| HpCB | |||||||
| 13 C 12 -2,2′,3,4′,5,6,6′-HpCB | 188L | 2,2′,3,3′,4,4′,5-HpCB | 170 | 13 C 12 -2,2′,3,4′,5,6,6′-HpCB | 188L | 2,2′,3,4,4′,5,6′-HpCB | 182 |
| 13 C 12 -2,2′,3,4′,5,6,6′-HpCB | 188L | 2,2′,3,3′,4,4′,6-HpCB | 171 | 13 C 12 -2,2′,3,4′,5,6,6′-HpCB | 188L | 2,2′,3,4,4′,5′,6-HpCB | 183 |
| 13 C 12 -2,2′,3,4′,5,6,6′-HpCB | 188L | 2,2′,3,3′,4,5,5′-HpCB | 172 | 13 C 12 -2,2′,3,4′,5,6,6′-HpCB | 188L | 2,2′,3,4,4′,5′,6-HpCB | 184 |
| 13 C 12 -2,2′,3,4′,5,6,6′-HpCB | 188L | 2,2′,3,3′,4,5,6-HpCB | 173 | 13 C 12 -2,2′,3,4′,5,6,6′-HpCB | 188L | 2,2′,3,4,4′,6,6′-HpCB | 185 |
| 13 C 12 -2,2′,3,4′,5,6,6′-HpCB | 188L | 2,2′,3,3′,4,5,6′-HpCB | 174 | 13 C 12 -2,2′,3,4′,5,6,6′-HpCB | 188L | 2,2′,3,4,5,5′,6-HpCB | 186 |
| 13 C 12 -2,2′,3,4′,5,6,6′-HpCB | 188L | 2,2′,3,3′,4,5′,6-HpCB | 175 | 13 C 12 -2,2′,3,4′,5,6,6′-HpCB | 188L | 2,2′,3,4′,5,5′,6-HpCB | 187 |
| 13 C 12 -2,2′,3,4′,5,6,6′-HpCB | 188L | 2,2′,3,3′,4,6,6′-HpCB | 176 | 13 C 12 -2,2′,3,4′,5,6,6′-HpCB | 188L | 2,2′,3,4′,5,6,6′-HpCB | 188 |
| 13 C 12 -2,2′,3,4′,5,6,6′-HpCB | 188L | 2,2′,3,3′,4′,5,6-HpCB | 177 | 13 C 12 -2,3,3′,4,4′,5,5′-HpCB | 189L | 2,3,3′,4,4′,5,5′-HpCB | 189 |
| 13 C 12 -2,2′,3,4′,5,6,6′-HpCB | 188L | 2,2′,3,3′,5,5′,6-HpCB | 178 | 13 C 12 -2,3,3′,4,4′,5,5′-HpCB | 189L | 2,3,3′,4,4′,5,6-HpCB | 190 |
| 13 C 12 -2,2′,3,4′,5,6,6′-HpCB | 188L | 2,2′,3,3′,5,6,6′-HpCB | 179 | 13 C 12 -2,3,3′,4,4′,5,5′-HpCB | 189L | 2,3,3′,4,4′,5′,6-HpCB | 191 |
| 13 C 12 -2,2′,3,4′,5,6,6′-HpCB | 188L | 2,2′,3,4,4′,5,5′-HpCB | 180 | 13 C 12 -2,3,3′,4,4′,5,5′-HpCB | 189L | 2,3,3′,4,5,5′,6-HpCB | 192 |
| 13 C 12 -2,2′,3,4′,5,6,6′-HpCB | 188L | 2,2′,3,4,4′,5,6-HpCB | 181 | 13 C 12 -2,3,3′,4,4′,5,5′-HpCB | 189L | 2,3,3′,4′,5,5′,6-HpCB | 193 |
| OcCB | NoCB | ||||||
| 13 C 12 -2,2′,3,3′,5,5′,6,6′-OcCB | 202L | 2,2′,3,3′,4,4′,5,5′-OcCB | 194 | 13 C 12 -2,2′,3,3′,4,4′,5,5′,6-NoCB | 206L | 2,2′,3,3′,4,4′,5,5′,6-NoCB | 206 |
| 13 C 12 -2,2′,3,3′,5,5′,6,6′-OcCB | 202L | 2,2′,3,3′,4,4′,5,6-OcCB | 195 | 13 C 12 -2,2′,3,3′,4,4′,5,5′,6-NoCB | 206L | 2,2′,3,3′,4,4′,5,6,6′-NoCB | 207 |
| 13 C 12 -2,2′,3,3′,5,5′,6,6′-OcCB | 202L | 2,2′,3,3′,4,4′,5,6′-OcCB | 196 | 13 C 12 -2,2′,3,3′,4,5,5′,6,6′-NoCB | 208L | 2,2′,3,3′,4,5,5′,6,6′- NoCB | 208 |
| 13 C 12 -2,2′,3,3′,5,5′,6,6′-OcCB | 202L | 2,2′,3,3′,4,4′,6,6′-OcCB | 197 | DeCB | |||
| 13 C 12 -2,2′,3,3′,5,5′,6,6′-OcCB | 202L | 2,2′,3,3′,4,5,5′,6-OcCB | 198 | 13 C 12 -DeCB | 209L | 2,2′,3,3′,4,4′,5,5′,6,6′-DeCB | 209 |
| 13 C 12 -2,2′,3,3′,5,5′,6,6′-OcCB | 202L | 2,2′,3,3′,4,5,5′,6′-OcCB | 199 | ||||
| 13 C 12 -2,2′,3,3′,5,5′,6,6′-OcCB | 202L | 2,2′,3,3′,4,5,6,6′-OcCB | 200 | ||||
| 13 C 12 -2,2′,3,3′,5,5′,6,6′-OcCB | 202L | 2,2′,3,3′,4,5′,6,6′-OcCB | 201 | ||||
| 13 C 12 -2,2′,3,3′,5,5′,6,6′-OcCB | 202L | 2,2′,3,3′,5,5′,6,6′-OcCB | 202 | ||||
| 13 C 12 -2,3′,3′,4,4′,5,5′,6-OcCB | 205L | 2,2′,3,4,4′,5,5′,6-OcCB | 203 | ||||
| 13 C 12 -2,3′,3′,4,4′,5,5′,6-OcCB | 205L | 2,2′,3,4,4′,5,6,6′-OcCB | 204 | ||||
| 13 C 12 -2,3′,3′,4,4′,5,5′,6-OcCB | 205L | 2,3,3′,4,4′,5,5′,6-OcCB | 205 | ||||
Appendix B to Method 23
Preparation of XAD-2 Adsorbent Resin
1.0 Scope and Application
XAD-2® resin, as supplied by the original manufacturer, is impregnated with a bicarbonate solution to inhibit microbial growth during storage. Remove both the salt solution and any residual extractable chemicals used in the polymerization process before use. Prepare the resin by a series of water and organic extractions, followed by careful drying.
2.0 Extraction
2.1 You may perform the extraction using a Soxhlet extractor or other apparatus that generates resin meeting the requirements in Section 13.1 of Method 23. Use an all-glass thimble containing an extra-coarse frit for extraction of the resin. The frit is recessed 10-15 mm above a crenellated ring at the bottom of the thimble to facilitate drainage. Because the resin floats on methylene chloride, carefully retain the resin in the extractor cup with a glass wool plug and stainless-steel screen. This process involves sequential extraction with the following recommended solvents in the listed order.
• Water initial rinse: Place resin in a suitable container, soak for approximately 5 min with Type II water, remove fine floating resin particles and discard the water. Fill with Type II water a second time, let stand overnight, remove fine floating resin particles, and discard the water.
• Hot water: Extract with water for 8 hr.
• Methyl alcohol: Extract for 22 hr.
• Methylene chloride: Extract for 22 hr.
• Toluene: Extract for 22 hr.
• Methylene chloride: Extract for 22 hr.
Note: You may store the resin in a sealed glass container filled with toluene prior to the final toluene extraction. It may be necessary to repeat the final methylene chloride extractions to meet the cleanliness requirements in Section 13.1 of Method 23.
2.2 You may use alternative extraction procedures to clean large batches of resin. Any size extractor may be constructed; the choice depends on the needs of the sampling programs. The resin is held in a glass or stainless-steel cylinder between a pair of coarse and fine screens. Spacers placed under the bottom screen allow for even distribution of clean solvent. Clean solvent is circulated through the resin for extraction. A flow rate is maintained upward through the resin to allow maximum solvent contact and prevent channeling.
2.2.1 Experience has shown that 1 mL/g of resin extracted is the minimum necessary to extract and clean the resin. The aqueous rinse is critical to the subsequent organic rinses and may be accomplished by simply flushing the canister with about 1 liter of distilled water for every 25 g of resin. A small pump may be useful for pumping the water through the canister. You should perform the water extraction at the rate of about 20 to 40 mL/min.
2.2.2 All materials of construction are glass, PTFE, or stainless steel. Pumps, if used, should not contain extractable materials.
3.0 Drying
3.1 Dry the adsorbent of extraction solvent before use. This section provides a recommended procedure to dry adsorbent that is wet with solvent. However, you may use other procedures if the cleanliness requirements in Section 13.1 of Method 23 are met.
3.2 Drying Column. A simple column with suitable retainers will hold all the XAD-2 from the extractor or the Soxhlet extractor, as shown in Figure B-1, with sufficient space for drying the bed while generating a minimum backpressure in the column.
3.3 Drying Procedure: Dry the adsorbent using clean inert gas. Liquid nitrogen from a standard commercial liquid nitrogen cylinder has proven to be a reliable source of large volumes of gas free from organic contaminants. You may use high-purity tank nitrogen to dry the resin. However, you should pass the high-purity nitrogen through a bed of activated charcoal approximately 150 mL in volume prior to entering the drying apparatus.
3.3.1 Connect the gas vent of a liquid nitrogen cylinder or the exit of the activated carbon scrubber to the column by a length of precleaned copper tubing ( e.g., 0.95 cm ID) coiled to pass through a heat source. A convenient heat source is a water bath heated from a steam line. The final nitrogen temperature should only be warm to the touch and not over 40 °C.
3.3.2 Allow the methylene chloride to drain from the resin prior to placing the resin in the drying apparatus.
3.3.3 Flow nitrogen through the drying apparatus at a rate that does not fluidize or agitate the resin. Continue the nitrogen flow until the residual solvent is removed.
Note: Experience has shown that about 500 g of resin may be dried overnight by consuming a full 160-L cylinder of liquid nitrogen.
Figure B-1. XAD-2 fluidized-bed drying apparatus
Method 24 - Determination of Volatile Matter Content, Water Content, Density, Volume Solids, and Weight Solids of Surface Coatings
1.0 Scope and Application
1.1 Analytes.
| Analyte | CAS No. |
|---|---|
| Volatile organic compounds Water | No CAS Number assigned 7732-18-5 |
1.2 Applicability. This method is applicable for the determination of volatile matter content, water content, density, volume solids, and weight solids of paint, varnish, lacquer, or other related surface coatings.
1.3 Precision and Bias. Intra-and inter-laboratory analytical precision statements are presented in section 13.1. No bias has been identified.
2.0 Summary of Method
2.1 Standard methods are used to determine the volatile matter content, water content, density, volume solids, and weight solids of paint, varnish, lacquer, or other related surface coatings.
3.0 Definitions
3.1 Waterborne coating means any coating which contains more than 5 percent water by weight in its volatile fraction.
3.2 Multicomponent coatings are coatings that are packaged in two or more parts, which are combined before application. Upon combination a coreactant from one part of the coating chemically reacts, at ambient conditions, with a coreactant from another part of the coating.
3.3 Ultraviolet (UV) radiation-cured coatings are coatings which contain unreacted monomers that are polymerized by exposure to ultraviolet light.
4.0 Interferences [Reserved]
5.0 Safety
5.1 Disclaimer. This method may involve hazardous materials, operations, and equipment. This test method may not address all of the safety problems associated with its use. It is the responsibility of the user of this test method to establish appropriate safety and health practices and to determine the applicability of regulatory limitations prior to performing this test method.
5.2 Hazardous Components. Several of the compounds that may be contained in the coatings analyzed by this method may be irritating or corrosive to tissues (e.g., heptane) or may be toxic (e.g., benzene, methyl alcohol). Nearly all are fire hazards. Appropriate precautions can be found in reference documents, such as Reference 3 of section 16.0.
6.0 Equipment and Supplies
The equipment and supplies specified in the ASTM methods listed in sections 6.1 through 6.6 (incorporated by reference - see §60.17 for acceptable versions of the methods) are required:
6.1 ASTM D 1475-60, 80, or 90, Standard Test Method for Density of Paint, Varnish, Lacquer, and Related Products.
6.2 ASTM D 2369-81, 87, 90, 92, 93, 95, or 10. Standard Test Method for Volatile Content of Coatings.
6.3 ASTM D 3792-79 or 91, Standard Test Method for Water Content of Water Reducible Paints by Direct Injection into a Gas Chromatograph.
6.4 ASTM D 4017-81, 90, or 96a, Standard Test Method for Water in Paints and Paint Materials by the Karl Fischer Titration Method.
6.5 ASTM 4457-85 91, Standard Test Method for Determination of Dichloromethane and 1,1,1-Trichloroethane in Paints and Coatings by Direct Injection into a Gas Chromatograph.
6.6 ASTM D 5403-93, Standard Test Methods for Volatile Content of Radiation Curable Materials.
6.7 ASTM D 6419-00, Test Method for Volatile Content of Sheet-Fed and Coldset Web Offset Printing Inks.
7.0 Reagents and Standards
7.1 The reagents and standards specified in the ASTM methods listed in sections 6.1 through 6.6 are required.
8.0 Sample Collection, Preservation, Storage, and Transport
8.1 Follow the sample collection, preservation, storage, and transport procedures described in Reference 1 of section 16.0.
9.0 Quality Control
9.1 Reproducibility
Note:
Not applicable to UV radiation-cured coatings). The variety of coatings that may be subject to analysis makes it necessary to verify the ability of the analyst and the analytical procedures to obtain reproducible results for the coatings tested. Verification is accomplished by running duplicate analyses on each sample tested (Sections 11.2 through 11.4) and comparing the results with the intra-laboratory precision statements (Section 13.1) for each parameter.
9.2 Confidence Limits for Waterborne Coatings. Because of the inherent increased imprecision in the determination of the VOC content of waterborne coatings as the weight percent of water increases, measured parameters for waterborne coatings are replaced with appropriate confidence limits (Section 12.6). These confidence limits are based on measured parameters and inter-laboratory precision statements.
10.0 Calibration and Standardization
10.1 Perform the calibration and standardization procedures specified in the ASTM methods listed in sections 6.1 through 6.6.
11.0 Analytical Procedure
Additional guidance can be found in Reference 2 of section 16.0.
11.1 Non Thin-film Ultraviolet Radiation-cured (UV radiation-cured) Coatings.
11.1.1 Volatile Content. Use the procedure in ASTM D 5403 to determine the volatile matter content of the coating except the curing test described in NOTE 2 of ASTM D 5403 is required.
11.1.2 Water Content. To determine water content, follow section 11.3.2.
11.1.3 Coating Density. To determine coating density, follow section 11.3.3.
11.1.4 Solids Content. To determine solids content, follow section 11.3.4.
11.1.5 To determine if a coating or ink can be classified as a thin-film UV cured coating or ink, use the equation in section 12.2. If C is less than 0.2 g and A is greater than or equal to 225 cm 2 (35 in 2) then the coating or ink is considered a thin-film UV radiation-cured coating and ASTM D 5403 is not applicable.
Note:
As noted in section 1.4 of ASTM D 5403, this method may not be applicable to radiation curable materials wherein the volatile material is water.
11.2 Multi-component Coatings.
11.2.1 Sample Preparation.
11.2.1.1 Prepare about 100 ml of sample by mixing the components in a storage container, such as a glass jar with a screw top or a metal can with a cap. The storage container should be just large enough to hold the mixture. Combine the components (by weight or volume) in the ratio recommended by the manufacturer. Tightly close the container between additions and during mixing to prevent loss of volatile materials. However, most manufacturers mixing instructions are by volume. Because of possible error caused by expansion of the liquid when measuring the volume, it is recommended that the components be combined by weight. When weight is used to combine the components and the manufacturer's recommended ratio is by volume, the density must be determined by section 11.3.3.
11.2.1.2 Immediately after mixing, take aliquots from this 100 ml sample for determination of the total volatile content, water content, and density.
11.2.2 Volatile Content. To determine total volatile content, use the apparatus and reagents described in ASTM D2369 (incorporated by reference; see §60.17 for the approved versions of the standard), respectively, and use the following procedures:
11.2.2.1 Weigh and record the weight of an aluminum foil weighing dish. Add 3 ±1 ml of suitable solvent as specified in ASTM D2369 to the weighing dish. Using a syringe as specified in ASTM D2369, weigh to 1 mg, by difference, a sample of coating into the weighing dish. For coatings believed to have a volatile content less than 40 weight percent, a suitable size is 0.3 0.10 g, but for coatings believed to have a volatile content greater than 40 weight percent, a suitable size is 0.5 ±0.1 g.
Note:
If the volatile content determined pursuant to section 12.4 is not in the range corresponding to the sample size chosen repeat the test with the appropriate sample size. Add the specimen dropwise, shaking (swirling) the dish to disperse the specimen completely in the solvent. If the material forms a lump that cannot be dispersed, discard the specimen and prepare a new one. Similarly, prepare a duplicate. The sample shall stand for a minimum of 1 hour, but no more than 24 hours prior to being oven cured at 110 ±5°C (230 ±9°F) for 1 hour.
11.2.2.2 Heat the aluminum foil dishes containing the dispersed specimens in the forced draft oven for 60 min at 110 ±5°C (230 ±9°F). Caution - provide adequate ventilation, consistent with accepted laboratory practice, to prevent solvent vapors from accumulating to a dangerous level.
11.2.2.3 Remove the dishes from the oven, place immediately in a desiccator, cool to ambient temperature, and weigh to within 1 mg.
11.2.2.4 Run analyses in pairs (duplicate sets) for each coating mixture until the criterion in section 11.4 is met. Calculate WV following Equation 24-2 and record the arithmetic average.
11.2.3 Water Content. To determine water content, follow section 11.3.2.
11.2.4 Coating Density. To determine coating density, follow section 11.3.3.
11.2.5 Solids Content. To determine solids content, follow section 11.3.4.
11.2.6 Exempt Solvent Content. To determine the exempt solvent content, follow section 11.3.5.
Note:
For all other coatings (i.e., water-or solvent-borne coatings) not covered by multicomponent or UV radiation-cured coatings, analyze as shown below:
11.3 Water-or Solvent-borne coatings.
11.3.1 Volatile Content. Use the procedure in ASTM D 2369 to determine the volatile matter content (may include water) of the coating.
11.3.1.1 Record the following information:
W1 = weight of dish and sample before heating, g
W2 = weight of dish and sample after heating, g
W3 = sample weight, g.
11.3.1.2 Calculate the weight fraction of the volatile matter (Wv) for each analysis as shown in section 12.3.
11.3.1.3 Run duplicate analyses until the difference between the two values in a set is less than or equal to the intra-laboratory precision statement in section 13.1.
11.3.1.4 Record the arithmetic average (Wv).
11.3.2 Water Content. For waterborne coatings only, determine the weight fraction of water (Ww) using either ASTM D 3792 or ASTM D 4017.
11.3.2.1 Run duplicate analyses until the difference between the two values in a set is less than or equal to the intra-laboratory precision statement in section 13.1.
11.3.2.2 Record the arithmetic average (ww).
11.3.3 Coating Density. Determine the density (Dc, kg/l) of the surface coating using the procedure in ASTM D 1475.
11.3.3.1 Run duplicate analyses until each value in a set deviates from the mean of the set by no more than the intra-laboratory precision statement in section 13.1.
11.3.3.2 Record the arithmetic average (Dc).
11.3.4 Solids Content. Determine the volume fraction (Vs) solids of the coating by calculation using the manufacturer's formulation.
11.3.5 Exempt Solvent Content. Determine the weight fraction of exempt solvents (WE) by using ASTM Method D4457. Run a duplicate set of determinations and record the arithmetic average (WE).
11.4 Sample Analysis Criteria. For Wv and Ww, run duplicate analyses until the difference between the two values in a set is less than or equal to the intra-laboratory precision statement for that parameter. For Dc, run duplicate analyses until each value in a set deviates from the mean of the set by no more than the intra-laboratory precision statement. If, after several attempts, it is concluded that the ASTM procedures cannot be used for the specific coating with the established intra-laboratory precision (excluding UV radiation-cured coatings), the U.S. Environmental Protection Agency (EPA) will assume responsibility for providing the necessary procedures for revising the method or precision statements upon written request to: Director, Emissions, Monitoring, and Analysis Division, MD-14, Office of Air Quality Planning and Standards, U.S. Environmental Protection Agency, Research Triangle Park, NC 27711.
12.0 Calculations and Data Analysis
12.1 Nomenclature.
A = Area of substrate, cm 2, (in 2).
C = Amount of coating or ink added to the substrate, g.
Dc = Density of coating or ink, g/cm 3 (g/in 3).
F = Manufacturer's recommended film thickness, cm (in).
Wo = Weight fraction of nonaqueous volatile matter, g/g.
Ws = Weight fraction of solids, g/g.
Wv = Weight fraction of the volatile matter, g/g.
Ww = Weight fraction of the water, g/g.
12.2 To determine if a coating or ink can be classified as a thin-film UV cured coating or ink, use the following equation:
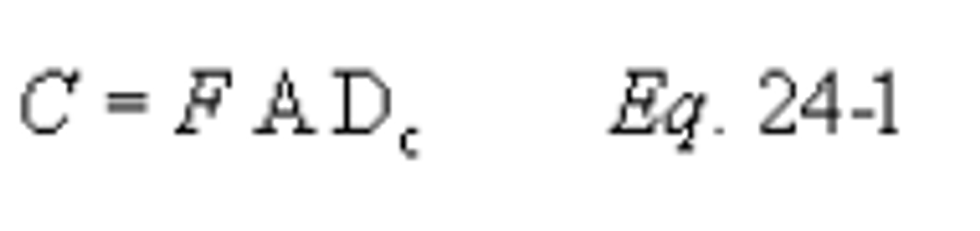
12.3 Calculate Wv for each analysis as shown below:
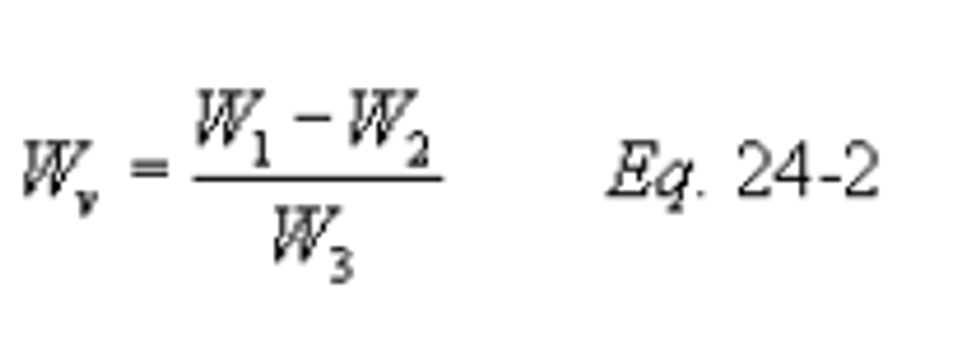
12.4 Nonaqueous Volatile Matter.
12.4.1 Solvent-borne Coatings.
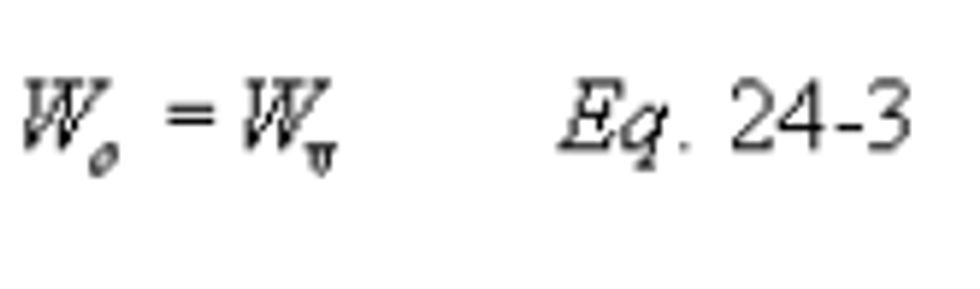
12.4.2 Waterborne Coatings.
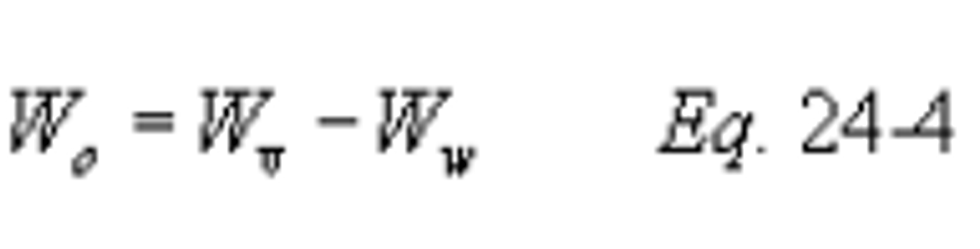
12.4.3 Coatings Containing Exempt Solvents.
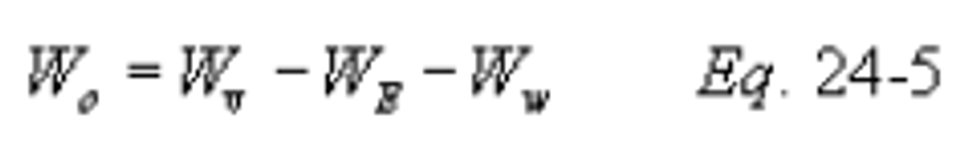
12.5 Weight Fraction Solids.
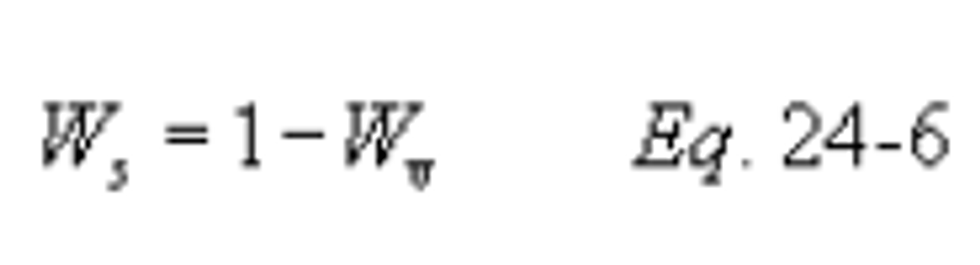
12.6 Confidence Limit Calculations for Waterborne Coatings. To calculate the lower confidence limit, subtract the appropriate inter-laboratory precision value from the measured mean value for that parameter. To calculate the upper confidence limit, add the appropriate inter-laboratory precision value to the measured mean value for that parameter. For Wv and Dc, use the lower confidence limits; for Ww, use the upper confidence limit. Because Ws is calculated, there is no adjustment for this parameter.
13.0 Method Performance
13.1 Analytical Precision Statements. The intra-and inter-laboratory precision statements are given in Table 24-1 in section 17.0.
14.0 Pollution Prevention [Reserved]
15.0 Waste Management [Reserved]
16.0 References
Same as specified in section 6.0, with the addition of the following:
1. Standard Procedure for Collection of Coating and Ink Samples for Analysis by Reference Methods 24 and 24A. EPA-340/1-91-010. U.S. Environmental Protection Agency, Stationary Source Compliance Division, Washington, D.C. September 1991.
2. Standard Operating Procedure for Analysis of Coating and Ink Samples by Reference Methods 24 and 24A.
EPA-340/1-91-011. U.S. Environmental Protection Agency, Stationary Source Compliance Division, Washington, D.C. September 1991.
3. Handbook of Hazardous Materials: Fire, Safety, Health. Alliance of American Insurers. Schaumberg, IL. 1983.
17.0 Tables, Diagrams, Flowcharts, and Validation Data
| Intra-laboratory | Inter-laboratory | |
|---|---|---|
| Volatile matter content, Wv | ±0.015 W v | ±0.047 W͞ v |
| Water content, Ww | ±0.029 W͞ w | ±0.075 W w |
| Density, Dc | ±0.001 kg/l | ±0.002 kg/l |
Method 24A - Determination of Volatile Matter Content and Density of Publication Rotogravure Inks and Related Publication Rotogravure Coatings
1.0 Scope and Application
1.1 Analytes.
| Analyte | CAS No. |
|---|---|
| Volatile organic compounds (VOC) | No CAS number assigned. |
1.2 Applicability. This method is applicable for the determination of the VOC content and density of solvent-borne (solvent-reducible) publication rotogravure inks and related publication rotogravure coatings.
2.0 Summary of Method
2.1 Separate procedures are used to determine the VOC weight fraction and density of the ink or related coating and the density of the solvent in the ink or related coating. The VOC weight fraction is determined by measuring the weight loss of a known sample quantity which has been heated for a specified length of time at a specified temperature. The density of both the ink or related coating and solvent are measured by a standard procedure. From this information, the VOC volume fraction is calculated.
3.0 Definitions [Reserved]
4.0 Interferences [Reserved]
5.0 Safety
5.1 Disclaimer. This method may involve hazardous materials, operations, and equipment. This test method does not purport to address all of the safety problems associated with its use. It is the responsibility of the user of this test method to establish appropriate safety and health practices and to determine the applicability of regulatory limitations prior to performing this test method.
5.2 Hazardous Components. Some of the compounds that may be contained in the inks or related coatings analyzed by this method may be irritating or corrosive to tissues or may be toxic. Nearly all are fire hazards. Appropriate precautions can be found in reference documents, such as Reference 6 of section 16.0.
6.0 Equipment and Supplies
The following equipment and supplies are required for sample analysis:
6.1 Weighing Dishes. Aluminum foil, 58 mm (2.3 in.) in diameter by 18 mm (0.7 in.) high, with a flat bottom. There must be at least three weighing dishes per sample.
6.2 Disposable Syringe. 5 ml.
6.3 Analytical Balance. To measure to within 0.1 mg.
6.4 Oven. Vacuum oven capable of maintaining a temperature of 120 ±2°C (248 ±4°F) and an absolute pressure of 510 ±51 mm Hg (20 ±2 in. Hg) for 4 hours. Alternatively, a forced draft oven capable of maintaining a temperature of 120 ±2°C (248 ±4°F) for 24 hours.
6.5 The equipment and supplies specified in ASTM D 1475-60, 80, or 90 (incorporated by reference - see §60.17).
7.0 Reagents and Standards
7.1 The reagents and standards specified in ASTM D 1475-60, 80, or 90 are required.
8.0 Sample Collection, Preservation, Storage, and Transport
8.1 Follow the sample collection, preservation, storage, and transport procedures described in Reference 4 of section 16.0.
9.0 Quality Control [Reserved]
10.0 Calibration and Standardization [Reserved]
11.0 Analytical Procedure
Additional guidance can be found in Reference 5 of section 16.0.
11.1 VOC Weight Fraction. Shake or mix the ink or related coating sample thoroughly to assure that all the solids are completely suspended. Label and weigh to the nearest 0.1 mg a weighing dish and record this weight (Mx1). Using a 5 ml syringe, without a needle, extract an aliquot from the ink or related coating sample. Weigh the syringe and aliquot to the nearest 0.1 mg and record this weight (Mcy1). Transfer 1 to 3 g of the aliquot to the tared weighing dish. Reweigh the syringe and remaining aliquot to the nearest 0.1 mg and record this weight (Mcy2). Heat the weighing dish with the transferred aliquot in a vacuum oven at an absolute pressure of 510 ±51 mm Hg (20 ±2 in. Hg) and a temperature of 120 ±2°C (248 ±4°F) for 4 hours. Alternatively, heat the weighing dish with the transferred aliquot in a forced draft oven at a temperature of 120 ±2°C for 24 hours. After the weighing dish has cooled, reweigh it to the nearest 0.1 mg and record the weight (Mx2). Repeat this procedure two times for each ink or related coating sample, for a total of three samples.
11.2 Ink or Related Coating Density. Determine the density of the ink or related coating (Dc) according to the procedure outlined in ASTM D 1475. Make a total of three determinations for each ink or related coating sample. Report the ink or related coating density as the arithmetic average (Dc) of the three determinations.
11.3 Solvent Density. Determine the density of the solvent (Do) according to the procedure outlined in ASTM D 1475. Make a total of three determinations for each ink or related coating sample. Report the solvent density as the arithmetic average (Do) of the three determinations.
12.0 Calculations and Data Analysis
12.1 VOC Weight Fraction. For each determination, calculate the volatile organic content weight fraction (Wo) using the following equation:
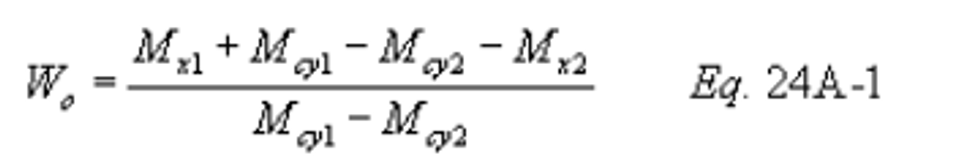
Make a total of three determinations. Report the VOC weight fraction as the arithmetic average (W͞ o) of the three determinations.
12.2 VOC Volume Fraction. Calculate the volume fraction volatile organic content (Vo) using the following equation:
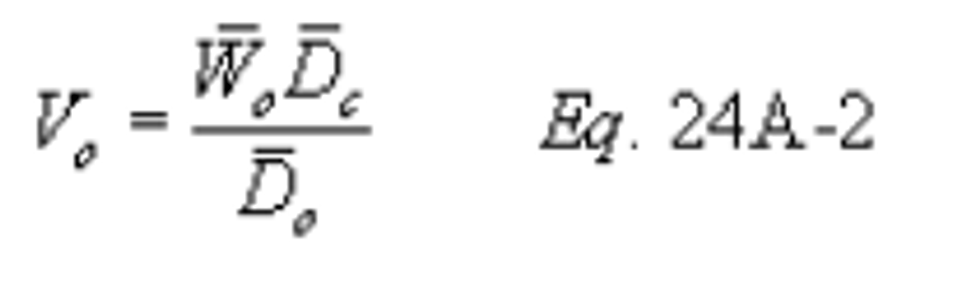
13.0 Method Performance [Reserved]
14.0 Pollution Prevention [Reserved]
15.0 Waste Management [Reserved]
16.0 References
1. Standard Test Method for Density of Paint, Varnish, Lacquer, and Related Products. ASTM Designation D 1475.
2. Teleconversation. Wright, Chuck, Inmont Corporation with Reich, R., A., Radian Corporation. September 25, 1979, Gravure Ink Analysis.
3. Teleconversation. Oppenheimer, Robert, Gravure Research Institute with Burt, Rick, Radian Corporation, November 5, 1979, Gravure Ink Analysis.
4. Standard Procedure for Collection of Coating and Ink Samples for Analysis by Reference Methods 24 and 24A. EPA-340/1-91-010. U.S. Environmental Protection Agency, Stationary Source Compliance Division, Washington, D.C. September 1991.
5. Standard Operating Procedure for Analysis of Coating and Ink Samples by Reference Methods 24 and 24A. EPA-340/1-91-011. U.S. Environmental Protection Agency, Stationary Source Compliance Division, Washington, D.C. September 1991.
6. Handbook of Hazardous Materials: Fire, Safety, Health. Alliance of American Insurers. Schaumberg, IL. 1983.
17.0 Tables, Diagrams, Flowcharts, and Validation Data [Reserved]
Method 25 - Determination of Total Gaseous Nonmethane Organic Emissions as Carbon
1.0 Scope and Application
1.1 Analytes.
| Analyte | CAS No. | Sensitivity |
|---|---|---|
| Total gaseous nonmethane organic compounds (TGNMO) | N/A | Dependent upon analytical equipment. |
1.2 Applicability.
1.2.1 This method is applicable for the determination of volatile organic compounds (VOC) (measured as total gaseous nonmethane organics (TGNMO) and reported as carbon) in stationary source emissions. This method is not applicable for the determination of organic particulate matter.
1.2.2 This method is not the only method that applies to the measurement of VOC. Costs, logistics, and other practicalities of source testing may make other test methods more desirable for measuring VOC contents of certain effluent streams. Proper judgment is required in determining the most applicable VOC test method. For example, depending upon the molecular composition of the organics in the effluent stream, a totally automated semicontinuous nonmethane organics (NMO) analyzer interfaced directly to the source may yield accurate results. This approach has the advantage of providing emission data semicontinuously over an extended time period.
1.2.3 Direct measurement of an effluent with a flame ionization detector (FID) analyzer may be appropriate with prior characterization of the gas stream and knowledge that the detector responds predictably to the organic compounds in the stream. If present, methane (CH4) will, of course, also be measured. The FID can be used under any of the following limited conditions: (1) Where only one compound is known to exist; (2) when the organic compounds consist of only hydrogen and carbon; (3) where the relative percentages of the compounds are known or can be determined, and the FID responses to the compounds are known; (4) where a consistent mixture of the compounds exists before and after emission control and only the relative concentrations are to be assessed; or (5) where the FID can be calibrated against mass standards of the compounds emitted (solvent emissions, for example).
1.2.4 Another example of the use of a direct FID is as a screening method. If there is enough information available to provide a rough estimate of the analyzer accuracy, the FID analyzer can be used to determine the VOC content of an uncharacterized gas stream. With a sufficient buffer to account for possible inaccuracies, the direct FID can be a useful tool to obtain the desired results without costly exact determination.
1.2.5 In situations where a qualitative/quantitative analysis of an effluent stream is desired or required, a gas chromatographic FID system may apply. However, for sources emitting numerous organics, the time and expense of this approach will be formidable.
2.0 Summary of Method
2.1 An emission sample is withdrawn from the stack at a constant rate through a heated filter and a chilled condensate trap by means of an evacuated sample tank. After sampling is completed, the TGNMO are determined by independently analyzing the condensate trap and sample tank fractions and combining the analytical results. The organic content of the condensate trap fraction is determined by oxidizing the NMO to carbon dioxide (CO2) and quantitatively collecting in the effluent in an evacuated vessel; then a portion of the CO2 is reduced to CH4 and measured by an FID. The organic content of the sample tank fraction is measured by injecting a portion of the sample into a gas chromatographic column to separate the NMO from carbon monoxide (CO), CO2, and CH4; the NMO are oxidized to CO2, reduced to CH4, and measured by an FID. In this manner, the variable response of the FID associated with different types of organics is eliminated.
3.0 Definitions [Reserved]
4.0 Interferences
4.1 Carbon Dioxide and Water Vapor. When carbon dioxide (CO2) and water vapor are present together in the stack, they can produce a positive bias in the sample. The magnitude of the bias depends on the concentrations of CO2 and water vapor. As a guideline, multiply the CO2 concentration, expressed as volume percent, times the water vapor concentration. If this product does not exceed 100, the bias can be considered insignificant. For example, the bias is not significant for a source having 10 percent CO2 and 10 percent water vapor, but it might be significant for a source having 10 percent CO2 and 20 percent water vapor.
4.2. Particulate Matter. Collection of organic particulate matter in the condensate trap would produce a positive bias. A filter is included in the sampling equipment to minimize this bias.
5.0 Safety
5.1 Disclaimer. This method may involve hazardous materials, operations, and equipment. This test method may not address all of the safety problems associated with its use. It is the responsibility of the user of this test method to establish appropriate safety and health practices and determine the applicability of regulatory limitations prior to performing this test method.
6.0 Equipment and Supplies
6.1 Sample Collection. The sampling system consists of a heated probe, heated filter, condensate trap, flow control system, and sample tank (see Figure 25-1). The TGNMO sampling equipment can be constructed from commercially available components and components fabricated in a machine shop. The following equipment is required:
6.1.1 Heated Probe. 6.4-mm ( 1/4-in.) OD stainless steel tubing with a heating system capable of maintaining a gas temperature at the exit end of at least 129°C (265°F). The probe shall be equipped with a temperature sensor at the exit end to monitor the gas temperature. A suitable probe is shown in Figure 25-1. The nozzle is an elbow fitting attached to the front end of the probe while the temperature sensor is inserted in the side arm of a tee fitting attached to the rear of the probe. The probe is wrapped with a suitable length of high temperature heating tape, and then covered with two layers of glass cloth insulation and one layer of aluminum foil or an equivalent wrapping.
Note:
If it is not possible to use a heating system for safety reasons, an unheated system with an in-stack filter is a suitable alternative.
6.1.2 Filter Holder. 25-mm ( 15/16-in.) ID Gelman filter holder with 303 stainless steel body and 316 stainless steel support screen with the Viton O-ring replaced by a Teflon O-ring.
6.1.3 Filter Heating System.
6.1.3.1 A metal box consisting of an inner and an outer shell separated by insulating material with a heating element in the inner shell capable of maintaining a gas temperature at the filter of 121 ±3°C (250 ±5°F). The heating box shall include temperature sensors to monitor the gas temperature immediately upstream and immediately downstream of the filter.
6.1.3.2 A suitable heating box is shown in Figure 25-2. The outer shell is a metal box that measures 102 mm × 280 mm × 292 mm (4 in. × 11 in. × 11 1/2 in.), while the inner shell is a metal box measuring 76 mm × 229 mm × 241 mm (3 in. × 9 in. × 9 1/2 in.). The inner box is supported by 13-mm ( 1/2-in.) phenolic rods. The void space between the boxes is filled with ceramic fiber insulation which is sealed in place by means of a silicon rubber bead around the upper sides of the box. A removable lid made in a similar manner, with a 25-mm (1-in.) gap between the parts is used to cover the heating chamber. The inner box is heated with a 250-watt cartridge heater, shielded by a stainless steel shroud. The heater is regulated by a thermostatic temperature controller which is set to maintain a gas temperature of 121°C (250°F) as measured by the temperature sensor upstream of the filter.
Note:
If it is not possible to use a heating system for safety reasons, an unheated system with an in-stack filter is a suitable alternative.
6.1.4 Condensate Trap. 9.5-mm ( 3/8-in.) OD 316 stainless steel tubing bent into a U-shape. Exact dimensions are shown in Figure 25-3. The tubing shall be packed with coarse quartz wool, to a density of approximately 0.11 g/cm 3 before bending. While the condensate trap is packed with dry ice in the Dewar, an ice bridge may form between the arms of the condensate trap making it difficult to remove the condensate trap. This problem can be prevented by attaching a steel plate between the arms of the condensate trap in the same plane as the arms to completely fill the intervening space.
6.1.5 Valve. Stainless steel control valve for starting and stopping sample flow.
6.1.6 Metering Valve. Stainless steel valve for regulating the sample flow rate through the sample train.
6.1.7 Rate Meter. Rotameter, or equivalent, capable of measuring sample flow in the range of 60 to 100 cm 3/min (0.13 to 0.21 ft 3/hr).
6.1.8 Sample Tank. Stainless steel or aluminum tank with a minimum volume of 4 liters (0.14 ft 3).
Note:
Sample volumes greater than 4 liters may be required for sources with low organic concentrations.
6.1.9 Mercury Manometer. U-tube manometer or absolute pressure gauge capable of measuring pressure to within 1 mm Hg in the range of 0 to 900 mm.
6.1.10 Vacuum Pump. Capable of evacuating to an absolute pressure of 10 mm Hg.
6.2 Condensate Recovery. The system for the recovery of the organics captured in the condensate trap consists of a heat source, an oxidation catalyst, a nondispersive infrared (NDIR) analyzer, and an intermediate collection vessel (ICV). Figure 25-4 is a schematic of a typical system. The system shall be capable of proper oxidation and recovery, as specified in section 10.1.1. The following major components are required:
6.2.1 Heat Source. Sufficient to heat the condensate trap (including probe) to a temperature of 200°C (390°F). A system using both a heat gun and an electric tube furnace is recommended.
6.2.2 Heat Tape. Sufficient to heat the connecting tubing between the water trap and the oxidation catalyst to 100°C (212°F).
6.2.3 Oxidation Catalyst. A suitable length of 9.5 mm ( 3/8-in.) OD Inconel 600 tubing packed with 15 cm (6 in.) of 3.2 mm ( 3/8-in.) diameter 19 percent chromia on alumina pellets. The catalyst material is packed in the center of the catalyst tube with quartz wool packed on either end to hold it in place.
6.2.4 Water Trap. Leak-proof, capable of removing moisture from the gas stream.
6.2.5 Syringe Port. A 6.4-mm ( 1/4-in.) OD stainless steel tee fitting with a rubber septum placed in the side arm.
6.2.6 NDIR Detector. Capable of indicating CO2 concentration in the range of zero to 5 percent, to monitor the progress of combustion of the organic compounds from the condensate trap.
6.2.7 Flow-Control Valve. Stainless steel, to maintain the trap conditioning system near atmospheric pressure.
6.2.8 Intermediate Collection Vessel. Stainless steel or aluminum, equipped with a female quick connect. Tanks with nominal volumes of at least 6 liters (0.2 ft 3) are recommended.
6.2.9 Mercury Manometer. Same as described in section 6.1.9.
6.2.10 Syringe. 10-ml gas-tight glass syringe equipped with an appropriate needle.
6.2.11 Syringes. 10-µl and 50-µl liquid injection syringes.
6.2.12 Liquid Sample Injection Unit. 316 Stainless steel U-tube fitted with an injection septum (see Figure 25-7).
6.3 Analysis.
6.3.1 NMO Analyzer. The NMO analyzer is a gas chromatograph (GC) with backflush capability for NMO analysis and is equipped with an oxidation catalyst, reduction catalyst, and FID. Figures 25-5 and 25-6 are schematics of a typical NMO analyzer. This semicontinuous GC/FID analyzer shall be capable of: (1) Separating CO, CO2, and CH4 from NMO, (2) reducing the CO2 to CH4 and quantifying as CH4, and (3) oxidizing the NMO to CO2, reducing the CO2 to CH4 and quantifying as CH4, according to section 10.1.2. The analyzer consists of the following major components:
6.3.1.1 Oxidation Catalyst. A suitable length of 9.5-mm ( 3/8-in.) OD Inconel 600 tubing packed with 5.1 cm (2 in.) of 19 percent chromia on 3.2-mm ( 1/8-in.) alumina pellets. The catalyst material is packed in the center of the tube supported on either side by quartz wool. The catalyst tube must be mounted vertically in a 650°C (1200°F) furnace. Longer catalysts mounted horizontally may be used, provided they can meet the specifications of section 10.1.2.1.
6.3.1.2 Reduction Catalyst. A 7.6-cm (3-in.) length of 6.4-mm ( 1/4-in.) OD Inconel tubing fully packed with 100-mesh pure nickel powder. The catalyst tube must be mounted vertically in a 400°C (750°F) furnace.
6.3.1.3 Separation Column(s). A 30-cm (1-ft) length of 3.2-mm ( 1/8-in.) OD stainless steel tubing packed with 60/80 mesh Unibeads 1S followed by a 61-cm (2-ft) length of 3.2-mm ( 1/8-in.) OD stainless steel tubing packed with 60/80 mesh Carbosieve G. The Carbosieve and Unibeads columns must be baked separately at 200°C (390°F) with carrier gas flowing through them for 24 hours before initial use.
6.3.1.4 Sample Injection System. A single 10-port GC sample injection valve or a group of valves with sufficient ports fitted with a sample loop properly sized to interface with the NMO analyzer (1-cc loop recommended).
6.3.1.5 FID. An FID meeting the following specifications is required:
6.3.1.5.1 Linearity. A linear response (±5 percent) over the operating range as demonstrated by the procedures established in section 10.1.2.3.
6.3.1.5.2 Range. A full scale range of 10 to 50,000 ppm CH4. Signal attenuators shall be available to produce a minimum signal response of 10 percent of full scale.
6.3.1.6 Data Recording System. Analog strip chart recorder or digital integration system compatible with the FID for permanently recording the analytical results.
6.3.2 Barometer. Mercury, aneroid, or other barometer capable of measuring atmospheric pressure to within 1 mm Hg.
6.3.3 Temperature Sensor. Capable of measuring the laboratory temperature within 1°C (2°F).
6.3.4 Vacuum Pump. Capable of evacuating to an absolute pressure of 10 mm Hg.
7.0 Reagents and Standards
7.1 Sample Collection. The following reagents are required for sample collection:
7.1.1 Dry Ice. Solid CO2, crushed.
7.1.2 Coarse Quartz Wool. 8 to 15 um.
7.1.3 Filters. Glass fiber filters, without organic binder, exhibiting at least 99.95 percent efficiency (<0.05 percent penetration) on 0.3 micron dioctyl phthalate smoke particles. The filter efficiency test shall be conducted in accordance with ASTM Method D2986-71, 78, or 95a (incorporated by reference - see §60.17). Test data from the supplier's quality control program are sufficient for this purpose.
7.2 NMO Analysis. The following gases are required for NMO analysis:
7.2.1 Carrier Gases. Helium (He) and oxygen (O2) containing less than 1 ppm CO2 and less than 0.1 ppm hydrocarbon.
7.2.2 Fuel Gas. Hydrogen (H2), at least 99.999 percent pure.
7.2.3 Combustion Gas. Either air (less than 0.1 ppm total hydrocarbon content) or O2 (purity 99.99 percent or greater), as required by the detector.
7.3 Condensate Analysis. The following are required for condensate analysis:
7.3.1 Gases. Containing less than 1 ppm carbon.
7.3.1.1 Air.
7.3.1.2 Oxygen.
7.3.2 Liquids. To conform to the specifications established by the Committee on Analytical Reagents of the American Chemical Society.
7.3.2.1 Hexane.
7.3.2.2 Decane.
7.4 Calibration. For all calibration gases, the manufacturer must recommend a maximum shelf life for each cylinder (i.e., the length of time the gas concentration is not expected to change more than ±5 percent from its certified value). The date of gas cylinder preparation, certified organic concentration, and recommended maximum shelf life must be affixed to each cylinder before shipment from the gas manufacturer to the buyer. The following calibration gases are required:
7.4.1 Oxidation Catalyst Efficiency Check Calibration Gas. Gas mixture standard with nominal concentration of 1 percent methane in air.
7.4.2 FID Linearity and NMO Calibration Gases. Three gas mixture standards with nominal propane concentrations of 20 ppm, 200 ppm, and 3000 ppm, in air.
7.4.3 CO2 Calibration Gases. Three gas mixture standards with nominal CO2 concentrations of 50 ppm, 500 ppm, and 1 percent, in air.
Note:
Total NMO less than 1 ppm required for 1 percent mixture.
7.4.4 NMO Analyzer System Check Calibration Gases. Four calibration gases are needed as follows:
7.4.4.1 Propane Mixture. Gas mixture standard containing (nominal) 50 ppm CO, 50 ppm CH4, 1 percent CO2, and 20 ppm C3H8, prepared in air.
7.4.4.2 Hexane. Gas mixture standard containing (nominal) 50 ppm hexane in air.
7.4.4.3 Toluene. Gas mixture standard containing (nominal) 20 ppm toluene in air.
7.4.4.4 Methanol. Gas mixture standard containing (nominal) 100 ppm methanol in air.
8.0 Sample Collection, Preservation, Transport, and Storage
8.1 Sampling Equipment Preparation.
8.1.1 Condensate Trap Cleaning. Before its initial use and after each use, a condensate trap should be thoroughly cleaned and checked to ensure that it is not contaminated. Both cleaning and checking can be accomplished by installing the trap in the condensate recovery system and treating it as if it were a sample. The trap should be heated as described in section 11.1.3. A trap may be considered clean when the CO2 concentration in its effluent gas drops below 10 ppm. This check is optional for traps that most recently have been used to collect samples which were then recovered according to the procedure in section 11.1.3.
8.1.2 Sample Tank Evacuation and Leak-Check. Evacuate the sample tank to 10 mm Hg absolute pressure or less. Then close the sample tank valve, and allow the tank to sit for 60 minutes. The tank is acceptable if a change in tank vacuum of less than 1 mm Hg is noted. The evacuation and leak-check may be conducted either in the laboratory or the field.
8.1.3 Sampling Train Assembly. Just before assembly, measure the tank vacuum using a mercury manometer. Record this vacuum, the ambient temperature, and the barometric pressure at this time. Close the sample tank valve and assemble the sampling system as shown in Figure 25-1. Immerse the condensate trap body in dry ice at least 30 minutes before commencing sampling to improve collection efficiency. The point where the inlet tube joins the trap body should be 2.5 to 5 cm (1 to 2 in.) above the top of the dry ice.
8.1.4 Pretest Leak-Check. A pretest leak-check is required. Calculate or measure the approximate volume of the sampling train from the probe tip to the sample tank valve. After assembling the sampling train, plug the probe tip, and make certain that the sample tank valve is closed. Turn on the vacuum pump, and evacuate the sampling system from the probe tip to the sample tank valve to an absolute pressure of 10 mm Hg or less. Close the purge valve, turn off the pump, wait a minimum period of 10 minutes, and recheck the indicated vacuum. Calculate the maximum allowable pressure change based on a leak rate of 1 percent of the sampling rate using Equation 25-1, section 12.2. If the measured pressure change exceeds the allowable, correct the problem and repeat the leak-check before beginning sampling.
8.2 Sample Collection.
8.2.1 Unplug the probe tip, and place the probe into the stack such that the probe is perpendicular to the duct or stack axis; locate the probe tip at a single preselected point of average velocity facing away from the direction of gas flow. For stacks having a negative static pressure, seal the sample port sufficiently to prevent air in-leakage around the probe. Set the probe temperature controller to 129°C (265°F) and the filter temperature controller to 121°C (250°F). Allow the probe and filter to heat for about 30 minutes before purging the sample train.
8.2.2 Close the sample valve, open the purge valve, and start the vacuum pump. Set the flow rate between 60 and 100 cm 3/min (0.13 and 0.21 ft 3/hr), and purge the train with stack gas for at least 10 minutes.
8.2.3 When the temperatures at the exit ends of the probe and filter are within the corresponding specified ranges, check the dry ice level around the condensate trap, and add dry ice if necessary. Record the clock time. To begin sampling, close the purge valve and stop the pump. Open the sample valve and the sample tank valve. Using the flow control valve, set the flow through the sample train to the proper rate. Adjust the flow rate as necessary to maintain a constant rate (±10 percent) throughout the duration of the sampling period. Record the sample tank vacuum and flowmeter setting at 5-minute intervals. (See Figure 25-8.) Select a total sample time greater than or equal to the minimum sampling time specified in the applicable subpart of the regulations; end the sampling when this time period is reached or when a constant flow rate can no longer be maintained because of reduced sample tank vacuum.
Note:
If sampling had to be stopped before obtaining the minimum sampling time (specified in the applicable subpart) because a constant flow rate could not be maintained, proceed as follows: After closing the sample tank valve, remove the used sample tank from the sampling train (without disconnecting other portions of the sampling train). Take another evacuated and leak-checked sample tank, measure and record the tank vacuum, and attach the new tank to the sampling train. After the new tank is attached to the sample train, proceed with the sampling until the required minimum sampling time has been exceeded.
8.3 Sample Recovery. After sampling is completed, close the flow control valve, and record the final tank vacuum; then record the tank temperature and barometric pressure. Close the sample tank valve, and disconnect the sample tank from the sample system. Disconnect the condensate trap at the inlet to the rate meter, and tightly seal both ends of the condensate trap. Do not include the probe from the stack to the filter as part of the condensate sample.
8.4 Sample Storage and Transport. Keep the trap packed in dry ice until the samples are returned to the laboratory for analysis. Ensure that run numbers are identified on the condensate trap and the sample tank(s).
9.0 Quality Control
| Section | Quality control measure | Effect |
|---|---|---|
| 10.1.1 | Initial performance check of condensate recovery apparatus | Ensure acceptable condensate recovery efficiency. |
| 10.1.2, 10.2 | NMO analyzer initial and daily performance checks | Ensure precision of analytical results. |
10.0 Calibration and Standardization
Note:
Maintain a record of performance of each item.
10.1 Initial Performance Checks.
10.1.1 Condensate Recovery Apparatus. Perform these tests before the system is first placed in operation, after any shutdown of 6 months or more, and after any major modification of the system, or at the frequency recommended by the manufacturer.
10.1.1.1 Carrier Gas and Auxiliary O2 Blank Check. Analyze each new tank of carrier gas or auxiliary O2 with the NMO analyzer to check for contamination. Treat the gas cylinders as noncondensible gas samples, and analyze according to the procedure in section 11.2.3. Add together any measured CH4, CO, CO2, or NMO. The total concentration must be less than 5 ppm.
10.1.1.2 Oxidation Catalyst Efficiency Check.
10.1.1.2.1 With a clean condensate trap installed in the recovery system or a 1/8″ stainless steel connector tube, replace the carrier gas cylinder with the high level methane standard gas cylinder (Section 7.4.1). Set the four-port valve to the recovery position, and attach an ICV to the recovery system. With the sample recovery valve in vent position and the flow-control and ICV valves fully open, evacuate the manometer or gauge, the connecting tubing, and the ICV to 10 mm Hg absolute pressure. Close the flow-control and vacuum pump valves.
10.1.1.2.2 After the NDIR response has stabilized, switch the sample recovery valve from vent to collect. When the manometer or pressure gauge begins to register a slight positive pressure, open the flow-control valve. Keep the flow adjusted such that the pressure in the system is maintained within 10 percent of atmospheric pressure. Continue collecting the sample in a normal manner until the ICV is filled to a nominal gauge pressure of 300 mm Hg. Close the ICV valve, and remove the ICV from the system. Place the sample recovery valve in the vent position, and return the recovery system to its normal carrier gas and normal operating conditions. Analyze the ICV for CO2 using the NMO analyzer; the catalyst efficiency is acceptable if the CO2 concentration is within 2 percent of the methane standard concentration.
10.1.1.3 System Performance Check. Construct a liquid sample injection unit similar in design to the unit shown in Figure 25-7. Insert this unit into the condensate recovery and conditioning system in place of a condensate trap, and set the carrier gas and auxiliary O2 flow rates to normal operating levels. Attach an evacuated ICV to the system, and switch from system vent to collect. With the carrier gas routed through the injection unit and the oxidation catalyst, inject a liquid sample (see sections 10.1.1.3.1 to 10.1.1.3.4) into the injection port. Operate the trap recovery system as described in section 11.1.3. Measure the final ICV pressure, and then analyze the vessel to determine the CO2 concentration. For each injection, calculate the percent recovery according to section 12.7. Calculate the relative standard deviation for each set of triplicate injections according to section 12.8. The performance test is acceptable if the average percent recovery is 100 ±5 percent and the relative standard deviation is less than 2 percent for each set of triplicate injections.
10.1.1.3.1 50 µl hexane.
10.1.1.3.2 10 µl hexane.
10.1.1.3.3 50 µl decane.
10.1.1.3.4 10 µl decane.
10.1.2 NMO Analyzer. Perform these tests before the system is first placed in operation, after any shutdown longer than 6 months, and after any major modification of the system.
10.1.2.1 Oxidation Catalyst Efficiency Check. Turn off or bypass the NMO analyzer reduction catalyst. Make triplicate injections of the high level methane standard (Section 7.4.1). The oxidation catalyst operation is acceptable if the FID response is less than 1 percent of the injected methane concentration.
10.1.2.2 Reduction Catalyst Efficiency Check. With the oxidation catalyst unheated or bypassed and the heated reduction catalyst bypassed, make triplicate injections of the high level methane standard (Section 7.4.1). Repeat this procedure with both catalysts operative. The reduction catalyst operation is acceptable if the responses under both conditions agree within 5 percent of their average.
10.1.2.3 NMO Analyzer Linearity Check Calibration. While operating both the oxidation and reduction catalysts, conduct a linearity check of the analyzer using the propane standards specified in section 7.4.2. Make triplicate injections of each calibration gas. For each gas (i.e., each set of triplicate injections), calculate the average response factor (area/ppm C) for each gas, as well as and the relative standard deviation (according to section 12.8). Then calculate the overall mean of the response factor values. The instrument linearity is acceptable if the average response factor of each calibration gas is within 2.5 percent of the overall mean value and if the relative standard deviation gas is less than 2 percent of the overall mean value. Record the overall mean of the propane response factor values as the NMO calibration response factor (RFNMO). Repeat the linearity check using the CO2 standards specified in section 7.4.3. Make triplicate injections of each gas, and then calculate the average response factor (area/ppm C) for each gas, as well as the overall mean of the response factor values. Record the overall mean of the response factor values as the CO2 calibration response factor (RFCO2). The RFCO2 must be within 10 percent of the RFNMO.
10.1.2.4 System Performance Check. Check the column separation and overall performance of the analyzer by making triplicate injections of the calibration gases listed in section 7.4.4. The analyzer performance is acceptable if the measured NMO value for each gas (average of triplicate injections) is within 5 percent of the expected value.
10.2 NMO Analyzer Daily Calibration. The following calibration procedures shall be performed before and immediately after the analysis of each set of samples, or on a daily basis, whichever is more stringent:
10.2.1 CO2 Response Factor. Inject triplicate samples of the high level CO2 calibration gas (Section 7.4.3), and calculate the average response factor. The system operation is adequate if the calculated response factor is within 5 percent of the RFCO2 calculated during the initial performance test (Section 10.1.2.3). Use the daily response factor (DRFCO2) for analyzer calibration and the calculation of measured CO2 concentrations in the ICV samples.
10.2.2 NMO Response Factors. Inject triplicate samples of the mixed propane calibration cylinder gas (Section 7.4.4.1), and calculate the average NMO response factor. The system operation is adequate if the calculated response factor is within 10 percent of the RFNMO calculated during the initial performance test (Section 10.1.2.4). Use the daily response factor (DRFNMO) for analyzer calibration and calculation of NMO concentrations in the sample tanks.
10.3 Sample Tank and ICV Volume. The volume of the gas sampling tanks used must be determined. Determine the tank and ICV volumes by weighing them empty and then filled with deionized distilled water; weigh to the nearest 5 g, and record the results. Alternatively, measure the volume of water used to fill them to the nearest 5 ml.
11.0 Analytical Procedure
11.1 Condensate Recovery. See Figure 25-9. Set the carrier gas flow rate, and heat the catalyst to its operating temperature to condition the apparatus.
11.1.1 Daily Performance Checks. Each day before analyzing any samples, perform the following tests:
11.1.1.1 Leak-Check. With the carrier gas inlets and the sample recovery valve closed, install a clean condensate trap in the system, and evacuate the system to 10 mm Hg absolute pressure or less. Monitor the system pressure for 10 minutes. The system is acceptable if the pressure change is less than 2 mm Hg.
11.1.1.2 System Background Test. Adjust the carrier gas and auxiliary oxygen flow rate to their normal values of 100 cc/min and 150 cc/min, respectively, with the sample recovery valve in vent position. Using a 10-ml syringe, withdraw a sample from the system effluent through the syringe port. Inject this sample into the NMO analyzer, and measure the CO2 content. The system background is acceptable if the CO2 concentration is less than 10 ppm.
11.1.1.3 Oxidation Catalyst Efficiency Check. Conduct a catalyst efficiency test as specified in section 10.1.1.2. If the criterion of this test cannot be met, make the necessary repairs to the system before proceeding.
11.1.2 Condensate Trap CO2 Purge and Sample Tank Pressurization.
11.1.2.1 After sampling is completed, the condensate trap will contain condensed water and organics and a small volume of sampled gas. This gas from the stack may contain a significant amount of CO2 which must be removed from the condensate trap before the sample is recovered. This is accomplished by purging the condensate trap with zero air and collecting the purged gas in the original sample tank.
11.1.2.2 Begin with the sample tank and condensate trap from the test run to be analyzed. Set the four-port valve of the condensate recovery system in the CO2 purge position as shown in Figure 25-9. With the sample tank valve closed, attach the sample tank to the sample recovery system. With the sample recovery valve in the vent position and the flow control valve fully open, evacuate the manometer or pressure gauge to the vacuum of the sample tank. Next, close the vacuum pump valve, open the sample tank valve, and record the tank pressure.
11.1.2.3 Attach the dry ice-cooled condensate trap to the recovery system, and initiate the purge by switching the sample recovery valve from vent to collect position. Adjust the flow control valve to maintain atmospheric pressure in the recovery system. Continue the purge until the CO2 concentration of the trap effluent is less than 5 ppm. CO2 concentration in the trap effluent should be measured by extracting syringe samples from the recovery system and analyzing the samples with the NMO analyzer. This procedure should be used only after the NDIR response has reached a minimum level. Using a 10-ml syringe, extract a sample from the syringe port prior to the NDIR, and inject this sample into the NMO analyzer.
11.1.2.4 After the completion of the CO2 purge, use the carrier gas bypass valve to pressurize the sample tank to approximately 1,060 mm Hg absolute pressure with zero air.
11.1.3 Recovery of the Condensate Trap Sample (See Figure 25-10).
11.1.3.1 Attach the ICV to the sample recovery system. With the sample recovery valve in a closed position, between vent and collect, and the flow control and ICV valves fully open, evacuate the manometer or gauge, the connecting tubing, and the ICV to 10 mm Hg absolute pressure. Close the flow-control and vacuum pump valves.
11.1.3.2 Begin auxiliary oxygen flow to the oxidation catalyst at a rate of 150 cc/min, then switch the four-way valve to the trap recovery position and the sample recovery valve to collect position. The system should now be set up to operate as indicated in Figure 25-10. After the manometer or pressure gauge begins to register a slight positive pressure, open the flow control valve. Adjust the flow-control valve to maintain atmospheric pressure in the system within 10 percent.
11.1.3.3 Remove the condensate trap from the dry ice, and allow it to warm to ambient temperature while monitoring the NDIR response. If, after 5 minutes, the CO2 concentration of the catalyst effluent is below 10,000 ppm, discontinue the auxiliary oxygen flow to the oxidation catalyst. Begin heating the trap by placing it in a furnace preheated to 200°C (390°F). Once heating has begun, carefully monitor the NDIR response to ensure that the catalyst effluent concentration does not exceed 50,000 ppm. Whenever the CO2 concentration exceeds 50,000 ppm, supply auxiliary oxygen to the catalyst at the rate of 150 cc/min. Begin heating the tubing that connected the heated sample box to the condensate trap only after the CO2 concentration falls below 10,000 ppm. This tubing may be heated in the same oven as the condensate trap or with an auxiliary heat source such as a heat gun. Heating temperature must not exceed 200°C (390°F). If a heat gun is used, heat the tubing slowly along its entire length from the upstream end to the downstream end, and repeat the pattern for a total of three times. Continue the recovery until the CO2 concentration drops to less than 10 ppm as determined by syringe injection as described under the condensate trap CO2 purge procedure (Section 11.1.2).
11.1.3.4 After the sample recovery is completed, use the carrier gas bypass valve to pressurize the ICV to approximately 1060 mm Hg absolute pressure with zero air.
11.2 Analysis. Once the initial performance test of the NMO analyzer has been successfully completed (see section 10.1.2) and the daily CO2 and NMO response factors have been determined (see section 10.2), proceed with sample analysis as follows:
11.2.1 Operating Conditions. The carrier gas flow rate is 29.5 cc/min He and 2.2 cc/min O2. The column oven is heated to 85°C (185°F). The order of elution for the sample from the column is CO, CH4, CO2, and NMO.
11.2.2 Analysis of Recovered Condensate Sample. Purge the sample loop with sample, and then inject the sample. Under the specified operating conditions, the CO2 in the sample will elute in approximately 100 seconds. As soon as the detector response returns to baseline following the CO2 peak, switch the carrier gas flow to backflush, and raise the column oven temperature to 195°C (380°F) as rapidly as possible. A rate of 30°C/min (90°F) has been shown to be adequate. Record the value obtained for the condensible organic material (Ccm) measured as CO2 and any measured NMO. Return the column oven temperature to 85°C (185°F) in preparation for the next analysis. Analyze each sample in triplicate, and report the average Ccm.
11.2.3 Analysis of Sample Tank. Perform the analysis as described in section 11.2.2, but record only the value measured for NMO (Ctm).
12.0 Data Analysis and Calculations
Carry out the calculations, retaining at least one extra significant figure beyond that of the acquired data. Round off figures after final calculations. All equations are written using absolute pressure; absolute pressures are determined by adding the measured barometric pressure to the measured gauge or manometer pressure.
12.1 Nomenclature.
C = TGNMO concentration of the effluent, ppm C equivalent.
Cc = Calculated condensible organic (condensate trap) concentration of the effluent, ppm C equivalent.
Ccm = Measured concentration (NMO analyzer) for the condensate trap ICV, ppm CO2.
Ct = Calculated noncondensible organic concentration (sample tank) of the effluent, ppm C equivalent.
Ctm = Measured concentration (NMO analyzer) for the sample tank, ppm NMO.
F = Sampling flow rate, cc/min.
L = Volume of liquid injected, µl.
M = Molecular weight of the liquid injected, g/g-mole.
Mc = TGNMO mass concentration of the effluent, mg C/dsm 3.
N = Carbon number of the liquid compound injected (N = 12 for decane, N = 6 for hexane).
n = Number of data points.
Pf = Final pressure of the intermediate collection vessel, mm Hg absolute.
Pb = Barometric pressure, cm Hg.
Pti = Gas sample tank pressure before sampling, mm Hg absolute.
Pt = Gas sample tank pressure after sampling, but before pressurizing, mm Hg absolute.
Ptf = Final gas sample tank pressure after pressurizing, mm Hg absolute.
q = Total number of analyzer injections of intermediate collection vessel during analysis (where k = injection number, 1 * * * q).
r = Total number of analyzer injections of sample tank during analysis (where j = injection number, 1 * * * r).
r = Density of liquid injected, g/cc.
Tf = Final temperature of intermediate collection vessel,°K.
Tti = Sample tank temperature before sampling,°K.
Tt = Sample tank temperature at completion of sampling,°K.
Ttf = Sample tank temperature after pressurizing,°K.
V = Sample tank volume, m 3.
Vt = Sample train volume, cc.
Vv = Intermediate collection vessel volume, m 3.
Vs = Gas volume sampled, dsm 3.
xi = Individual measurements.
x͞ = Mean value.
ΔP = Allowable pressure change, cm Hg.
Θ = Leak-check period, min.
12.2 Allowable Pressure Change. For the pretest leak-check, calculate the allowable pressure change using Equation 25-1:
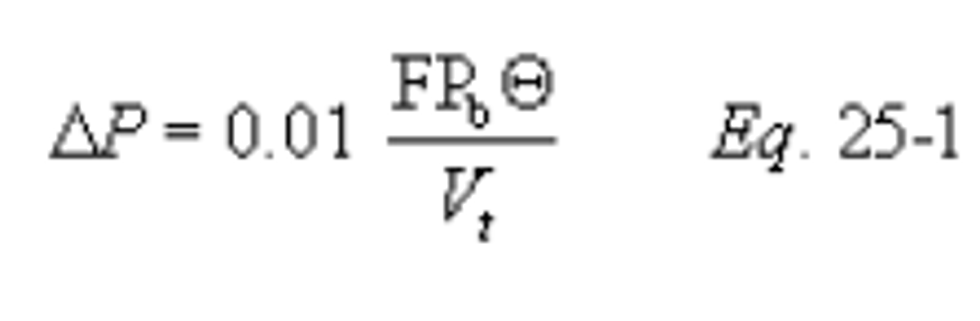
12.3 Sample Volume. For each test run, calculate the gas volume sampled using Equation 25-2:
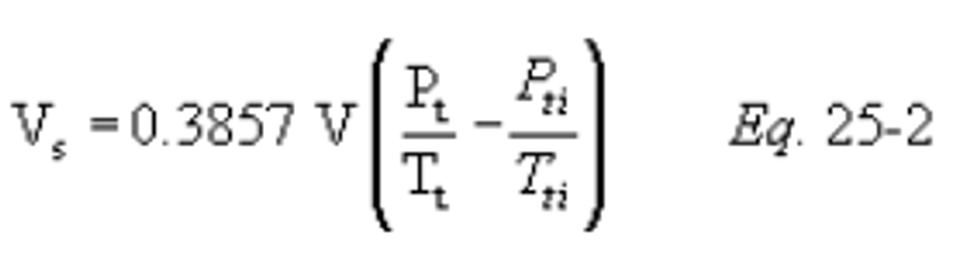
12.4 Noncondensible Organics. For each sample tank, determine the concentration of nonmethane organics (ppm C) using Equation 25-3:
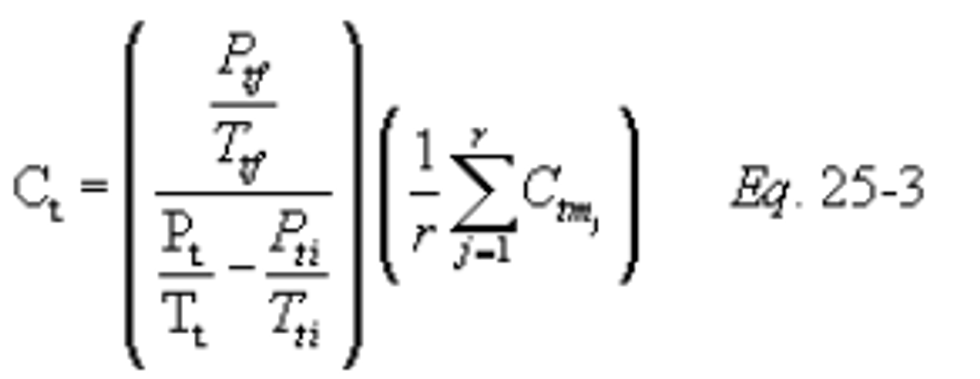
12.5 Condensible Organics. For each condensate trap determine the concentration of organics (ppm C) using Equation 25-4:
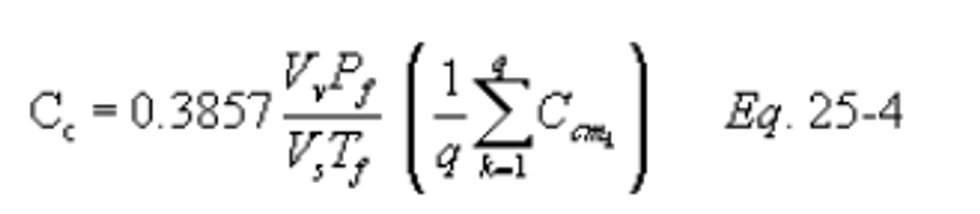
12.6 TGNMO Mass Concentration. Determine the TGNMO mass concentration as carbon for each test run, using Equation 25-5:
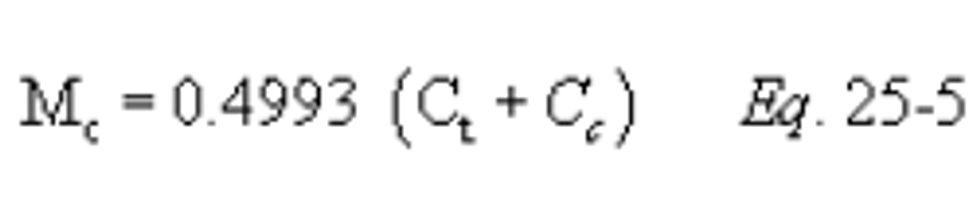
12.7 Percent Recovery. Calculate the percent recovery for the liquid injections to the condensate recovery and conditioning system using Equation 25-6:
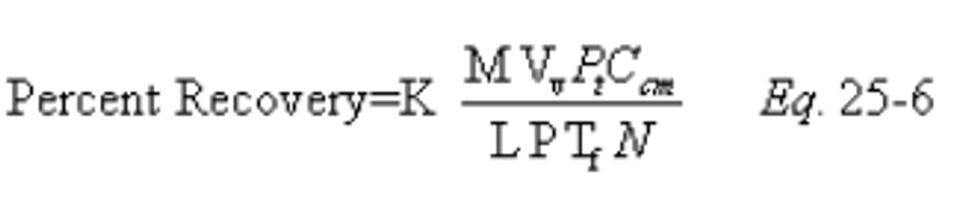
where K = 1.604 (°K)(g-mole)(%)/(mm Hg)(ml)(m 3)(ppm).
12.8 Relative Standard Deviation. Use Equation 25-7 to calculate the relative standard deviation (RSD) of percent recovery and analyzer linearity.
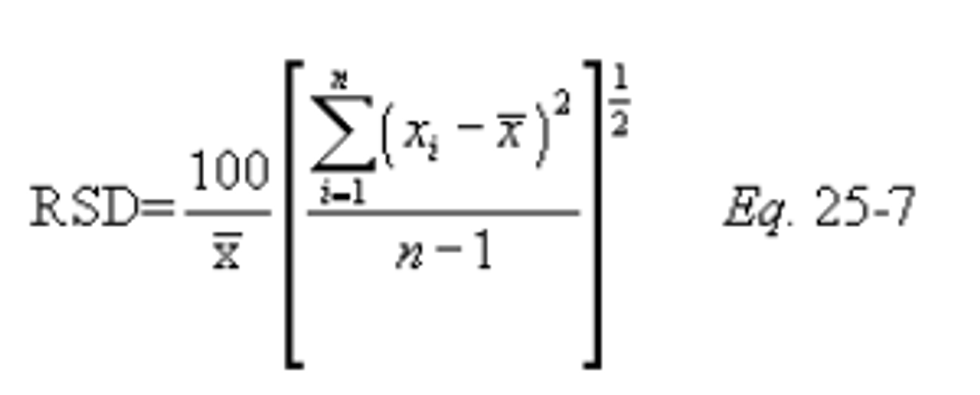
12.9 Record and Report Initial Method Checks as follows:
12.9.1 Calibration and Linearity Check Gas Certifications (sections 7.2 and 7.4 of this method).
12.9.2 Condensate Trap Blank Check (section 8.1.1 of this method).
12.9.3 Pretest Leak-Check (section 8.1.4 of this method).
12.9.4 Condensate Recovery Apparatus (section 10.1.1 of this method).
12.9.5 Carrier Gas and Auxiliary O 2 Blank Check (section 10.1.1.1 of this method).
12.9.6 Oxidation Catalyst Efficiency Check (section 10.1.1.2 of this method).
12.9.7 System Performance Check (section 10.1.1.3 of this method).
12.9.8 Oxidation Catalyst Efficiency Check (section 10.1.2.1 of this method).
12.9.9 Reduction Catalyst Efficiency Check (section 10.1.2.2 of this method).
12.9.10 NMO Analyzer Linearity Check Calibration (section 10.1.2.3 of this method).
12.9.11 NMO Analyzer Daily Calibration (section 10.2 of this method).
12.9.12 Condensate Recovery (section 11.1 of this method).
12.9.13 Daily Performance Checks (section 11.1.1 of this method).
12.9.14 Leak-Check (section 11.1.1.1 of this method).
12.9.15 System Background Test (section 11.1.1.2 of this method).
12.9.16 Oxidation Catalyst Efficiency Check (section 11.1.1.3 of this method).
13.0 Method Performance
13.1 Range. The minimum detectable limit of the method has been determined to be 50 parts per million by volume (ppm). No upper limit has been established.
14.0 Pollution Prevention [Reserved]
15.0 Waste Management [Reserved]
16.0 References
1. Salo, A.E., S. Witz, and R.D. MacPhee. Determination of Solvent Vapor Concentrations by Total Combustion Analysis: A Comparison of Infrared with Flame Ionization Detectors. Paper No. 75-33.2. (Presented at the 68th Annual Meeting of the Air Pollution Control Association. Boston, MA. June 15-20, 1975.) 14 p.
2. Salo, A.E., W.L. Oaks, and R.D. MacPhee. Measuring the Organic Carbon Content of Source Emissions for Air Pollution Control. Paper No. 74-190. (Presented at the 67th Annual Meeting of the Air Pollution Control Association. Denver, CO. June 9-13, 1974.) 25 p.
17.0 Tables, Diagrams, Flowcharts, and Validation Data
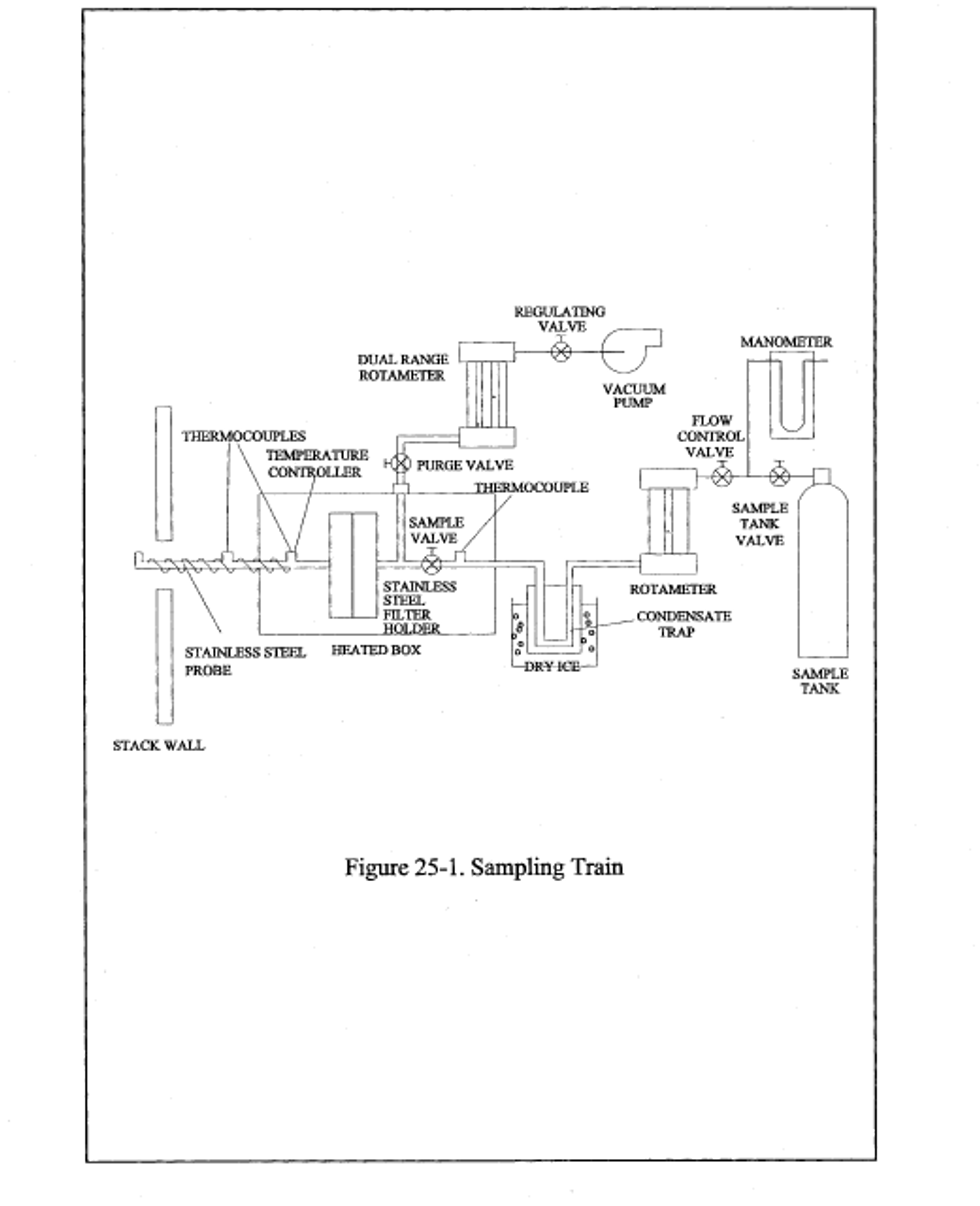
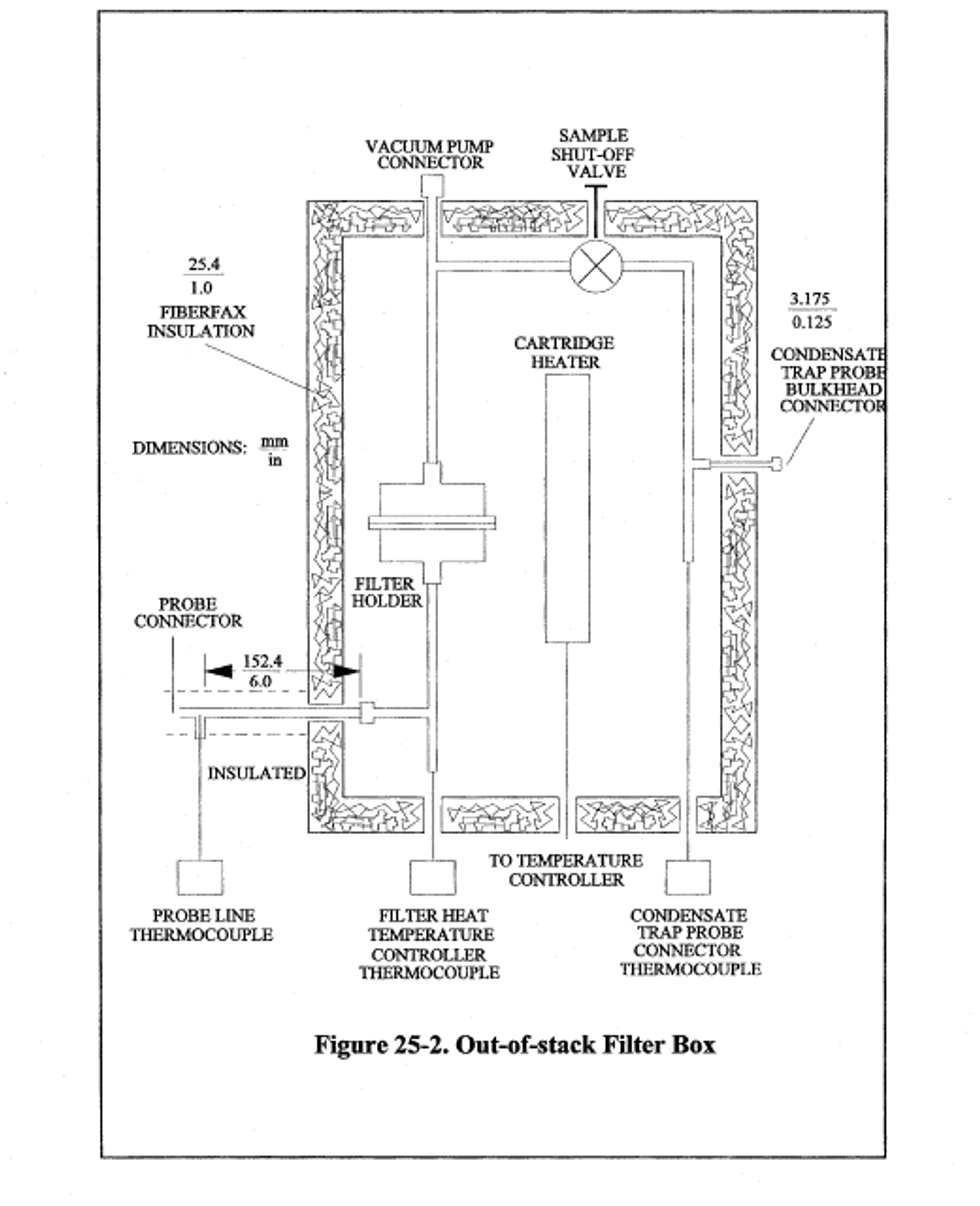
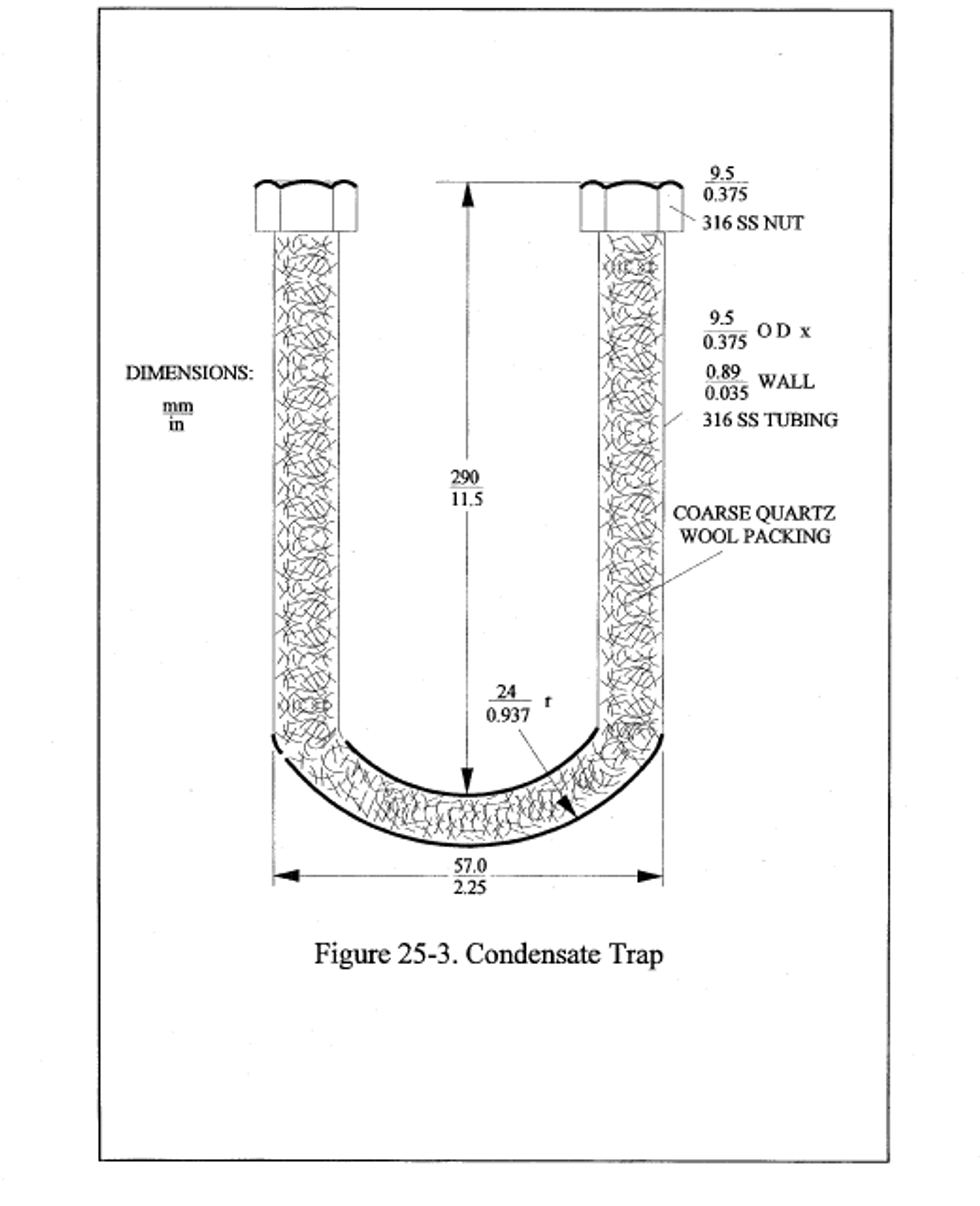
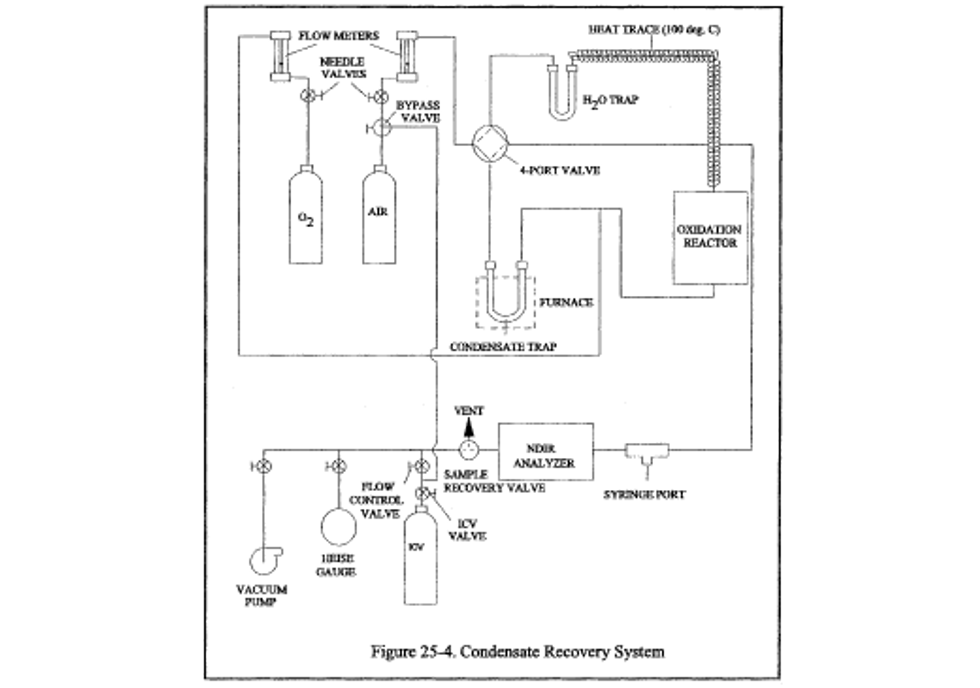
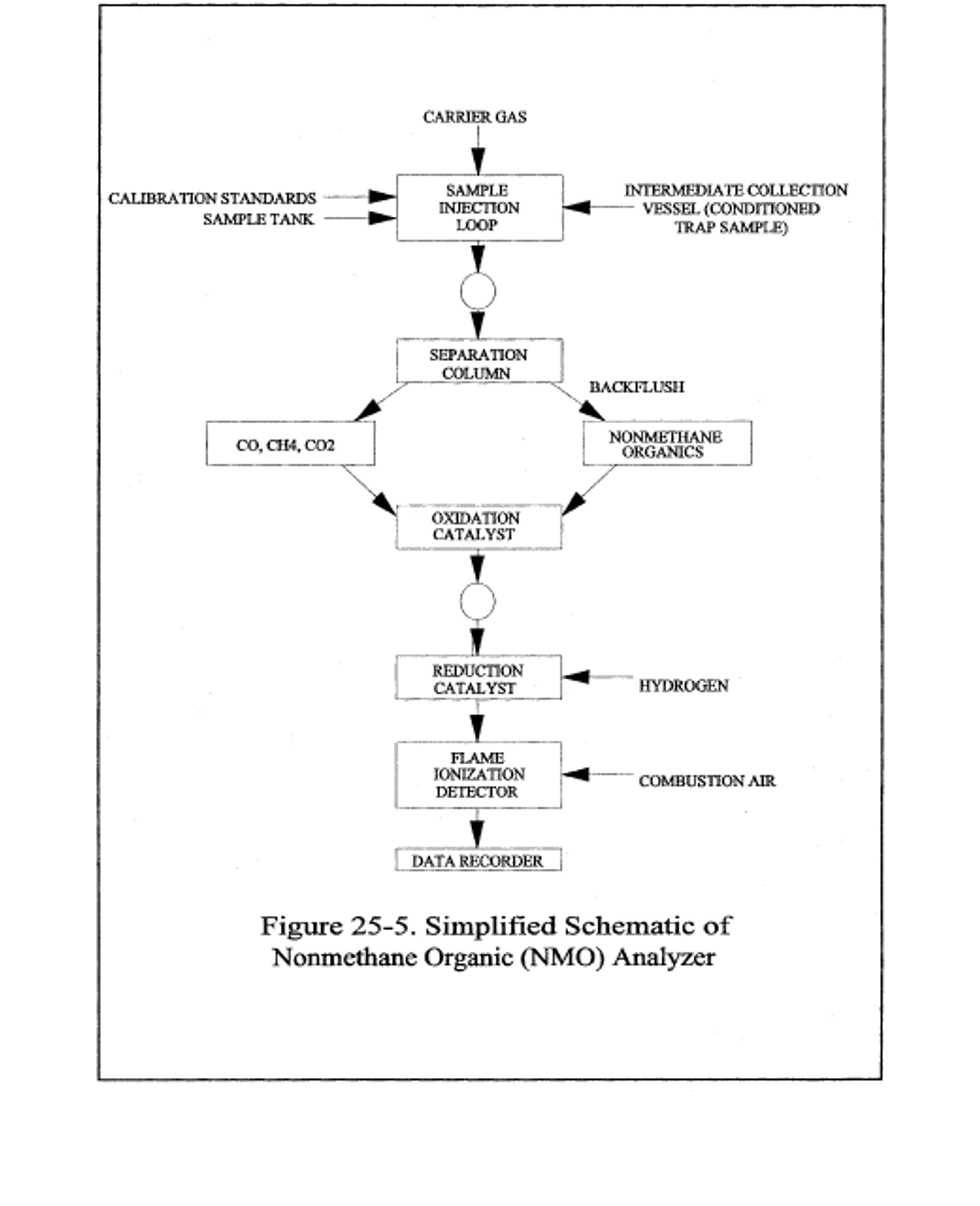
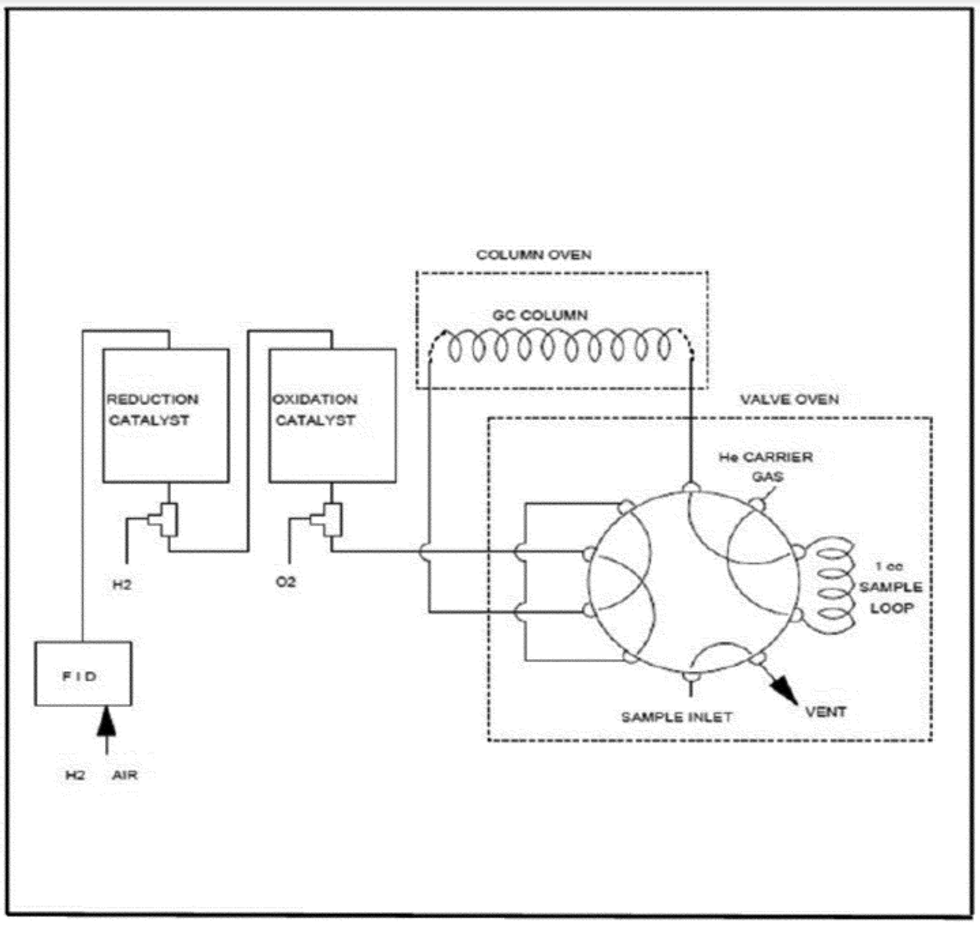
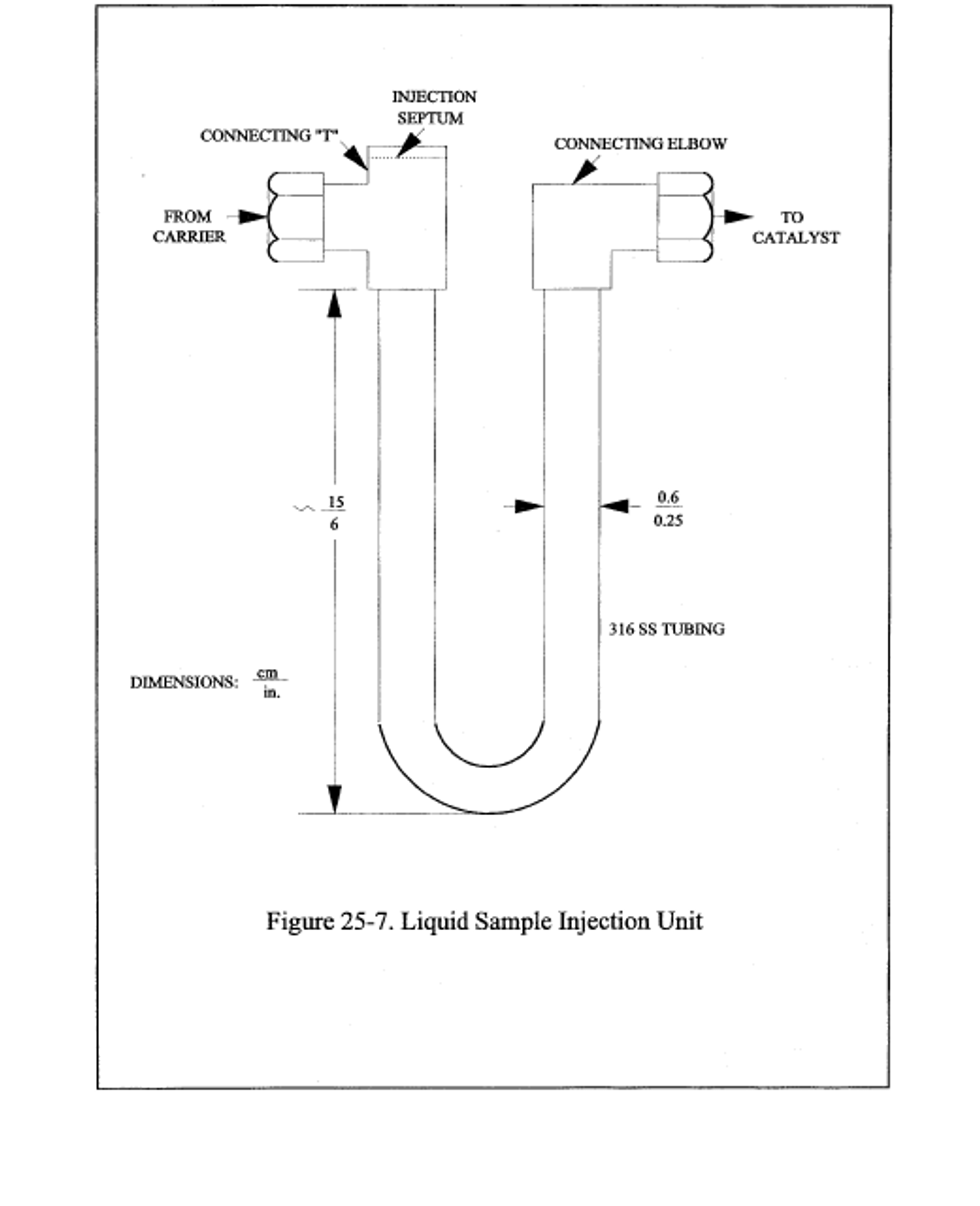

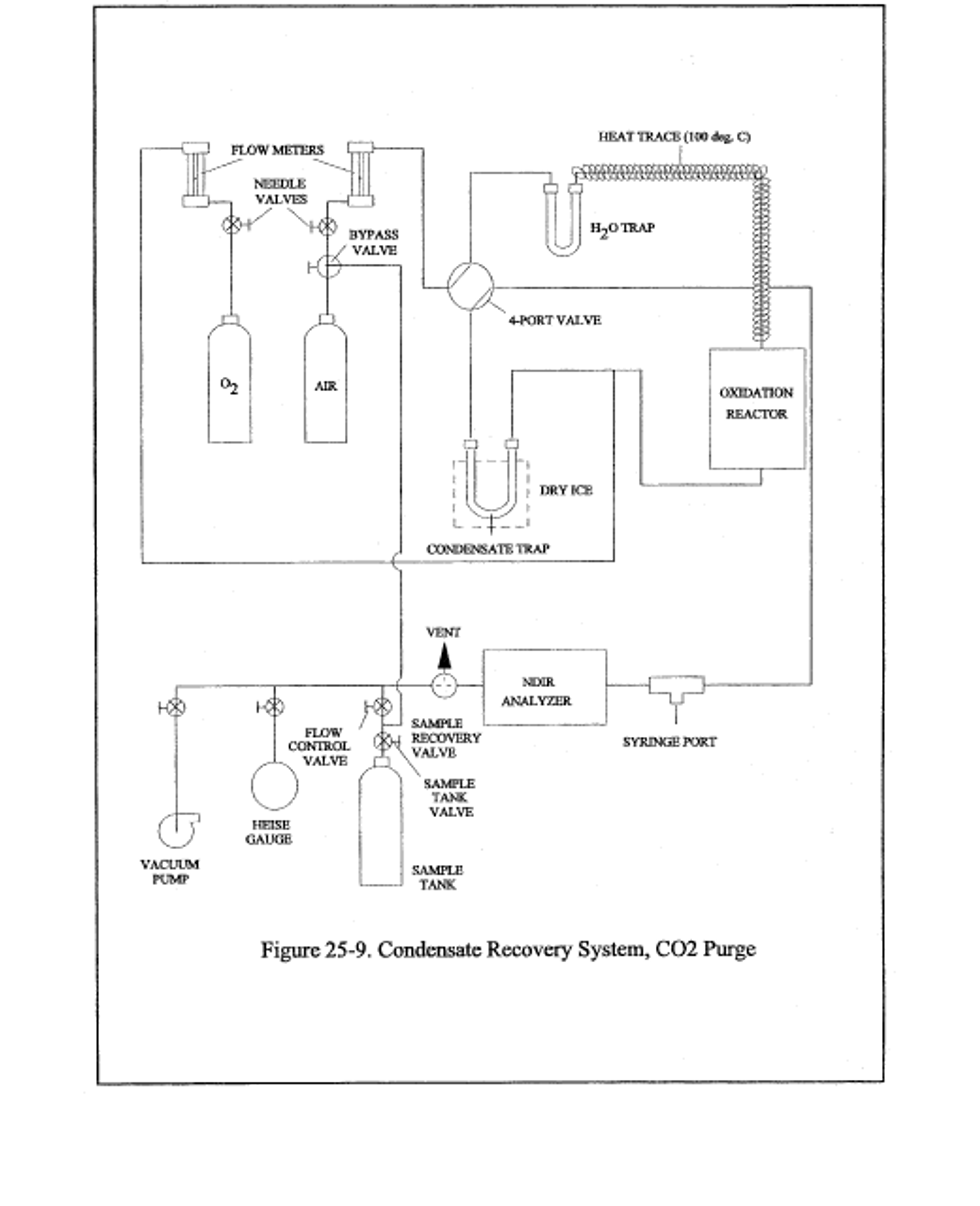
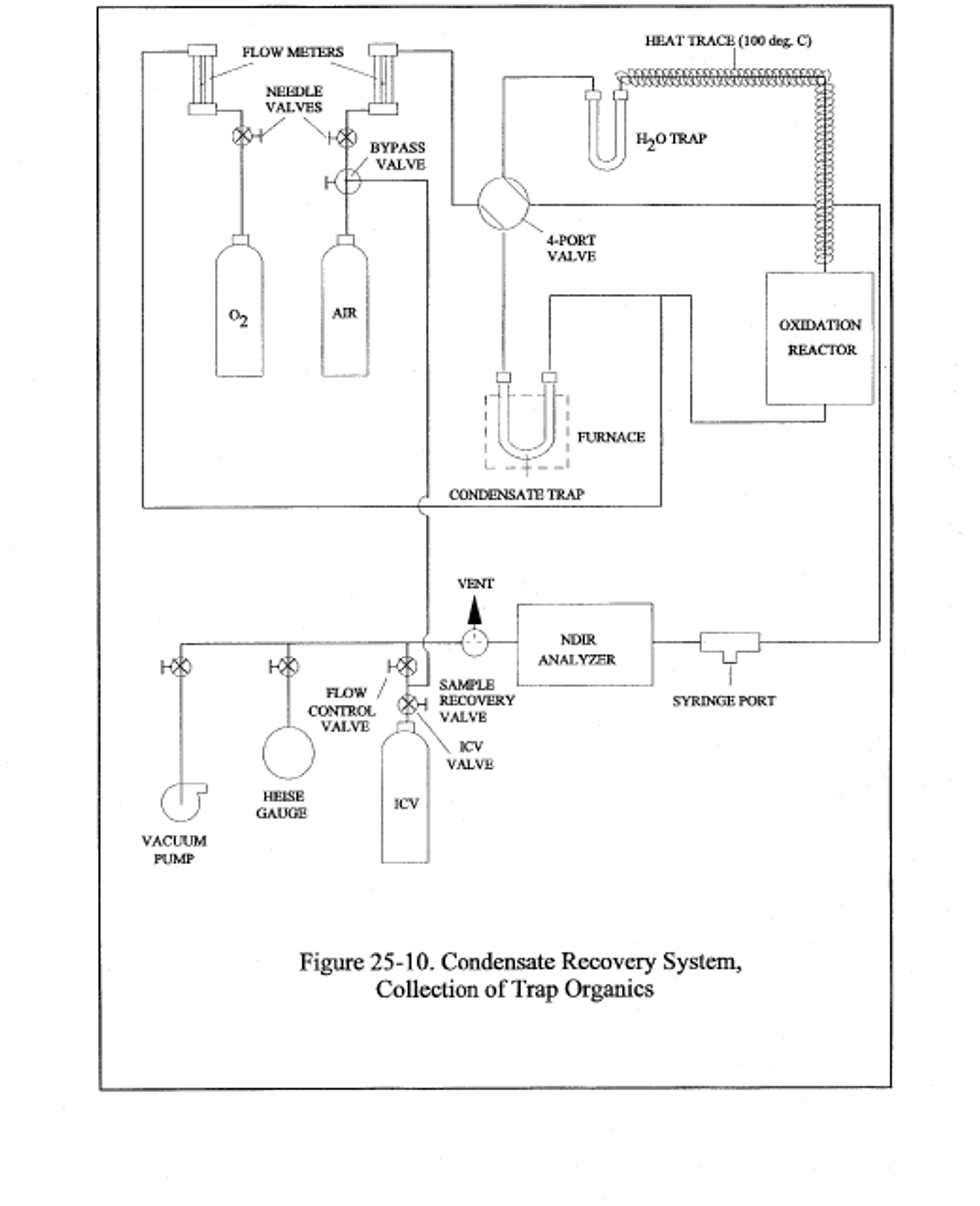
Method 25A - Determination of Total Gaseous Organic Concentration Using a Flame Ionization Analyzer
1.0 Scope and Application
1.1 Analytes.
| Analyte | CAS No. | Sensitivity |
|---|---|---|
| Total Organic Compounds | N/A | <2% of span. |
1.2 Applicability. This method is applicable for the determination of total gaseous organic concentration of vapors consisting primarily of alkanes, alkenes, and/or arenes (aromatic hydrocarbons). The concentration is expressed in terms of propane (or other appropriate organic calibration gas) or in terms of carbon.
1.3 Data Quality Objectives. Adherence to the requirements of this method will enhance the quality of the data obtained from air pollutant sampling methods.
2.0 Summary of Method
2.1 A gas sample is extracted from the source through a heated sample line and glass fiber filter to a flame ionization analyzer (FIA). Results are reported as volume concentration equivalents of the calibration gas or as carbon equivalents.
3.0 Definitions
3.1 Calibration drift means the difference in the measurement system response to a mid-level calibration gas before and after a stated period of operation during which no unscheduled maintenance, repair, or adjustment took place.
3.2 Calibration error means the difference between the gas concentration indicated by the measurement system and the know concentration of the calibration gas.
3.3 Calibration gas means a known concentration of a gas in an appropriate diluent gas.
3.4 Measurement system means the total equipment required for the determination of the gas concentration. The system consists of the following major subsystems:
3.4.1 Sample interface means that portion of a system used for one or more of the following: sample acquisition, sample transportation, sample conditioning, or protection of the analyzer(s) from the effects of the stack effluent.
3.4.2 Organic analyzer means that portion of the measurement system that senses the gas to be measured and generates an output proportional to its concentration.
3.5 Response time means the time interval from a step change in pollutant concentration at the inlet to the emission measurement system to the time at which 95 percent of the corresponding final value is reached as displayed on the recorder.
3.6 Span Value means the upper limit of a gas concentration measurement range that is specified for affected source categories in the applicable part of the regulations. The span value is established in the applicable regulation and is usually 1.5 to 2.5 times the applicable emission limit. If no span value is provided, use a span value equivalent to 1.5 to 2.5 times the expected concentration. For convenience, the span value should correspond to 100 percent of the recorder scale.
3.7 Zero drift means the difference in the measurement system response to a zero level calibration gas before or after a stated period of operation during which no unscheduled maintenance, repair, or adjustment took place.
4.0 Interferences [Reserved]
5.0 Safety
5.1 Disclaimer. This method may involve hazardous materials, operations, and equipment. This test method may not address all of the safety problems associated with its use. It is the responsibility of the user of this test method to establish appropriate safety and health practices and determine the applicability of regulatory limitations prior to performing this test method. The analyzer users manual should be consulted for specific precautions to be taken with regard to the analytical procedure.
5.2 Explosive Atmosphere. This method is often applied in highly explosive areas. Caution and care should be exercised in choice of equipment and installation.
6.0 Equipment and Supplies
6.1 Measurement System. Any measurement system for total organic concentration that meets the specifications of this method. A schematic of an acceptable measurement system is shown in Figure 25A-1. All sampling components leading to the analyzer shall be heated ≥110°C (220°F) throughout the sampling period, unless safety reasons are cited (Section 5.2) The essential components of the measurement system are described below:
6.1.1 Organic Concentration Analyzer. A flame ionization analyzer (FIA) capable of meeting or exceeding the specifications of this method. The flame ionization detector block shall be heated >120°C (250°F).
6.1.2 Sample Probe. Stainless steel, or equivalent, three-hole rake type. Sample holes shall be 4 mm (0.16-in.) in diameter or smaller and located at 16.7, 50, and 83.3 percent of the equivalent stack diameter. Alternatively, a single opening probe may be used so that a gas sample is collected from the centrally located 10 percent area of the stack cross-section.
6.1.3 Heated Sample Line. Stainless steel or Teflon” tubing to transport the sample gas to the analyzer. The sample line should be heated (≥110°C) to prevent any condensation.
6.1.4 Calibration Valve Assembly. A three-way valve assembly to direct the zero and calibration gases to the analyzers is recommended. Other methods, such as quick-connect lines, to route calibration gas to the analyzers are applicable.
6.1.5 Particulate Filter. An in-stack or an out-of-stack glass fiber filter is recommended if exhaust gas particulate loading is significant. An out-of-stack filter should be heated to prevent any condensation.
6.1.6 Recorder. A strip-chart recorder, analog computer, or digital recorder for recording measurement data. The minimum data recording requirement is one measurement value per minute.
7.0 Reagents and Standards
7.1 Calibration Gases. The calibration gases for the gas analyzer shall be propane in air or propane in nitrogen. Alternatively, organic compounds other than propane can be used; the appropriate corrections for response factor must be made. Calibration gases shall be prepared in accordance with the procedure listed in Citation 2 of section 16. Additionally, the manufacturer of the cylinder should provide a recommended shelf life for each calibration gas cylinder over which the concentration does not change more than ±2 percent from the certified value. For calibration gas values not generally available (i.e., organics between 1 and 10 percent by volume), alternative methods for preparing calibration gas mixtures, such as dilution systems (Test Method 205, 40 CFR Part 51, Appendix M), may be used with prior approval of the Administrator.
7.1.1 Fuel. A 40 percent H2/60 percent N2 gas mixture is recommended to avoid an oxygen synergism effect that reportedly occurs when oxygen concentration varies significantly from a mean value.
7.1.2 Zero Gas. High purity air with less than 0.1 part per million by volume (ppmv) of organic material (propane or carbon equivalent) or less than 0.1 percent of the span value, whichever is greater.
7.1.3 Low-level Calibration Gas. An organic calibration gas with a concentration equivalent to 25 to 35 percent of the applicable span value.
7.1.4 Mid-level Calibration Gas. An organic calibration gas with a concentration equivalent to 45 to 55 percent of the applicable span value.
7.1.5 High-level Calibration Gas. An organic calibration gas with a concentration equivalent to 80 to 90 percent of the applicable span value.
8.0 Sample Collection, Preservation, Storage, and Transport
8.1 Selection of Sampling Site. The location of the sampling site is generally specified by the applicable regulation or purpose of the test (i.e., exhaust stack, inlet line, etc.). The sample port shall be located to meet the testing requirements of Method 1.
8.2 Location of Sample Probe. Install the sample probe so that the probe is centrally located in the stack, pipe, or duct and is sealed tightly at the stack port connection.
8.3 Measurement System Preparation. Prior to the emission test, assemble the measurement system by following the manufacturer's written instructions for preparing sample interface and the organic analyzer. Make the system operable (Section 10.1).
8.4 Calibration Error Test. Immediately prior to the test series (within 2 hours of the start of the test), introduce zero gas and high-level calibration gas at the calibration valve assembly. Adjust the analyzer output to the appropriate levels, if necessary. Calculate the predicted response for the low-level and mid-level gases based on a linear response line between the zero and high-level response. Then introduce low-level and mid-level calibration gases successively to the measurement system. Record the analyzer responses for low-level and mid-level calibration gases and determine the differences between the measurement system responses and the predicted responses. These differences must be less than 5 percent of the respective calibration gas value. If not, the measurement system is not acceptable and must be replaced or repaired prior to testing. No adjustments to the measurement system shall be conducted after the calibration and before the drift check (Section 8.6.2). If adjustments are necessary before the completion of the test series, perform the drift checks prior to the required adjustments and repeat the calibration following the adjustments. If multiple electronic ranges are to be used, each additional range must be checked with a mid-level calibration gas to verify the multiplication factor.
8.5 Response Time Test. Introduce zero gas into the measurement system at the calibration valve assembly. When the system output has stabilized, switch quickly to the high-level calibration gas. Record the time from the concentration change to the measurement system response equivalent to 95 percent of the step change. Repeat the test three times and average the results.
8.6 Emission Measurement Test Procedure.
8.6.1 Organic Measurement. Begin sampling at the start of the test period, recording time and any required process information as appropriate. In particulate, note on the recording chart, periods of process interruption or cyclic operation.
8.6.2 Drift Determination. Immediately following the completion of the test period and hourly during the test period, reintroduce the zero and mid-level calibration gases, one at a time, to the measurement system at the calibration valve assembly. (Make no adjustments to the measurement system until both the zero and calibration drift checks are made.) Record the analyzer response. If the drift values exceed the specified limits, invalidate the test results preceding the check and repeat the test following corrections to the measurement system. Alternatively, recalibrate the test measurement system as in section 8.4 and report the results using both sets of calibration data (i.e., data determined prior to the test period and data determined following the test period).
Note:
Note on the recording chart periods of process interruption or cyclic operation.
9.0 Quality Control
| Method section | Quality control measure | Effect |
|---|---|---|
| 8.4 | Zero and calibration drift tests | Ensures that bias introduced by drift in the measurement system output during the run is no greater than 3 percent of span. |
10.0 Calibration and Standardization
10.1 FIA equipment can be calibrated for almost any range of total organic concentrations. For high concentrations of organics (>1.0 percent by volume as propane), modifications to most commonly available analyzers are necessary. One accepted method of equipment modification is to decrease the size of the sample to the analyzer through the use of a smaller diameter sample capillary. Direct and continuous measurement of organic concentration is a necessary consideration when determining any modification design.
11.0 Analytical Procedure
The sample collection and analysis are concurrent for this method (see section 8.0).
12.0 Calculations and Data Analysis
12.1 Determine the average organic concentration in terms of ppmv as propane or other calibration gas. The average shall be determined by integration of the output recording over the period specified in the applicable regulation. If results are required in terms of ppmv as carbon, adjust measured concentrations using Equation 25A-1.

Where:
Cc = Organic concentration as carbon, ppmv.
Cmeas = Organic concentration as measured, ppmv.
K = Carbon equivalent correction factor.
= 2 for ethane.
= 3 for propane.
= 4 for butane.
= Appropriate response factor for other organic calibration gases.
13.0 Method Performance
13.1 Measurement System Performance Specifications.
13.1.1 Zero Drift. Less than ±3 percent of the span value.
13.1.2 Calibration Drift. Less than ±3 percent of span value.
13.1.3 Calibration Error. Less than ±5 percent of the calibration gas value.
14.0 Pollution Prevention [Reserved]
15.0 Waste Management [Reserved]
16.0 References
1. Measurement of Volatile Organic Compounds - Guideline Series. U.S. Environmental Protection Agency. Research Triangle Park, NC. Publication No. EPA-450/2-78-041. June 1978. p. 46-54.
2. EPA Traceability Protocol for Assay and Certification of Gaseous Calibration Standards. U.S. Environmental Protection Agency, Quality Assurance and Technical Support Division. Research Triangle Park, N.C. September 1993.
3. Gasoline Vapor Emission Laboratory Evaluation - Part 2. U.S. Environmental Protection Agency, Office of Air Quality Planning and Standards. Research Triangle Park, NC. EMB Report No. 75-GAS-6. August 1975.
17.0 Tables, Diagrams, Flowcharts, and Validation Data
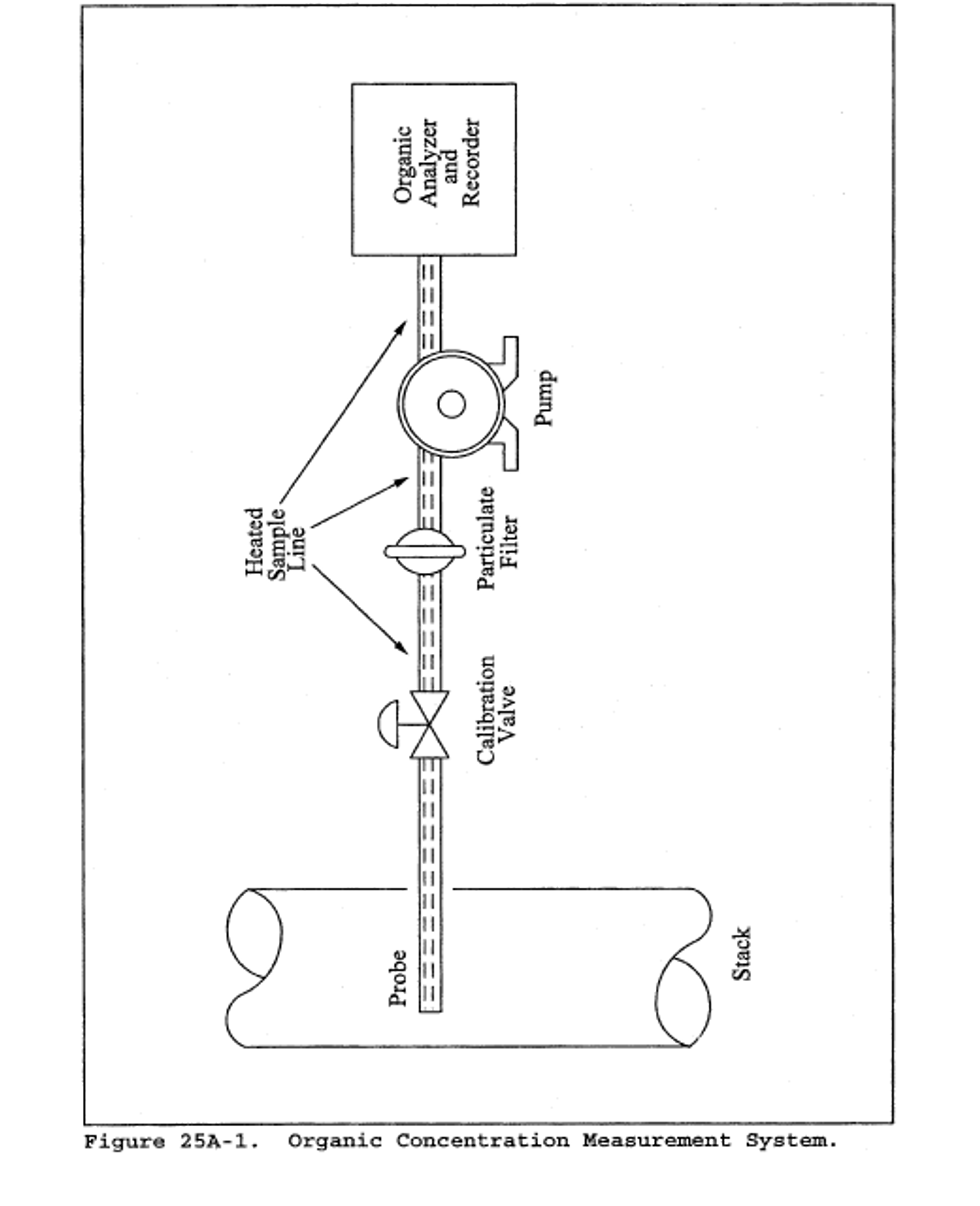
Method 25B - Determination of Total Gaseous Organic Concentration Using a Nondispersive Infrared Analyzer
Note:
This method does not include all of the specifications (e.g., equipment and supplies) and procedures (e.g., sampling) essential to its performance. Some material is incorporated by reference from other methods in this part. Therefore, to obtain reliable results, persons using this method should have a thorough knowledge of at least the following additional test methods: Method 1, Method 6C, and Method 25A.
1.0 Scope and Application
1.1 Analytes.
| Analyte | CAS No. | Sensitivity |
|---|---|---|
| Total Organic Compounds | N/A | <2% of span. |
1.2 Applicability. This method is applicable for the determination of total gaseous organic concentration of vapors consisting primarily of alkanes. Other organic materials may be measured using the general procedure in this method, the appropriate calibration gas, and an analyzer set to the appropriate absorption band.
1.3 Data Quality Objectives. Adherence to the requirements of this method will enhance the quality of the data obtained from air pollutant sampling methods.
2.0 Summary of Method
A gas sample is extracted from the source through a heated sample line, if necessary, and glass fiber filter to a nondispersive infrared analyzer (NDIR). Results are reported as volume concentration equivalents of the calibration gas or as carbon equivalents.
3.0 Definitions
Same as Method 25A, section 3.0.
4.0 Interferences [Reserved]
5.0 Safety
5.1 Disclaimer. This method may involve hazardous materials, operations, and equipment. This test method may not address all of the safety problems associated with its use. It is the responsibility of the user of this test method to establish appropriate safety and health practices and determine the applicability of regulatory limitations prior to performing this test method. The analyzer users manual should be consulted for specific precautions to be taken with regard to the analytical procedure.
5.2 Explosive Atmosphere. This method is often applied in highly explosive areas. Caution and care should be exercised in choice of equipment and installation.
6.0 Equipment and Supplies
Same as Method 25A, section 6.0, with the exception of the following:
6.1 Organic Concentration Analyzer. A nondispersive infrared analyzer designed to measure alkane organics and capable of meeting or exceeding the specifications in this method.
7.0 Reagents and Standards
Same as Method 25A, section 7.1. No fuel gas is required for an NDIR.
8.0 Sample Collection, Preservation, Storage, and Transport
Same as Method 25A, section 8.0.
9.0 Quality Control
Same as Method 25A, section 9.0.
10.0 Calibration and Standardization
Same as Method 25A, section 10.0.
11.0 Analytical Procedure
The sample collection and analysis are concurrent for this method (see section 8.0).
12.0 Calculations and Data Analysis
Same as Method 25A, section 12.0.
13.0 Method Performance [Reserved]
14.0 Pollution Prevention [Reserved]
15.0 Waste Management [Reserved]
16.0 References
Same as Method 25A, section 16.0.
17.0 Tables, Diagrams, Flowcharts, and Validation Data [Reserved]
Method 25C - Determination of Nonmethane Organic Compounds (NMOC) in Landfill Gases
Note:
This method does not include all of the specifications (e.g., equipment and supplies) and procedures (e.g., sampling and analytical) essential to its performance. Some material is incorporated by reference from other methods in this part. Therefore, to obtain reliable results, persons using this method should also have a thorough knowledge of EPA Method 25.
1.0 Scope and Application
1.1 Analytes.
| Analyte | CAS No. |
|---|---|
| Nonmethane organic compounds (NMOC) | No CAS number assigned. |
1.2 Applicability. This method is applicable to the sampling and measurement of NMOC as carbon in landfill gases (LFG).
1.3 Data Quality Objectives. Adherence to the requirements of this method will enhance the quality of the data obtained from air pollutant sampling methods.
2.0 Summary of Method
2.1 A sample probe that has been perforated at one end is driven or augured to a depth of 0.9 m (3 ft) below the bottom of the landfill cover. A sample of the landfill gas is extracted with an evacuated cylinder. The NMOC content of the gas is determined by injecting a portion of the gas into a gas chromatographic column to separate the NMOC from carbon monoxide (CO), carbon dioxide (CO2), and methane (CH4); the NMOC are oxidized to CO2, reduced to CH4, and measured by a flame ionization detector (FID). In this manner, the variable response of the FID associated with different types of organics is eliminated.
3.0 Definitions [Reserved]
4.0 Interferences [Reserved]
5.0 Safety
5.1 Since this method is complex, only experienced personnel should perform this test. LFG contains methane, therefore explosive mixtures may exist on or near the landfill. It is advisable to take appropriate safety precautions when testing landfills, such as refraining from smoking and installing explosion-proof equipment.
6.0 Equipment and Supplies
6.1 Sample Probe. Stainless steel, with the bottom third perforated. Teflon probe liners and sampling lines are also allowed. Non-perforated probes are allowed as long as they are withdrawn to create a gap equivalent to having the bottom third perforated. The sample probe must be capped at the bottom and must have a threaded cap with a sampling attachment at the top. The sample probe must be long enough to go through and extend no less than 0.9 m (3 ft) below the landfill cover. If the sample probe is to be driven into the landfill, the bottom cap should be designed to facilitate driving the probe into the landfill.
6.2 Sampling Train.
6.2.1 Rotameter with Flow Control Valve. Capable of measuring a sample flow rate of 100 ±10 ml/min. The control valve must be made of stainless steel.
6.2.2 Sampling Valve. Stainless steel.
6.2.3 Pressure Gauge. U-tube mercury manometer, or equivalent, capable of measuring pressure to within 1 mm Hg (0.5 in H2O) in the range of 0 to 1,100 mm Hg (0 to 590 in H2O).
6.2.4 Sample Tank. Stainless steel or aluminum cylinder, equipped with a stainless steel sample tank valve.
6.3 Vacuum Pump. Capable of evacuating to an absolute pressure of 10 mm Hg (5.4 in H2O).
6.4 Purging Pump. Portable, explosion proof, and suitable for sampling NMOC.
6.5 Pilot Probe Procedure. The following are needed only if the tester chooses to use the procedure described in section 8.2.1.
6.5.1 Pilot Probe. Tubing of sufficient strength to withstand being driven into the landfill by a post driver and an outside diameter of at least 6 mm (0.25 in.) smaller than the sample probe. The pilot probe shall be capped on both ends and long enough to go through the landfill cover and extend no less than 0.9 m (3 ft) into the landfill.
6.5.2 Post Driver and Compressor. Capable of driving the pilot probe and the sampling probe into the landfill. The Kitty Hawk portable post driver has been found to be acceptable.
6.6 Auger Procedure. The following are needed only if the tester chooses to use the procedure described in section 8.2.2.
6.6.1 Auger. Capable of drilling through the landfill cover and to a depth of no less than 0.9 m (3 ft) into the landfill.
6.6.2 Pea Gravel.
6.6.3 Bentonite.
6.7 NMOC Analyzer, Barometer, Thermometer, and Syringes. Same as in sections 6.3.1, 6.3.2, 6.33, and 6.2.10, respectively, of Method 25.
7.0 Reagents and Standards
7.1 NMOC Analysis. Same as in Method 25, section 7.2.
7.2 Calibration. Same as in Method 25, section 7.4, except omit section 7.4.3.
8.0 Sample Collection, Preservation, Storage, and Transport
8.1 Sample Tank Evacuation and Leak-Check. Conduct the sample tank evacuation and leak-check either in the laboratory or the field. Connect the pressure gauge and sampling valve to the sample tank. Evacuate the sample tank to 10 mm Hg (5.4 in H2O) absolute pressure or less. Close the sampling valve, and allow the tank to sit for 30 minutes. The tank is acceptable if no change more than ±2 mm is noted. Include the results of the leak-check in the test report.
8.2 Sample Probe Installation. The tester may use the procedure in section 8.2.1 or 8.2.2.
8.2.1 Pilot Probe Procedure. Use the post driver to drive the pilot probe at least 0.9 m (3 ft) below the landfill cover. Alternative procedures to drive the probe into the landfill may be used subject to the approval of the Administrator's designated representative.
8.2.1.1 Remove the pilot probe and drive the sample probe into the hole left by the pilot probe. The sample probe shall extend at least 0.9 m (3 ft) below the landfill cover and shall protrude about 0.3 m (1 ft) above the landfill cover. Seal around the sampling probe with bentonite and cap the sampling probe with the sampling probe cap.
8.2.2 Auger Procedure. Use an auger to drill a hole to at least 0.9 m (3 ft) below the landfill cover. Place the sample probe in the hole and backfill with pea gravel to a level 0.6 m (2 ft) from the surface. The sample probe shall protrude at least 0.3 m (1 ft) above the landfill cover. Seal the remaining area around the probe with bentonite. Allow 24 hours for the landfill gases to equilibrate inside the augured probe before sampling.
8.2.3 Driven Probes. Closed-point probes may be driven directly into the landfill in a single step. This method may not require backfilling if the probe is adequately sealed by its insertion. Unperforated probes that are inserted in this manner and withdrawn at a distance from a detachable tip to create an open space are also acceptable.
8.3 Sample Train Assembly. Just before assembling the sample train, measure the sample tank vacuum using the pressure gauge. Record the vacuum, the ambient temperature, and the barometric pressure at this time. Assemble the sampling probe purging system as shown in Figure 25C-1.
8.4 Sampling Procedure. Open the sampling valve and use the purge pump and the flow control valve to evacuate at least two sample probe volumes from the system at a flow rate of 500 ml/min or less. Close the sampling valve and replace the purge pump with the sample tank apparatus as shown in Figure 25C-2. Open the sampling valve and the sample tank valve and, using the flow control valve, sample at a flow rate of 500 ml/min or less until either a constant flow rate can no longer be maintained because of reduced sample tank vacuum or the appropriate composite volume is attained. Disconnect the sampling tank apparatus and pressurize the sample cylinder to approximately 1,060 mm Hg (567 in. H2O) absolute pressure with helium, and record the final pressure. Alternatively, the sample tank may be pressurized in the lab.
8.4.1 The following restrictions apply to compositing samples from different probe sites into a single cylinder: (1) Individual composite samples per cylinder must be of equal volume; this must be verified by recording the flow rate, sampling time, vacuum readings, or other appropriate volume measuring data, (2) individual composite samples must have a minimum volume of 1 liter unless data is provided showing smaller volumes can be accurately measured, and (3) composite samples must not be collected using the final cylinder vacuum as it diminishes to ambient pressure.
8.4.2 Use Method 3C to determine the percent N2 and O2 in each cylinder. The presence of N2 and O2 indicate either infiltration of ambient air into the landfill gas sample or an inappropriate testing site has been chosen where anaerobic decomposition has not begun. The landfill gas sample is acceptable if the concentration of N2 is less than 20 percent. Alternatively, the oxygen content of each cylinder must be less than 5 percent. Landfills with 3-year average annual rainfalls equal to or less than 20 inches annual rainfalls samples are acceptable when the N2 to O2 concentration ratio is greater than 3.71.
9.0 Quality Control
9.1 Miscellaneous Quality Control Measures.
| Section | Quality control measure | Effect |
|---|---|---|
| 8.4.2 | Verify that landfill gas sample contains less than 20 percent N 2 or 5 percent O 2 . Landfills with 3-year average annual rainfalls equal to or less than 20 inches annual rainfalls samples are acceptable when the N 2 to O 2 concentration ratio is greater than 3.71 | Ensures that ambient air was not drawn into the landfill gas sample and gas was sampled from an appropriate location. |
| 10.1, 10.2 | NMOC analyzer initial and daily performance checks | Ensures precision of analytical results. |
10.0 Calibration and Standardization
Note:
Maintain a record of performance of each item.
10.1 Initial NMOC Analyzer Performance Test. Same as in Method 25, section 10.1, except omit the linearity checks for CO2 standards.
10.2 NMOC Analyzer Daily Calibration.
10.2.1 NMOC Response Factors. Same as in Method 25, section 10.2.2.
10.3 Sample Tank Volume. The volume of the gas sampling tanks must be determined. Determine the tank volumes by weighing them empty and then filled with deionized water; weigh to the nearest 5 g, and record the results. Alternatively, measure the volume of water used to fill them to the nearest 5 ml.
11.0 Analytical Procedures
11.1 The oxidation, reduction, and measurement of NMOC's is similar to Method 25. Before putting the NMOC analyzer into routine operation, conduct an initial performance test. Start the analyzer, and perform all the necessary functions in order to put the analyzer into proper working order. Conduct the performance test according to the procedures established in section 10.1. Once the performance test has been successfully completed and the NMOC calibration response factor has been determined, proceed with sample analysis as follows:
11.1.1 Daily Operations and Calibration Checks. Before and immediately after the analysis of each set of samples or on a daily basis (whichever occurs first), conduct a calibration test according to the procedures established in section 10.2. If the criteria of the daily calibration test cannot be met, repeat the NMOC analyzer performance test (Section 10.1) before proceeding.
11.1.2 Operating Conditions. Same as in Method 25, section 11.2.1.
11.1.3 Analysis of Sample Tank. Purge the sample loop with sample, and then inject the sample. Under the specified operating conditions, the CO2 in the sample will elute in approximately 100 seconds. As soon as the detector response returns to baseline following the CO2 peak, switch the carrier gas flow to backflush, and raise the column oven temperature to 195°C (383°F) as rapidly as possible. A rate of 30°C/min (54°F/min) has been shown to be adequate. Record the value obtained for any measured NMOC. Return the column oven temperature to 85°C (185°F) in preparation for the next analysis. Analyze each sample in triplicate, and report the average as Ctm.
12.0 Data Analysis and Calculations
Note:
All equations are written using absolute pressure; absolute pressures are determined by adding the measured barometric pressure to the measured gauge or manometer pressure.
12.1 Nomenclature
Bw = Moisture content in the sample, fraction.
C N2 = N 2 concentration in the landfill gas sample.
C mN2 = Measured N 2 concentration, diluted landfill gas sample.
CmOx = Measured Oxygen concentration, fraction in landfill gas.
COx = Oxygen concentration in the diluted sample gas.
Ct = Calculated NMOC concentration, ppmv C equivalent.
Ctm = Measured NMOC concentration, ppmv C equivalent.
Pb = Barometric pressure, mm Hg.
Pt = Gas sample tank pressure after sampling, but before pressurizing, mm Hg absolute.
Ptf = Final gas sample tank pressure after pressurizing, mm Hg absolute.
Pti = Gas sample tank pressure after evacuation, mm Hg absolute.
Pw = Vapor pressure of H2O (from Table 25C-1), mm Hg.
r = Total number of analyzer injections of sample tank during analysis (where j = injection number, 1 . . . r).
Tt = Sample tank temperature at completion of sampling,°K.
Tti = Sample tank temperature before sampling,°K.
Ttf = Sample tank temperature after pressuring,°K.
12.2 Water Correction. Use Table 25C-1 (Section 17.0), the LFG temperature, and barometric pressure at the sampling site to calculate Bw.
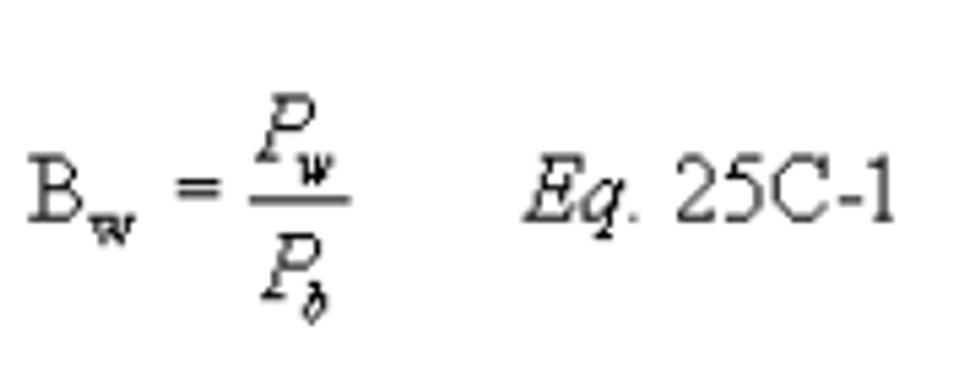
12.3 Nitrogen Concentration in the landfill gas. Use equation 25C-2 to calculate the measured concentration of nitrogen in the original landfill gas.

12.4 Oxygen Concentration in the landfill gas. Use equation 25C-3 to calculate the measured concentration of oxygen in the original landfill gas.

12.5 You must correct the NMOC Concentration for the concentration of nitrogen or oxygen based on which gas or gases passes the requirements in section 9.1 or based on the 3-year average annual rainfall based on the closest NOAA land-based station.
12.5.1 NMOC Concentration with nitrogen correction. Use Equation 25C-4 to calculate the concentration of NMOC for each sample tank when the nitrogen concentration is less than 20 percent.

12.5.2 NMOC Concentration with oxygen correction. Use Equation 25C-5 to calculate the concentration of NMOC for each sample tank if the landfill gas oxygen is less than 5 percent and the landfill gas nitrogen concentration is greater than 20 percent, or 3-year average annual rainfall based annual rainfall of less than 20 inches.

13.0 Method Performance [Reserved]
14.0 Pollution Prevention [Reserved]
15.0 Waste Management [Reserved]
16.0 References
1. Salo, Albert E., Samuel Witz, and Robert D. MacPhee. Determination of Solvent Vapor Concentrations by Total Combustion Analysis: A Comparison of Infrared with Flame Ionization Detectors. Paper No. 75-33.2. (Presented at the 68th Annual Meeting of the Air Pollution Control Association. Boston, Massachusetts. June 15-20, 1975.) 14 p.
2. Salo, Albert E., William L. Oaks, and Robert D. MacPhee. Measuring the Organic Carbon Content of Source Emissions for Air Pollution Control. Paper No. 74-190. (Presented at the 67th Annual Meeting of the Air Pollution Control Association. Denver, Colorado. June 9-13, 1974.) 25 p.
17.0 Tables, Diagrams, Flowcharts, and Validation Data
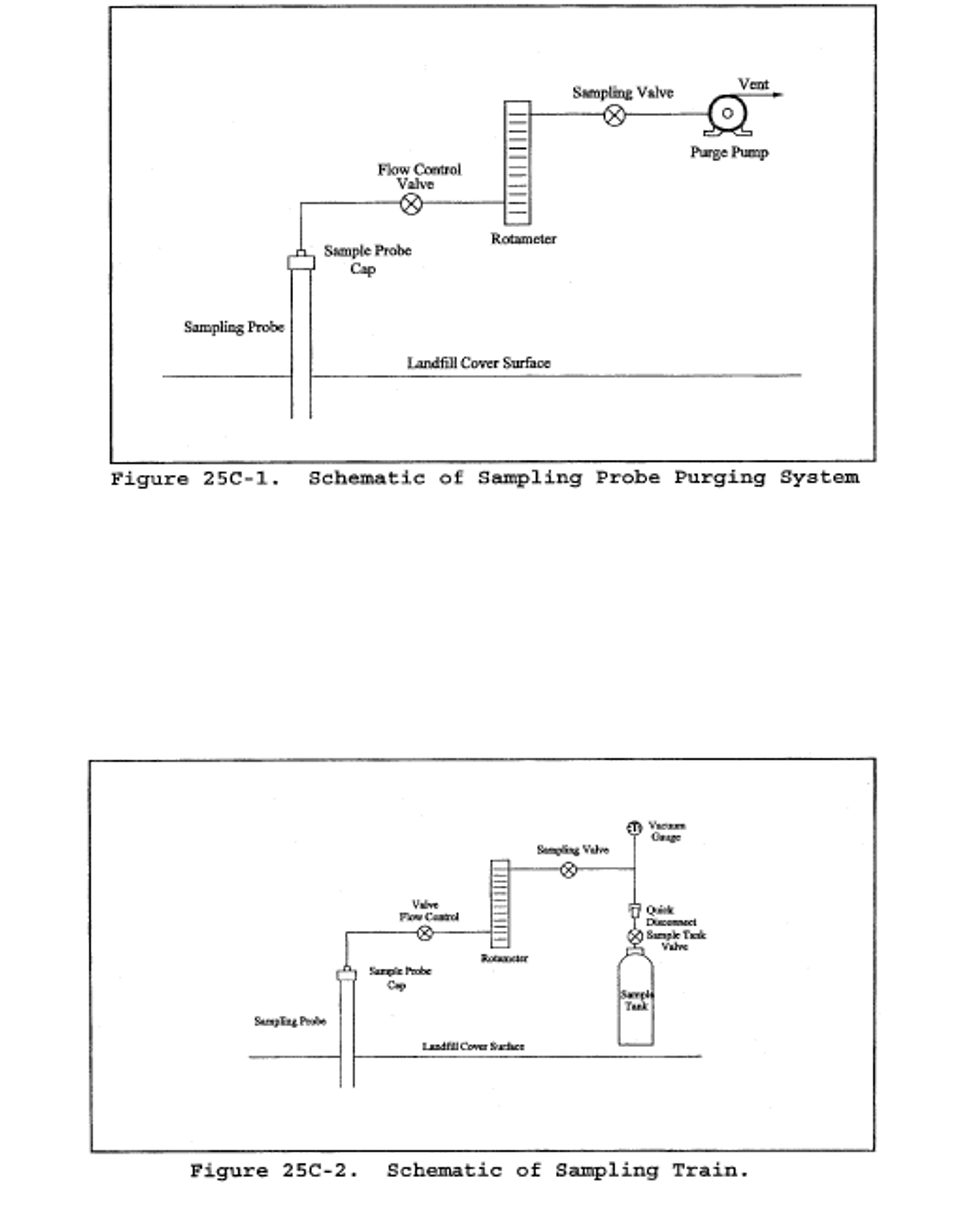
| Temperature,°C | Vapor Pressure of H2O, mm Hg | Temperature,°C | Vapor Pressure of H2O, mm Hg |
|---|---|---|---|
| 4 | 6.1 | 18 | 15.5 |
| 6 | 7.0 | 20 | 17.5 |
| 8 | 8.0 | 22 | 19.8 |
| 10 | 9.2 | 24 | 22.4 |
| 12 | 10.5 | 26 | 25.2 |
| 14 | 12.0 | 28 | 28.3 |
| 16 | 13.6 | 30 | 31.8 |
Method 25D - Determination of the Volatile Organic Concentration of Waste Samples
Note:
Performance of this method should not be attempted by persons unfamiliar with the operation of a flame ionization detector (FID) or an electrolytic conductivity detector (ELCD) because knowledge beyond the scope of this presentation is required.
1.0 Scope and Application
1.1 Analyte. Volatile Organic Compounds. No CAS No. assigned.
1.2 Applicability. This method is applicable for determining the volatile organic (VO) concentration of a waste sample.
2.0 Summary of Method
2.1 Principle. A sample of waste is obtained at a point which is most representative of the unexposed waste (where the waste has had minimum opportunity to volatilize to the atmosphere). The sample is suspended in an organic/aqueous matrix, then heated and purged with nitrogen for 30 min. in order to separate certain organic compounds. Part of the sample is analyzed for carbon concentration, as methane, with an FID, and part of the sample is analyzed for chlorine concentration, as chloride, with an ELCD. The VO concentration is the sum of the carbon and chlorine content of the sample.
3.0 Definitions
3.1 Well-mixed in the context of this method refers to turbulent flow which results in multiple-phase waste in effect behaving as single-phase waste due to good mixing.
4.0 Interferences [Reserved]
5.0 Safety
5.1 Disclaimer. This method may involve hazardous materials, operations, and equipment. This test method may not address all of the safety problems associated with its use. It is the responsibility of the user of this test method to establish appropriate safety and health practices and to determine the applicability of regulatory limitations prior to performing this test method.
6.0 Equipment and Supplies
Note:
Mention of trade names or specific products does not constitute endorsement by the Environmental Protection Agency.
6.1 Sampling. The following equipment is required:
6.1.1 Sampling Tube. Flexible Teflon, 0.25 in. ID (6.35 mm).
6.1.2 Sample Container. Borosilicate glass, 40-mL, and a Teflon-lined screw cap capable of forming an air tight seal.
6.1.3 Cooling Coil. Fabricated from 0.25 in (6.35 mm). ID 304 stainless steel tubing with a thermocouple at the coil outlet.
6.2 Analysis. The following equipment is required.
6.2.1 Purging Apparatus. For separating the VO from the waste sample. A schematic of the system is shown in Figure 25D-1. The purging apparatus consists of the following major components.
6.2.1.1 Purging Flask. A glass container to hold the sample while it is heated and purged with dry nitrogen. The cap of the purging flask is equipped with three fittings: one for a purging lance (fitting with the #7 Ace-thread), one for the Teflon exit tubing (side fitting, also a #7 Ace-thread), and a third (a 50-mm Ace-thread) to attach the base of the purging flask as shown in Figure 25D-2. The base of the purging flask is a 50-mm ID (2 in) cylindrical glass tube. One end of the tube is open while the other end is sealed. Exact dimensions are shown in Figure 25D-2.
6.2.1.2 Purging Lance. Glass tube, 6-mm OD (0.2 in) by 30 cm (12 in) long. The purging end of the tube is fitted with a four-arm bubbler with each tip drawn to an opening 1 mm (0.04 in) in diameter. Details and exact dimensions are shown in Figure 25D-2.
6.2.1.3 Coalescing Filter. Porous fritted disc incorporated into a container with the same dimensions as the purging flask. The details of the design are shown in Figure 25D-3.
6.2.1.4 Constant Temperature Chamber. A forced draft oven capable of maintaining a uniform temperature around the purging flask and coalescing filter of 75 ±2°C (167 ±3.6°F).
6.2.1.5 Three-way Valve. Manually operated, stainless steel. To introduce calibration gas into system.
6.2.1.6 Flow Controllers. Two, adjustable. One capable of maintaining a purge gas flow rate of 6 ±0.06 L/min (0.2 ±0.002 ft 3/min) The other capable of maintaining a calibration gas flow rate of 1-100 mL/min (0.00004-0.004 ft 3/min).
6.2.1.7 Rotameter. For monitoring the air flow through the purging system (0-10 L/min)(0-0.4 ft 3/min).
6.2.1.8 Sample Splitters. Two heated flow restrictors (placed inside oven or heated to 120 ±10°C (248 ±18°F) ). At a purge rate of 6 L/min (0.2 ft 3/min), one will supply a constant flow to the first detector (the rest of the flow will be directed to the second sample splitter). The second splitter will split the analytical flow between the second detector and the flow restrictor. The approximate flow to the FID will be 40 mL/min (0.0014 ft 3/min) and to the ELCD will be 15 mL/min (0.0005 ft 3/min), but the exact flow must be adjusted to be compatible with the individual detector and to meet its linearity requirement. The two sample splitters will be connected to each other by 1/8′ OD (3.175 mm) stainless steel tubing.
6.2.1.9 Flow Restrictor. Stainless steel tubing, 1/8′ OD (3.175 mm), connecting the second sample splitter to the ice bath. Length is determined by the resulting pressure in the purging flask (as measured by the pressure gauge). The resulting pressure from the use of the flow restrictor shall be 6-7 psig.
6.2.1.10 Filter Flask. With one-hole stopper. Used to hold ice bath. Excess purge gas is vented through the flask to prevent condensation in the flowmeter and to trap volatile organic compounds.
6.2.1.11 Four-way Valve. Manually operated, stainless steel. Placed inside oven, used to bypass purging flask.
6.2.1.12 On/Off Valves. Two, stainless steel. One heat resistant up to 130°C (266°F) and placed between oven and ELCD. The other a toggle valve used to control purge gas flow.
6.2.1.13 Pressure Gauge. Range 0-40 psi. To monitor pressure in purging flask and coalescing filter.
6.2.1.14 Sample Lines. Teflon, 1/4′ OD (6.35 mm), used inside the oven to carry purge gas to and from purging chamber and to and from coalescing filter to four-way valve. Also used to carry sample from four-way valve to first sample splitter.
6.2.1.15 Detector Tubing. Stainless steel, 1/8′ OD (3.175 mm), heated to 120 ±10°C (248 ±18°F) . Used to carry sample gas from each sample splitter to a detector. Each piece of tubing must be wrapped with heat tape and insulating tape in order to insure that no cold spots exist. The tubing leading to the ELCD will also contain a heat-resistant on-off valve (Section 6.2.1.12) which shall also be wrapped with heat-tape and insulation.
6.2.2 Volatile Organic Measurement System. Consisting of an FID to measure the carbon concentration of the sample and an ELCD to measure the chlorine concentration.
6.2.2.1 FID. A heated FID meeting the following specifications is required.
6.2.2.1.1 Linearity. A linear response (±5 percent) over the operating range as demonstrated by the procedures established in section 10.1.1.
6.2.2.1.2 Range. A full scale range of 50 pg carbon/sec to 50 µg carbon/sec. Signal attenuators shall be available to produce a minimum signal response of 10 percent of full scale.
6.2.2.1.3 Data Recording System. A digital integration system compatible with the FID for permanently recording the output of the detector. The recorder shall have the capability to start and stop integration at points selected by the operator or it shall be capable of the “integration by slices” technique (this technique involves breaking down the chromatogram into smaller increments, integrating the area under the curve for each portion, subtracting the background for each portion, and then adding all of the areas together for the final area count).
6.2.2.2 ELCD. An ELCD meeting the following specifications is required. 1-propanol must be used as the electrolyte. The electrolyte flow through the conductivity cell shall be 1 to 2 mL/min (0.00004 to 0.00007 ft 3/min).
Note:
A 1/4-in. ID (6.35 mm) quartz reactor tube is strongly recommended to reduce carbon buildup and the resulting detector maintenance.
6.2.2.2.1 Linearity. A linear response (±10 percent) over the response range as demonstrated by the procedures in section 10.1.2.
6.2.2.2.2 Range. A full scale range of 5.0 pg/sec to 500 ng/sec chloride. Signal attenuators shall be available to produce a minimum signal response of 10 percent of full scale.
6.2.2.2.3 Data Recording System. A digital integration system compatible with the output voltage range of the ELCD. The recorder must have the capability to start and stop integration at points selected by the operator or it shall be capable of performing the “integration by slices” technique.
7.0 Reagents and Standards
7.1 Sampling.
7.1.1 Polyethylene Glycol (PEG). Ninety-eight percent pure with an average molecular weight of 400. Before using the PEG, remove any organic compounds that might be detected as volatile organics by heating it to 120°C (248°F) and purging it with nitrogen at a flow rate of 1 to 2 L/min (0.04 to 0.07 ft 3/min) for 2 hours. The cleaned PEG must be stored under a 1 to 2 L/min (0.04 to 0.07 ft 3/min) nitrogen purge until use. The purge apparatus is shown in Figure 25D-4.
7.2 Analysis.
7.2.1 Sample Separation. The following are required for the sample purging step.
7.2.1.1 PEG. Same as section 7.1.1.
7.2.1.2 Purge Gas. Zero grade nitrogen (N2), containing less than 1 ppm carbon.
7.2.2 Volatile Organics Measurement. The following are required for measuring the VO concentration.
7.2.2.1 Hydrogen (H2). Zero grade H2, 99.999 percent pure.
7.2.2.2 Combustion Gas. Zero grade air or oxygen as required by the FID.
7.2.2.3 Calibration Gas. Pressurized gas cylinder containing 10 percent propane and 1 percent 1,1-dichloroethylene by volume in nitrogen.
7.2.2.4 Water. Deionized distilled water that conforms to American Society for Testing and Materials Specification D 1193-74, Type 3, is required for analysis. At the option of the analyst, the KMnO4 test for oxidizable organic matter may be omitted when high concentrations are not expected to be present.
7.2.2.5 1-Propanol. ACS grade or better. Electrolyte Solution. For use in the ELCD.
8.0 Sample Collection, Preservation, Storage, and Transport
8.1 Sampling.
8.1.1 Sampling Plan Design and Development. Use the procedures in chapter nine of Reference 1 in section 16 as guidance in developing a sampling plan.
8.1.2 Single Phase or Well-mixed Waste.
8.1.2.1 Install a sampling tap to obtain the sample at a point which is most representative of the unexposed waste (where the waste has had minimum opportunity to volatilize to the atmosphere). Assemble the sampling apparatus as shown in Figure 25D-5.
8.1.2.2 Prepare the sampling containers as follows: Pour 30 mL of clean PEG into the container. PEG will reduce but not eliminate the loss of organics during sample collection. Weigh the sample container with the screw cap, the PEG, and any labels to the nearest 0.01 g and record the weight (mst). Store the containers in an ice bath until 1 hour before sampling (PEG will solidify at ice bath temperatures; allow the containers to reach room temperature before sampling).
8.1.2.3 Begin sampling by purging the sample lines and cooling coil with at least four volumes of waste. Collect the purged material in a separate container and dispose of it properly.
8.1.2.4 After purging, stop the sample flow and direct the sampling tube to a preweighed sample container, prepared as described in section 8.1.2.2. Keep the tip of the tube below the surface of the PEG during sampling to minimize contact with the atmosphere. Sample at a flow rate such that the temperature of the waste is less than 10°C (50°F). Fill the sample container and immediately cap it (within 5 seconds) so that a minimum headspace exists in the container. Store immediately in a cooler and cover with ice.
8.1.3 Multiple-phase Waste. Collect a 10 g sample of each phase of waste generated using the procedures described in section 8.1.2 or 8.1.5. Each phase of the waste shall be analyzed as a separate sample. Calculate the weighted average VO concentration of the waste using Equation 25D-13 (Section 12.14).
8.1.4 Solid waste. Add approximately 10 g of the solid waste to a container prepared in the manner described in section 8.1.2.2, minimizing headspace. Cap and chill immediately.
8.1.5 Alternative to Tap Installation. If tap installation is impractical or impossible, fill a large, clean, empty container by submerging the container into the waste below the surface of the waste. Immediately fill a container prepared in the manner described in section 8.1.2.2 with approximately 10 g of the waste collected in the large container. Minimize headspace, cap and chill immediately.
8.1.6 Alternative sampling techniques may be used upon the approval of the Administrator.
8.2 Sample Recovery.
8.2.1 Assemble the purging apparatus as shown in Figures 25D-1 and 25D-2. The oven shall be heated to 75 ±2°C (167 ±3.6°F). The sampling lines leading from the oven to the detectors shall be heated to 120 ±10°C (248 ±18°F) with no cold spots. The flame ionization detector shall be operated with a heated block. Adjust the purging lance so that it reaches the bottom of the chamber.
8.2.2 Remove the sample container from the cooler, and wipe the exterior of the container to remove any extraneous ice, water, or other debris. Reweigh the sample container to the nearest 0.01 g, and record the weight (msf). Pour the contents of the sample container into the purging flask, rinse the sample container three times with a total of 20 mL of PEG (since the sample container originally held 30 mL of PEG, the total volume of PEG added to the purging flask will be 50 mL), transferring the rinsings to the purging flask after each rinse. Cap purging flask between rinses. The total volume of PEG in the purging flask shall be 50 mL. Add 50 mL of water to the purging flask.
9.0 Quality Control
9.1 Quality Control Samples. If audit samples are not available, prepare and analyze the two types of quality control samples (QCS) listed in Sections 9.1.1 and 9.1.2. Before placing the system in operation, after a shutdown of greater than six months, and after any major modifications, analyze each QCS in triplicate. For each detector, calculate the percent recovery by dividing measured concentration by theoretical concentration and multiplying by 100. Determine the mean percent recovery for each detector for each QCS triplicate analysis. The RSD for any triplicate analysis shall be ≤10 percent. For QCS 1 (methylene chloride), the percent recovery shall be ≥90 percent for carbon as methane, and ≥55 percent for chlorine as chloride. For QCS 2 (1,3-dichloro-2-propanol), the percent recovery shall be ≤15 percent for carbon as methane, and ≤6 percent for chlorine as chloride. If the analytical system does not meet the above-mentioned criteria for both detectors, check the system parameters (temperature, system pressure, purge rate, etc.), correct the problem, and repeat the triplicate analysis of each QCS.
9.1.1 QCS 1, Methylene Chloride. Prepare a stock solution by weighing, to the nearest 0.1 mg, 55 µL of HPLC grade methylene chloride in a tared 5 mL volumetric flask. Record the weight in milligrams, dilute to 5 mL with cleaned PEG, and inject 100 µL of the stock solution into a sample prepared as a water blank (50 mL of cleaned PEG and 60 mL of water in the purging flask). Analyze the QCS according to the procedures described in sections 10.2 and 10.3, excluding section 10.2.2. To calculate the theoretical carbon concentration (in mg) in QCS 1, multiply mg of methylene chloride in the stock solution by 3.777 × 10−3. To calculate the theoretical chlorine concentration (in mg) in QCS 1, multiply mg of methylene chloride in the stock solution by 1.670 × 10−2.
9.1.2 QCS 2, 1,3-dichloro-2-propanol. Prepare a stock solution by weighing, to the nearest 0.1 mg, 60 µL of high purity grade 1,3-dichloro-2-propanol in a tared 5 mL volumetric flask. Record the weight in milligrams, dilute to 5 mL with cleaned PEG, and inject 100 µL of the stock solution into a sample prepared as a water blank (50 mL of cleaned PEG and 60 mL of water in the purging flask). Analyze the QCS according to the procedures described in sections 10.2 and 10.3, excluding section 10.2.2. To calculate the theoretical carbon concentration (in mg) in QCS 2, multiply mg of 1,3-dichloro-2-propanol in the stock solution by 7.461 × 10−3. To calculate the theoretical chlorine concentration (in mg) in QCS 2, multiply mg of 1,3-dichloro-2-propanol in the stock solution by 1.099 × 10−2.
9.1.3 Routine QCS Analysis. For each set of compliance samples (in this context, set is per facility, per compliance test), analyze one QCS 1 and one QCS 2 sample. The percent recovery for each sample for each detector shall be ±13 percent of the mean recovery established for the most recent set of QCS triplicate analysis (Section 9.4). If the sample does not meet this criteria, check the system components and analyze another QCS 1 and 2 until a single set of QCS meet the ±13 percent criteria.
10.0 Calibration and Standardization
10.1 Initial Performance Check of Purging System. Before placing the system in operation, after a shutdown of greater than six months, after any major modifications, and at least once per month during continuous operation, conduct the linearity checks described in sections 10.1.1 and 10.1.2. Install calibration gas at the three-way calibration gas valve. See Figure 25D-1.
10.1.1 Linearity Check Procedure. Using the calibration standard described in section 7.2.2.3 and by varying the injection time, it is possible to calibrate at multiple concentration levels. Use Equation 25D-3 to calculate three sets of calibration gas flow rates and run times needed to introduce a total mass of carbon, as methane, (mc) of 1, 5, and 10 mg into the system (low, medium and high FID calibration, respectively). Use Equation 25D-4 to calculate three sets of calibration gas flow rates and run times needed to introduce a total chloride mass (mch) of 1, 5, and 10 mg into the system (low, medium and high ELCD calibration, respectively). With the system operating in standby mode, allow the FID and the ELCD to establish a stable baseline. Set the secondary pressure regulator of the calibration gas cylinder to the same pressure as the purge gas cylinder and set the proper flow rate with the calibration flow controller (see Figure 25D-1). The calibration gas flow rate can be measured with a flowmeter attached to the vent position of the calibration gas valve. Set the four-way bypass valve to standby position so that the calibration gas flows through the coalescing filter only. Inject the calibration gas by turning the calibration gas valve from vent position to inject position. Continue the calibration gas flow for the appropriate period of time before switching the calibration valve to vent position. Continue recording the response of the FID and the ELCD for 5 min after switching off calibration gas flow. Make triplicate injections of all six levels of calibration.
10.1.2 Linearity Criteria. Calculate the average response factor (Equations 25D-5 and 25D-6) and the relative standard deviation (RSD) (Equation 25D-10) at each level of the calibration curve for both detectors. Calculate the overall mean of the three response factor averages for each detector. The FID linearity is acceptable if each response factor is within 5 percent of the overall mean and if the RSD for each set of triplicate injections is less than 5 percent. The ELCD linearity is acceptable if each response factor is within 10 percent of the overall mean and if the RSD for each set of triplicate injections is less than 10 percent. Record the overall mean value of the response factors for the FID and the ELCD. If the calibration for either the FID or the ELCD does not meet the criteria, correct the detector/system problem and repeat sections 10.1.1 and 10.1.2.
10.2 Daily Calibrations.
10.2.1 Daily Linearity Check. Follow the procedures outlined in section 10.1.1 to analyze the medium level calibration for both the FID and the ELCD in duplicate at the start of the day. Calculate the response factors and the RSDs for each detector. For the FID, the calibration is acceptable if the average response factor is within 5 percent of the overall mean response factor (Section 10.1.2) and if the RSD for the duplicate injection is less than 5 percent. For the ELCD, the calibration is acceptable if the average response factor is within 10 percent of the overall mean response factor (Section 10.1.2) and if the RSD for the duplicate injection is less than 10 percent. If the calibration for either the FID or the ELCD does not meet the criteria, correct the detector/system problem and repeat sections 10.1.1 and 10.1.2.
10.2.2 Calibration Range Check.
10.2.2.1 If the waste concentration for either detector falls below the range of calibration for that detector, use the procedure outlined in section 10.1.1 to choose two calibration points that bracket the new target concentration. Analyze each of these points in triplicate (as outlined in section 10.1.1) and use the criteria in section 10.1.2 to determine the linearity of the detector in this “mini-calibration” range.
10.2.2.2 After the initial linearity check of the mini-calibration curve, it is only necessary to test one of the points in duplicate for the daily calibration check (in addition to the points specified in section 10.2.1). The average daily mini-calibration point should fit the linearity criteria specified in section 10.2.1. If the calibration for either the FID or the ELCD does not meet the criteria, correct the detector/system problem and repeat the calibration procedure mentioned in the first paragraph of section 10.2.2. A mini-calibration curve for waste concentrations above the calibration curve for either detector is optional.
10.3 Analytical Balance. Calibrate against standard weights.
11.0 Analysis
11.1 Sample Analysis.
11.1.1 Turn on the constant temperature chamber and allow the temperature to equilibrate at 75 ±2°C (167 ±3.6°F). Turn the four-way valve so that the purge gas bypasses the purging flask, the purge gas flowing through the coalescing filter and to the detectors (standby mode). Turn on the purge gas. Allow both the FID and the ELCD to warm up until a stable baseline is achieved on each detector. Pack the filter flask with ice. Replace ice after each run and dispose of the waste water properly. When the temperature of the oven reaches 75 ±2°C (167 ±3.6°F), start both integrators and record baseline. After 1 min, turn the four-way valve so that the purge gas flows through the purging flask, to the coalescing filter and to the sample splitters (purge mode). Continue recording the response of the FID and the ELCD. Monitor the readings of the pressure gauge and the rotameter. If the readings fall below established setpoints, stop the purging, determine the source of the leak, and resolve the problem before resuming. Leaks detected during a sampling period invalidate that sample.
11.1.2 As the purging continues, monitor the output of the detectors to make certain that the analysis is proceeding correctly and that the results are being properly recorded. Every 10 minutes read and record the purge flow rate, the pressure and the chamber temperature. Continue the purging for 30 minutes.
11.1.3 For each detector output, integrate over the entire area of the peak starting at 1 minute and continuing until the end of the run. Subtract the established baseline area from the peak area. Record the corrected area of the peak. See Figure 25D-6 for an example integration.
11.2 Water Blank. A water blank shall be analyzed for each batch of cleaned PEG prepared. Transfer about 60 mL of water into the purging flask. Add 50 mL of the cleaned PEG to the purging flask. Treat the blank as described in sections 8.2 and 8.3, excluding section 8.2.2. Calculate the concentration of carbon and chlorine in the blank sample (assume 10 g of waste as the mass). A VO concentration equivalent to ≤10 percent of the applicable standard may be subtracted from the measured VO concentration of the waste samples. Include all blank results and documentation in the test report.
12.0 Data Analysis and Calculations
12.1 Nomenclature.
Ab = Area under the water blank response curve, counts.
Ac = Area under the calibration response curve, counts.
As = Area under the sample response curve, counts.
C = Concentration of volatile organics in the sample, ppmw.
Cc = Concentration of carbon, as methane, in the calibration gas, mg/L.
Cch = Concentration of chloride in the calibration gas, mg/L.
Cj = VO concentration of phase j, ppmw.
DRt = Average daily response factor of the FID, mg CH4/counts.
Drth = Average daily response factor of the ELCD, mg Cl−/counts.
Fj = Weight fraction of phase j present in the waste.
mc = Mass of carbon, as methane, in a calibration run, mg.
mch = Mass of chloride in a calibration run, mg.
ms = Mass of the waste sample, g.
msc = Mass of carbon, as methane, in the sample, mg.
msf = Mass of sample container and waste sample, g.
msh = Mass of chloride in the sample, mg.
mst = Mass of sample container prior to sampling, g.
mVO = Mass of volatile organics in the sample, mg.
n = Total number of phases present in the waste.
Pp = Percent propane in calibration gas (L/L).
Pvc = Percent 1,1-dichloroethylene in calibration gas (L/L).
Qc = Flow rate of calibration gas, L/min.
tc = Length of time standard gas is delivered to the analyzer, min.
W = Weighted average VO concentration, ppmw.
12.2 Concentration of Carbon, as Methane, in the Calibration Gas.
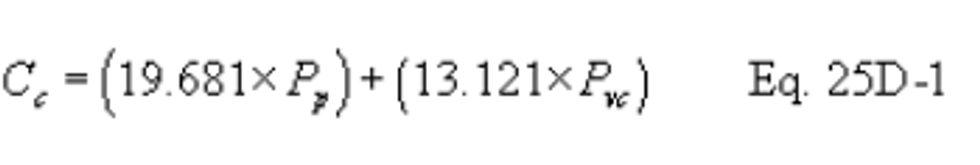
12.3 Concentration of Chloride in the Calibration Gas.
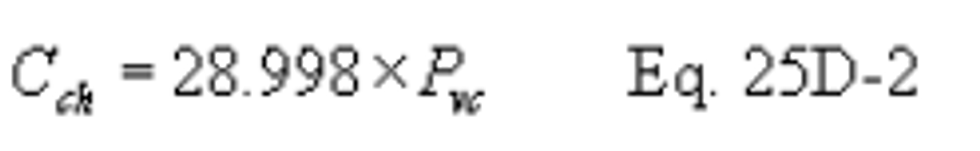
12.4 Mass of Carbon, as Methane, in a Calibration Run.
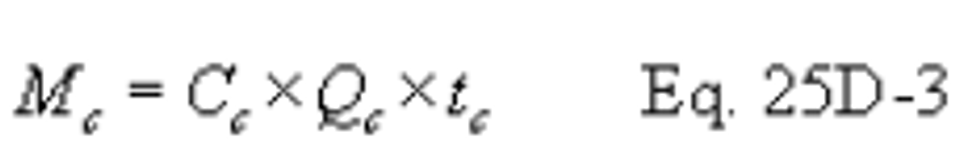
12.5 Mass of Chloride in a Calibration Run.
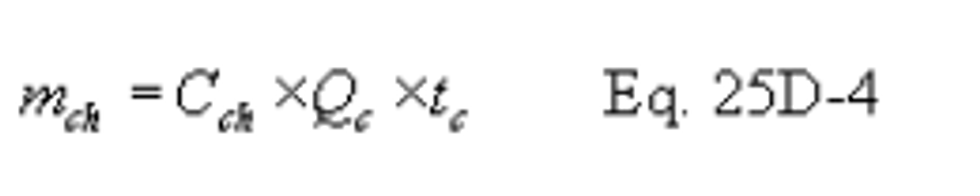
12.6 FID Response Factor, mg/counts.
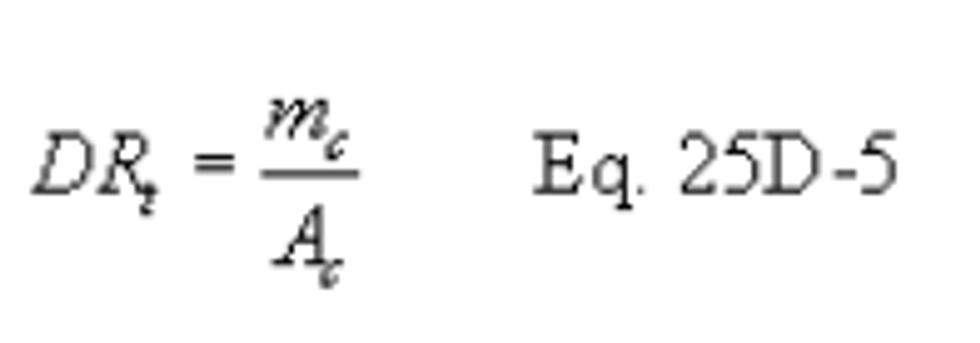
12.7 ELCD Response Factor, mg/counts.
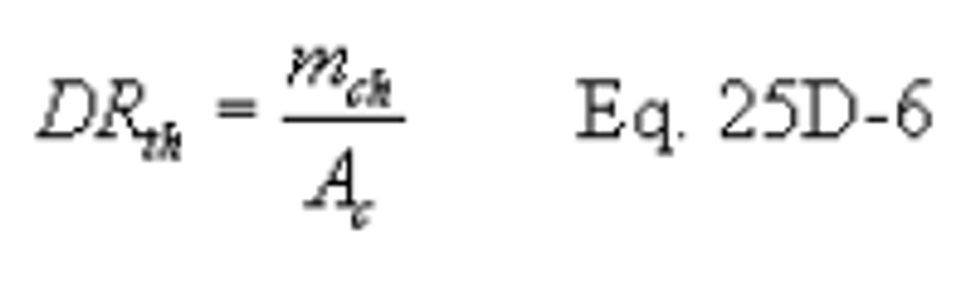
12.8 Mass of Carbon in the Sample.
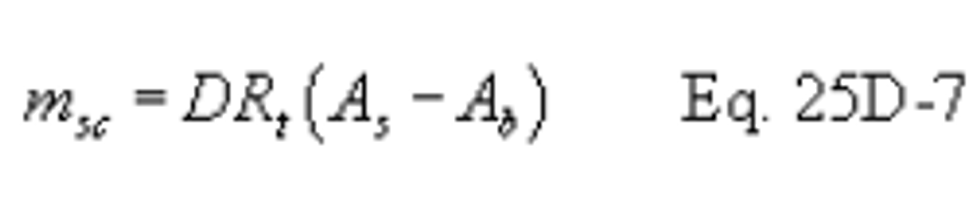
12.9 Mass of Chloride in the Sample.
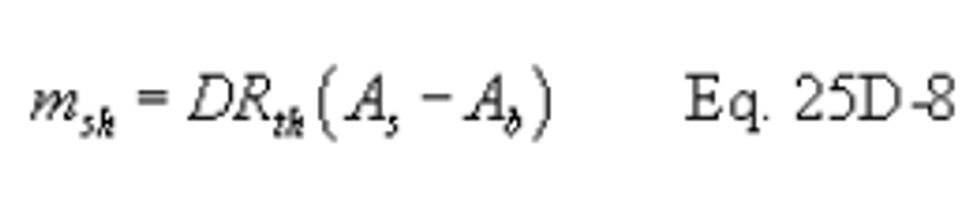
12.10 Mass of Volatile Organics in the Sample.
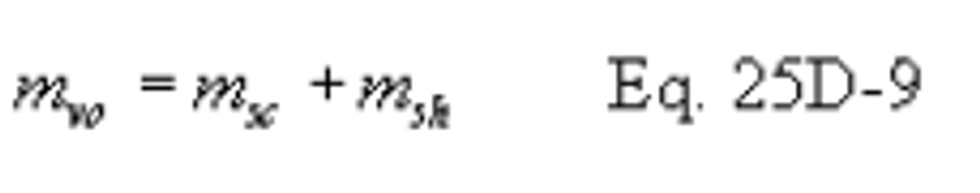
12.11 Relative Standard Deviation.
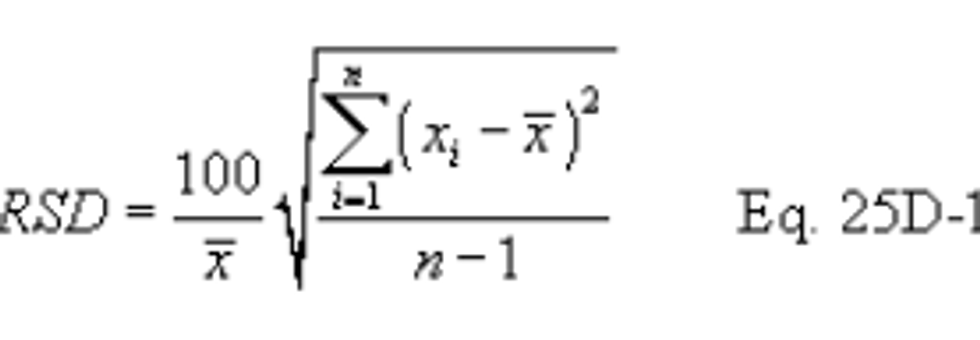
12.12 Mass of Sample.
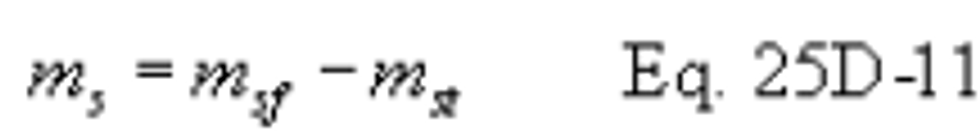
12.13 Concentration of Volatile Organics in Waste.
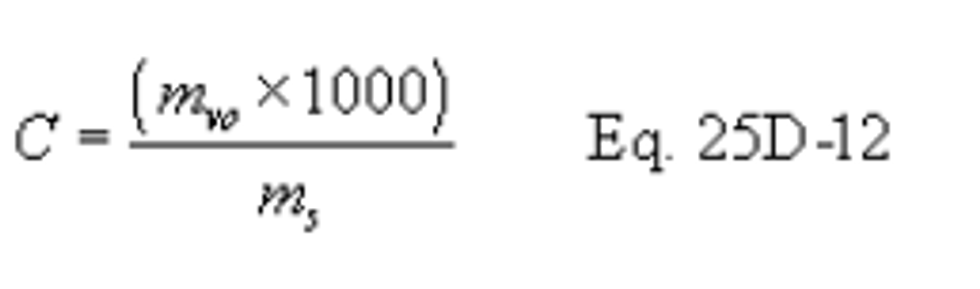
12.14 Weighted Average VO Concentration of Multi-phase Waste.
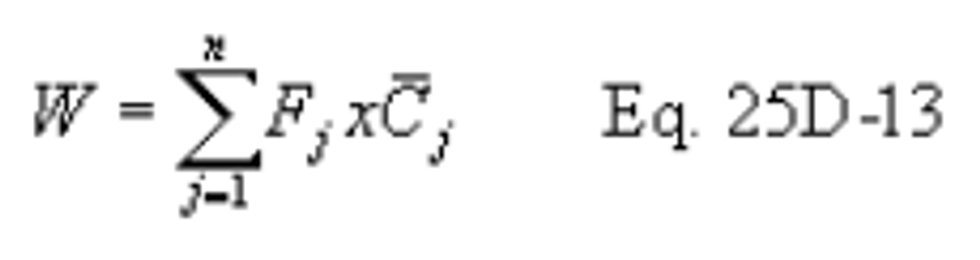
13.0 Method Performance [Reserved]
14.0 Pollution Prevention [Reserved]
15.0 Waste Management [Reserved]
16.0 References
1. “Test Methods for Evaluating Solid Waste, Physical/Chemistry Methods”, U.S. Environmental Protection Agency. Publication SW-846, 3rd Edition, November 1986 as amended by Update I, November 1990.
17.0 Tables, Diagrams, Flowcharts, and Validation Data
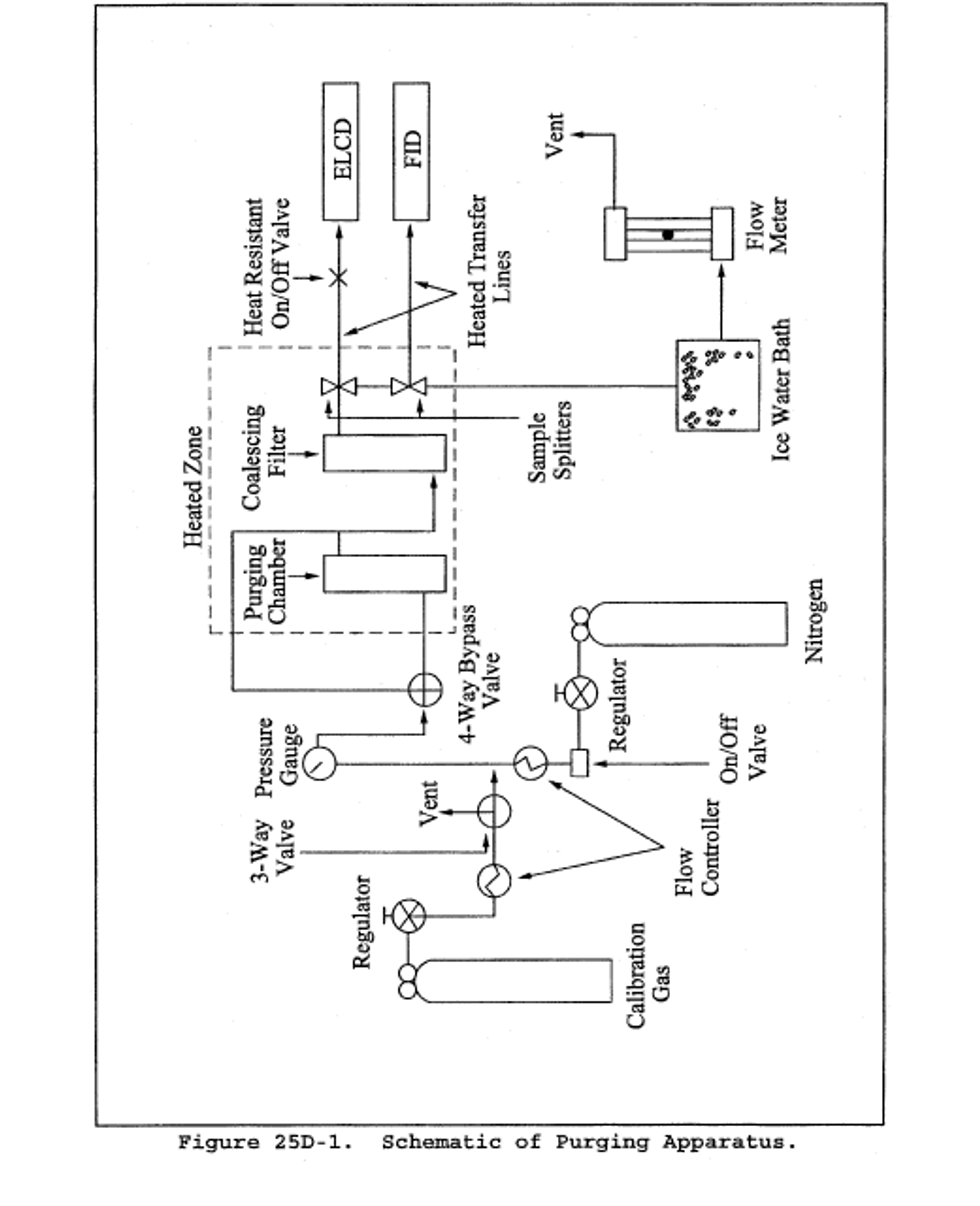
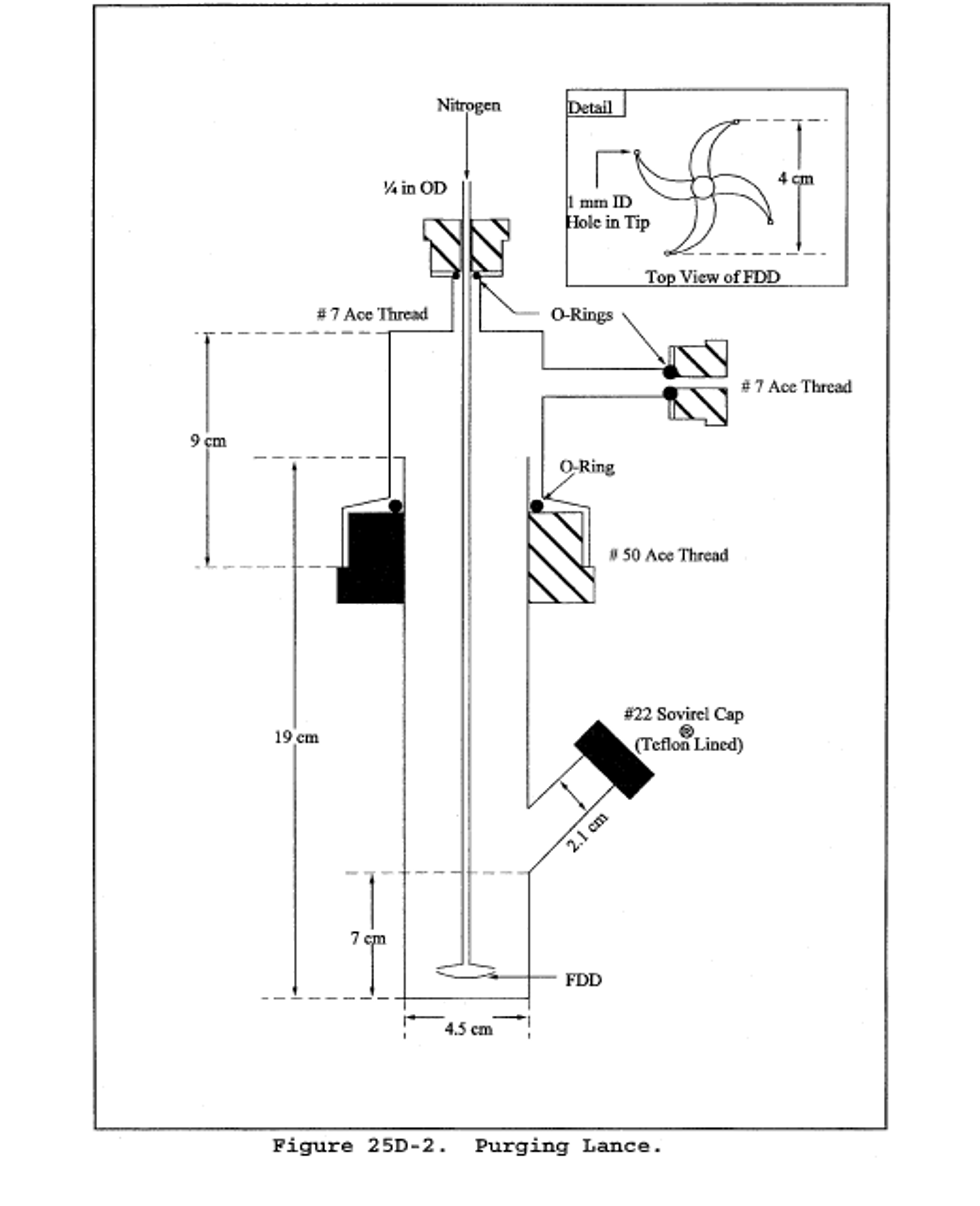
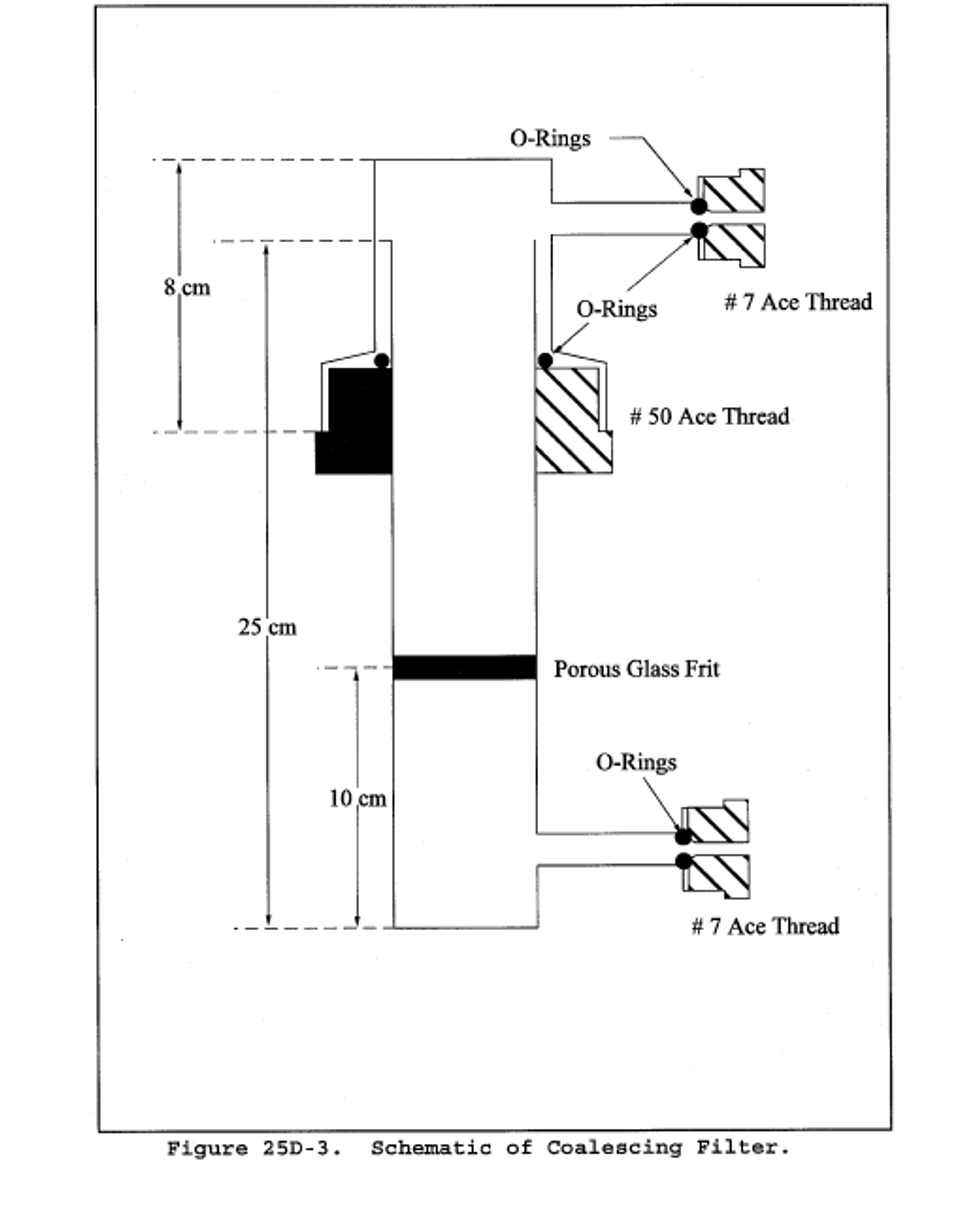
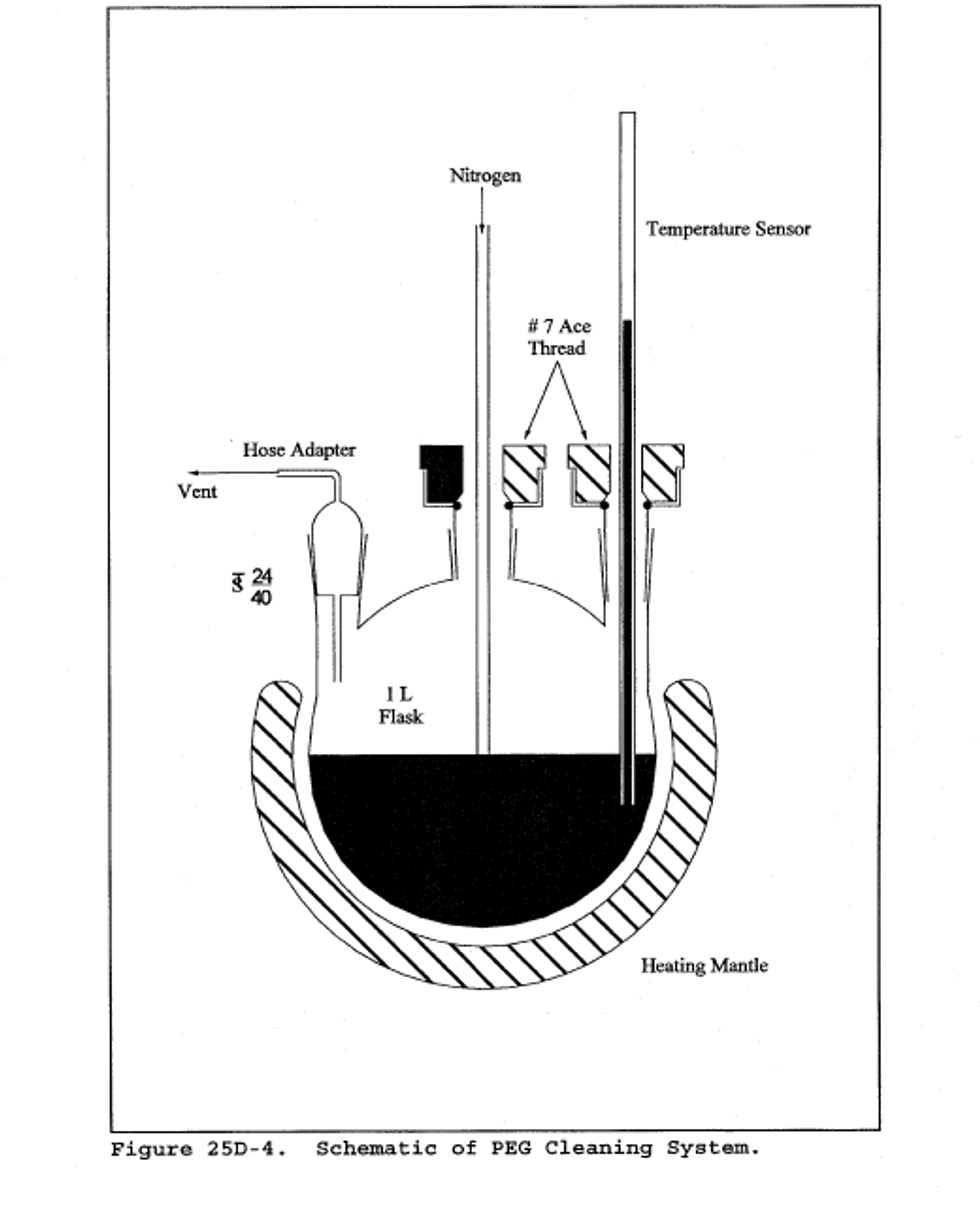
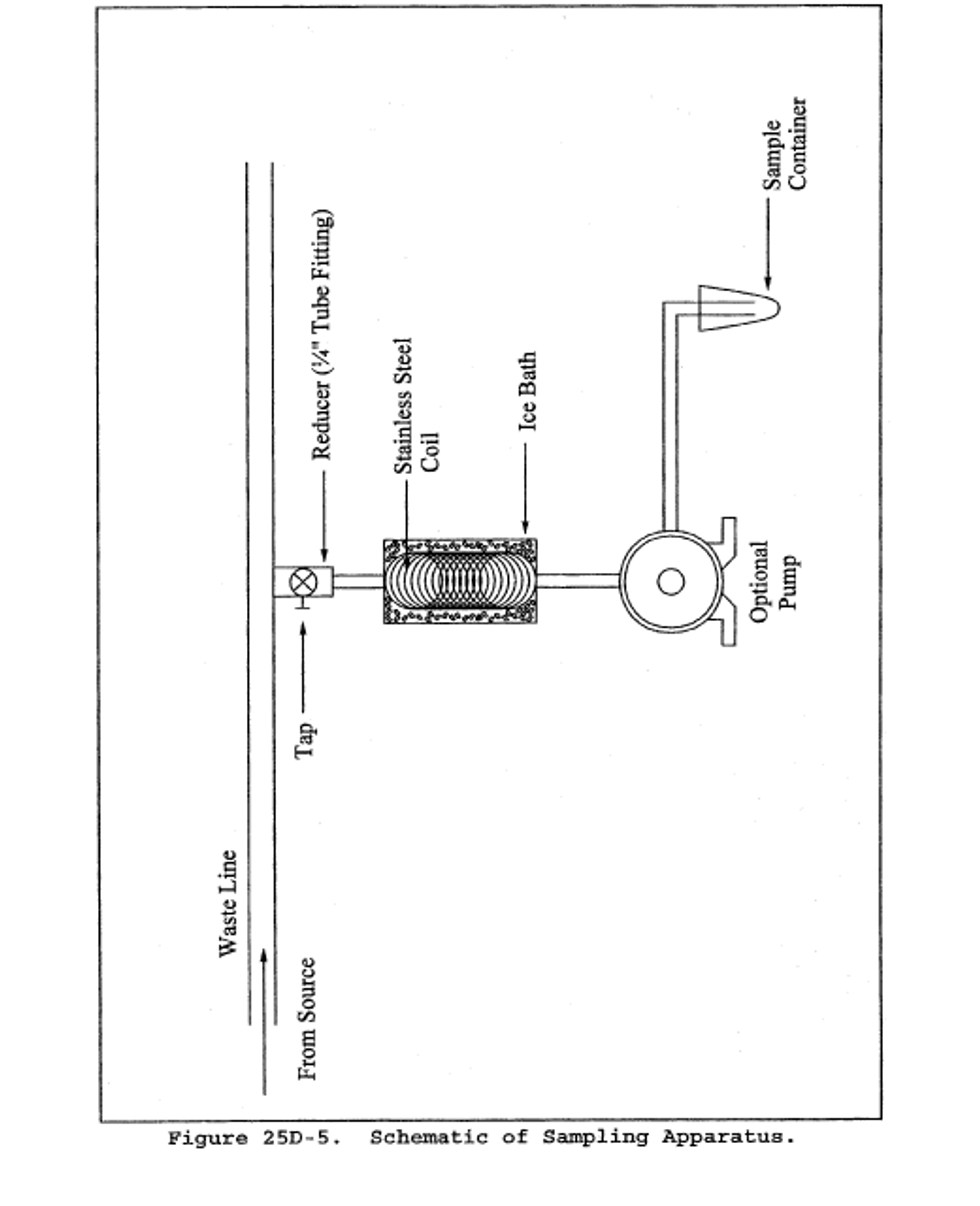
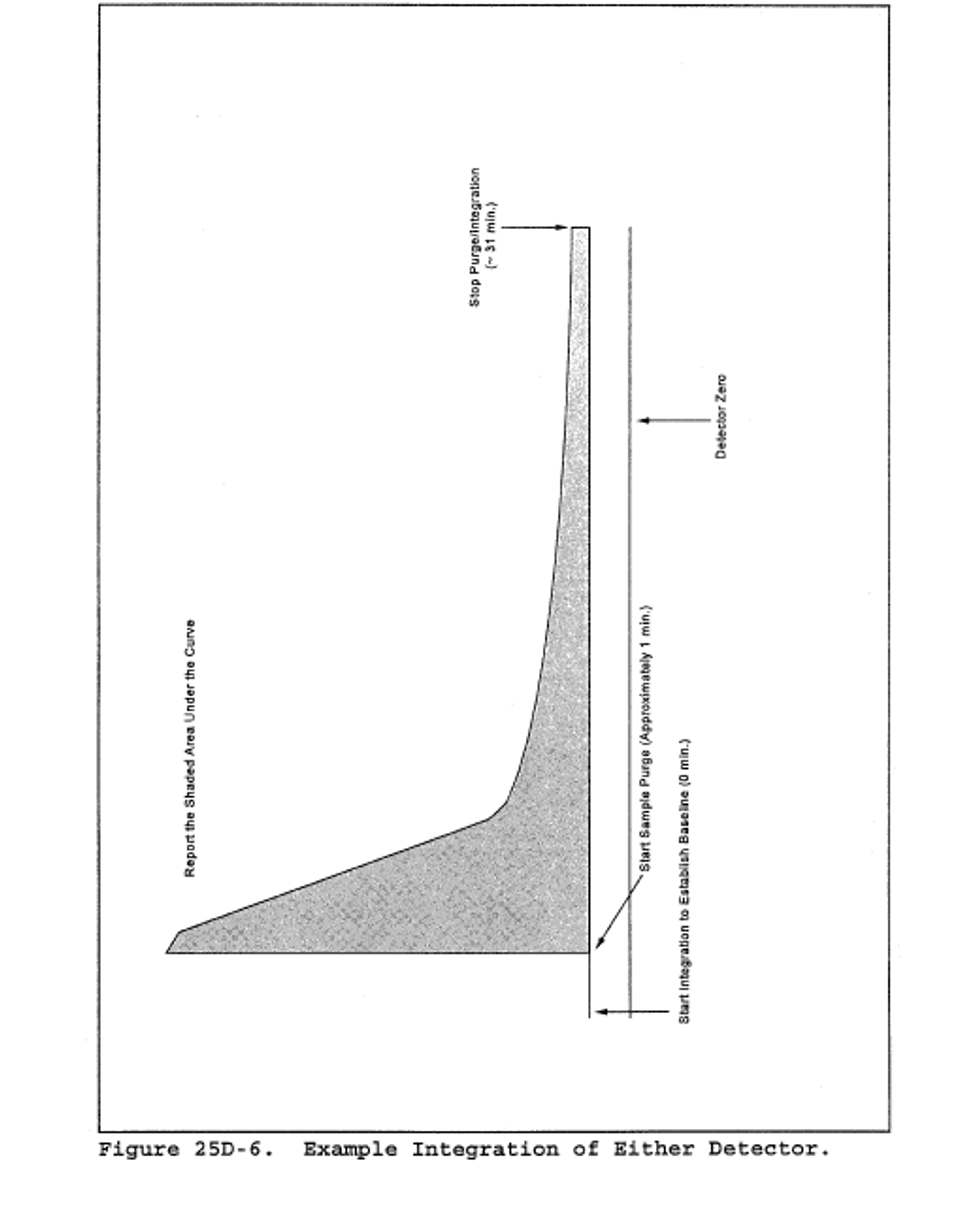
Method 25E - Determination of Vapor Phase Organic Concentration in Waste Samples
Note:
Performance of this method should not be attempted by persons unfamiliar with the operation of a flame ionization detector (FID) nor by those who are unfamiliar with source sampling because knowledge beyond the scope of this presentation is required. This method is not inclusive with respect to specifications (e.g., reagents and standards) and calibration procedures. Some material is incorporated by reference from other methods. Therefore, to obtain reliable results, persons using this method should have a thorough knowledge of at least the following additional test methods: Method 106, part 61, Appendix B, and Method 18, part 60, Appendix A.
1.0 Scope and Application
1.1 Applicability. This method is applicable for determining the vapor pressure of waste cited by an applicable regulation.
1.2 Data Quality Objectives. Adherence to the requirements of this method will enhance the quality of the data obtained from air pollutant sampling methods.
2.0 Summary of Method
2.1 The headspace vapor of the sample is analyzed for carbon content by a headspace analyzer, which uses an FID.
3.0 Definitions [Reserved]
4.0 Interferences
4.1 The analyst shall select the operating parameters best suited to the requirements for a particular analysis. The analyst shall produce confirming data through an adequate supplemental analytical technique and have the data available for review by the Administrator.
5.0 Safety [Reserved]
6.0 Equipment and Supplies
6.1 Sampling. The following equipment is required:
6.1.1 Sample Containers. Vials, glass, with butyl rubber septa, Perkin-Elmer Corporation Numbers 0105-0129 (glass vials), B001-0728 (gray butyl rubber septum, plug style), 0105-0131 (butyl rubber septa), or equivalent. The seal must be made from butyl rubber. Silicone rubber seals are not acceptable.
6.1.2 Vial Sealer. Perkin-Elmer Number 105-0106, or equivalent.
6.1.3 Gas-Tight Syringe. Perkin-Elmer Number 00230117, or equivalent.
6.1.4 The following equipment is required for sampling.
6.1.4.1 Tap.
6.1.4.2 Tubing. Teflon, 0.25-in. ID.
Note:
Mention of trade names or specific products does not constitute endorsement by the Environmental Protection Agency.
6.1.4.3 Cooling Coil. Stainless steel (304), 0.25 in.-ID, equipped with a thermocouple at the coil outlet.
6.2 Analysis. The following equipment is required.
6.2.1 Balanced Pressure Headspace Sampler. Perkin-Elmer HS-6, HS-100, or equivalent, equipped with a glass bead column instead of a chromatographic column.
6.2.2 FID. An FID meeting the following specifications is required.
6.2.2.1 Linearity. A linear response (±5 percent) over the operating range as demonstrated by the procedures established in section 10.2.
6.2.2.2 Range. A full scale range of 1 to 10,000 parts per million (ppm) propane (C3H8). Signal attenuators shall be available to produce a minimum signal response of 10 percent of full scale.
6.2.3 Data Recording System. Analog strip chart recorder or digital integration system compatible with the FID for permanently recording the output of the detector.
6.2.4 Temperature Sensor. Capable of reading temperatures in the range of 30 to 60°C (86 to 140°F) with an accuracy of ±0.1°C (±0.2°F).
7.0 Reagents and Standards
7.1 Analysis. The following items are required for analysis.
7.1.1 Hydrogen (H2). Zero grade hydrogen, as required by the FID.
7.1.2 Carrier Gas. Zero grade nitrogen, containing less than 1 ppm carbon (C) and less than 1 ppm carbon dioxide.
7.1.3 Combustion Gas. Zero grade air or oxygen as required by the FID.
7.2 Calibration and Linearity Check.
7.2.1 Stock Cylinder Gas Standard. 100 percent propane. The manufacturer shall: (a) Certify the gas composition to be accurate to ±3 percent or better (see section 7.2.1.1); (b) recommend a maximum shelf life over which the gas concentration does not change by greater than ±5 percent from the certified value; and (c) affix the date of gas cylinder preparation, certified propane concentration, and recommended maximum shelf life to the cylinder before shipment to the buyer.
7.2.1.1 Cylinder Standards Certification. The manufacturer shall certify the concentration of the calibration gas in the cylinder by (a) directly analyzing the cylinder and (b) calibrating his analytical procedure on the day of cylinder analysis. To calibrate his analytical procedure, the manufacturer shall use, as a minimum, a three-point calibration curve.
7.2.1.2 Verification of Manufacturer's Calibration Standards. Before using, the manufacturer shall verify each calibration standard by (a) comparing it to gas mixtures prepared in accordance with the procedure described in section 7.1 of Method 106 of Part 61, Appendix B, or by (b) calibrating it against Standard Reference Materials (SRM's) prepared by the National Bureau of Standards, if such SRM's are available. The agreement between the initially determined concentration value and the verification concentration value must be within ±5 percent. The manufacturer must reverify all calibration standards on a time interval consistent with the shelf life of the cylinder standards sold.
8.0 Sampling Collection, Preservation, Storage, and Transport
8.1 Install a sampling tap to obtain a sample at a point which is most representative of the unexposed waste (where the waste has had minimum opportunity to volatilize to the atmosphere). Assemble the sampling apparatus as shown in Figure 25E-1.
8.2 Begin sampling by purging the sample lines and cooling coil with at least four volumes of waste. Collect the purged material in a separate container and dispose of it properly.
8.3 After purging, stop the sample flow and transfer the Teflon sampling tube to a sample container. Sample at a flow rate such that the temperature of the waste is <10°C (<50°F). Fill the sample container halfway (±5 percent) and cap it within 5 seconds. Store immediately in a cooler and cover with ice.
8.4 Alternative sampling techniques may be used upon the approval of the Administrator.
9.0 Quality Control
9.1 Miscellaneous Quality Control Measures.
| Section | Quality control measure | Effect |
|---|---|---|
| 10.2, 10.3 | FID calibration and response check | Ensure precision of analytical results. |
10.0 Calibration and Standardization
Note:
Maintain a record of performance of each item.
10.1 Use the procedures in sections 10.2 to calibrate the headspace analyzer and FID and check for linearity before the system is first placed in operation, after any shutdown longer than 6 months, and after any modification of the system.
10.2 Calibration and Linearity. Use the procedures in section 10 of Method 18 of Part 60, Appendix A, to prepare the standards and calibrate the flowmeters, using propane as the standard gas. Fill the calibration standard vials halfway (±5 percent) with deionized water. Purge and fill the airspace with calibration standard. Prepare a minimum of three concentrations of calibration standards in triplicate at concentrations that will bracket the applicable cutoff. For a cutoff of 5.2 kPa (0.75 psi), prepare nominal concentrations of 30,000, 50,000, and 70,000 ppm as propane. For a cutoff of 27.6 kPa (4.0 psi), prepare nominal concentrations of 200,000, 300,000, and 400,000 ppm as propane.
10.2.1 Use the procedures in section 11.3 to measure the FID response of each standard. Use a linear regression analysis to calculate the values for the slope (k) and the y-intercept (b). Use the procedures in sections 12.3 and 12.2 to test the calibration and the linearity.
10.3 Daily FID Calibration Check. Check the calibration at the beginning and at the end of the daily runs by using the following procedures. Prepare 2 calibration standards at the nominal cutoff concentration using the procedures in section 10.2. Place one at the beginning and one at the end of the daily run. Measure the FID response of the daily calibration standard and use the values for k and b from the most recent calibration to calculate the concentration of the daily standard. Use an equation similar to 25E-2 to calculate the percent difference between the daily standard and Cs. If the difference is within 5 percent, then the previous values for k and b can be used. Otherwise, use the procedures in section 10.2 to recalibrate the FID.
11.0 Analytical Procedures
11.1 Allow one hour for the headspace vials to equilibrate at the temperature specified in the regulation. Allow the FID to warm up until a stable baseline is achieved on the detector.
11.2 Check the calibration of the FID daily using the procedures in section 10.3.
11.3 Follow the manufacturer's recommended procedures for the normal operation of the headspace sampler and FID.
11.4 Use the procedures in sections 12.4 and 12.5 to calculate the vapor phase organic vapor pressure in the samples.
11.5 Monitor the output of the detector to make certain that the results are being properly recorded.
12.0 Data Analysis and Calculations
12.1 Nomenclature.
A = Measurement of the area under the response curve, counts.
b = y-intercept of the linear regression line.
Ca = Measured vapor phase organic concentration of sample, ppm as propane.
Cma = Average measured vapor phase organic concentration of standard, ppm as propane.
Cm = Measured vapor phase organic concentration of standard, ppm as propane.
Cs = Calculated standard concentration, ppm as propane.
k = Slope of the linear regression line.
Pbar = Atmospheric pressure at analysis conditions, mm Hg (in. Hg).
P* = Organic vapor pressure in the sample, kPa (psi).
PD = Percent difference between the average measured vapor phase organic concentration (Cm) and the calculated standard concentration (Cs).
RSD = Relative standard deviation.
β = 1.333 × 10−7 kPa/[(mm Hg)(ppm)], (4.91 × 10−7 psi/[(in. Hg)(ppm)])
12.2 Linearity. Use the following equation to calculate the measured standard concentration for each standard vial.
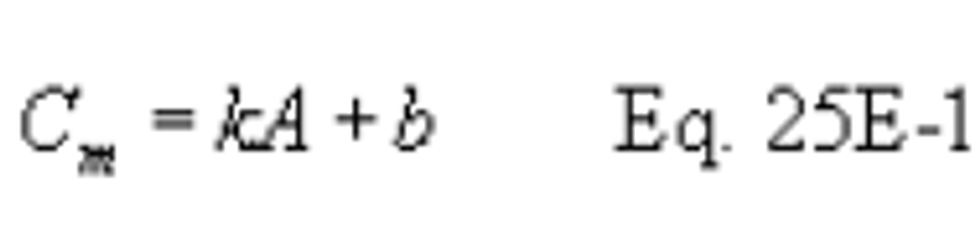
12.2.1 Calculate the average measured standard concentration (Cma) for each set of triplicate standards and use the following equation to calculate PD between Cma and Cs. The instrument linearity is acceptable if the PD is within five for each standard.
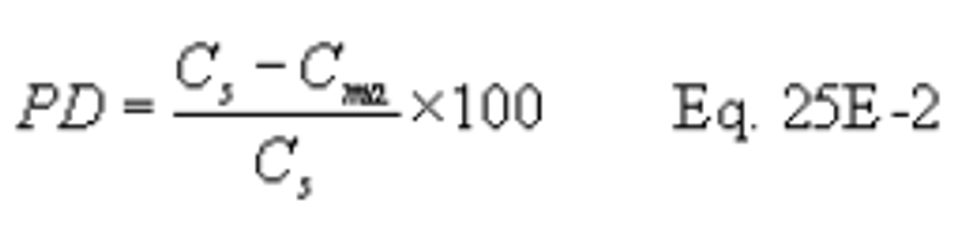
12.3. Relative Standard Deviation (RSD). Use the following equation to calculate the RSD for each triplicate set of standards.
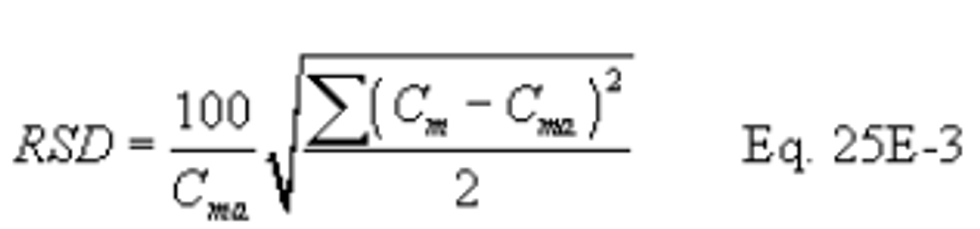
The calibration is acceptable if the RSD is within five for each standard concentration.
12.4 Concentration of organics in the headspace. Use the following equation to calculate the concentration of vapor phase organics in each sample.
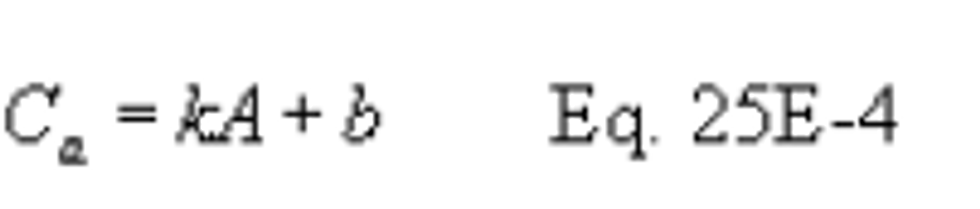
12.5 Vapor Pressure of Organics in the Headspace Sample. Use the following equation to calculate the vapor pressure of organics in the sample.
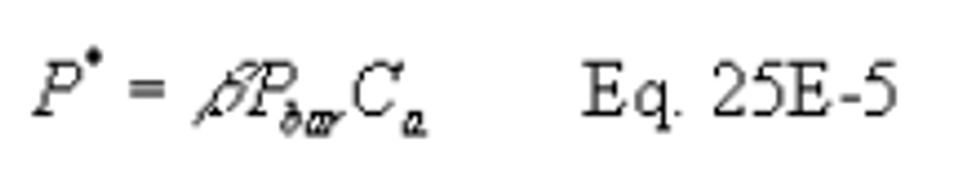
13.0 Method Performance [Reserved]
14.0 Pollution Prevention [Reserved]
15.0 Waste Management [Reserved]
16.0 References
1. Salo, Albert E., Samuel Witz, and Robert D. MacPhee. “Determination of Solvent Vapor Concentrations by Total Combustion Analysis: a Comparison of Infared with Flame Ionization Detectors. Paper No. 75-33.2. (Presented at the 68th Annual Meeting of the Air Pollution Control Association. Boston, Massachusetts.
2. Salo, Albert E., William L. Oaks, and Robert D. MacPhee. “Measuring the Organic Carbon Content of Source Emissions for Air Pollution Control. Paper No. 74-190. (Presented at the 67th Annual Meeting of the Air Pollution Control Association. Denver, Colorado. June 9-13, 1974.) p. 25.
17.0 Tables, Diagrams, Flowcharts, and Validation Data
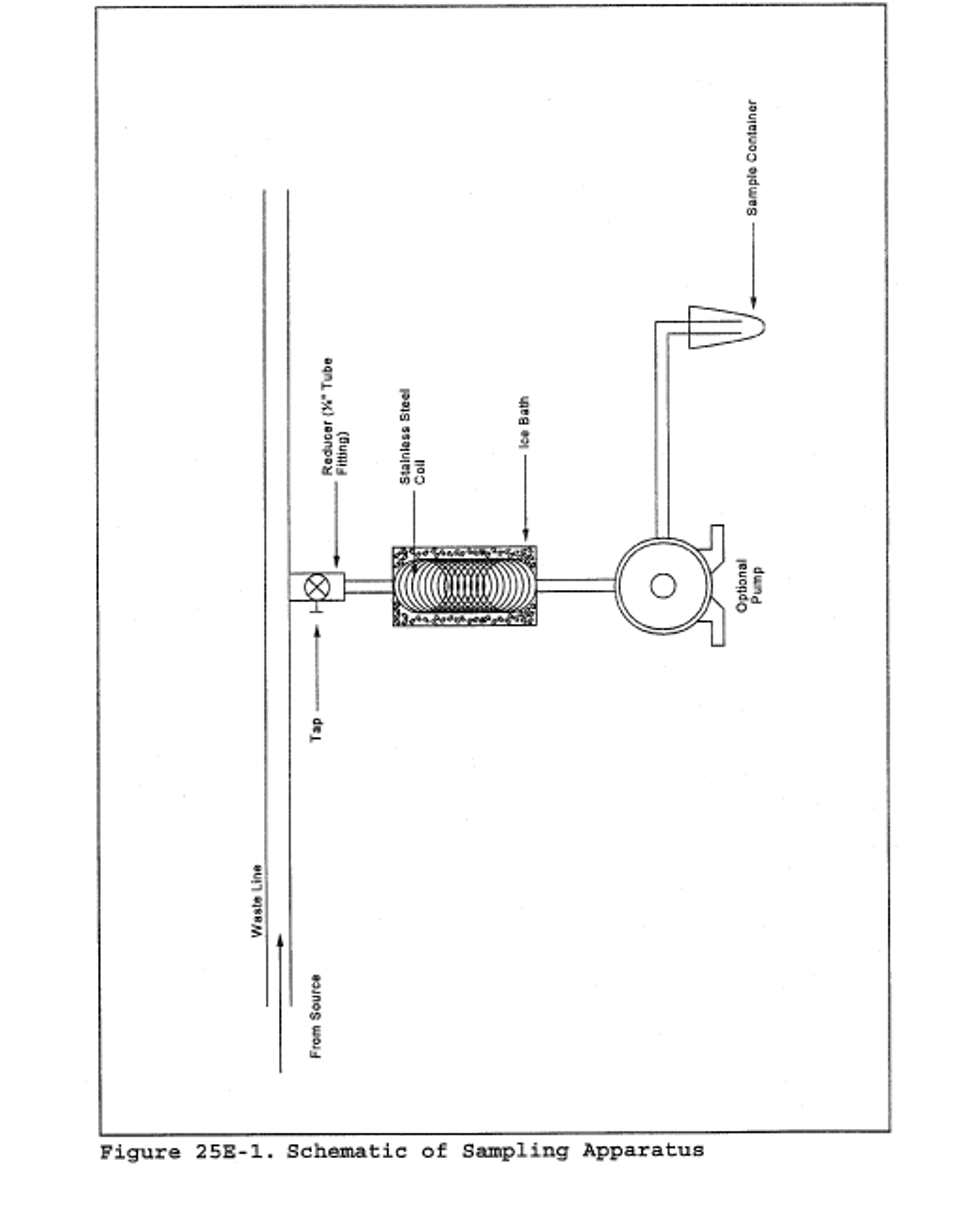
Editorial Note: For Federal Register citations affecting appendix A-7 to part 60, see the List of CFR sections Affected, which appears in the Finding Aids section of the printed volume and at www.govinfo.gov.
[36 FR 24877, Dec. 23, 1971; 85 FR 63415, Oct. 7, 2020; 88 FR 16742, March 20, 2023; 88 FR 18405, March 29, 2023]
['Air Programs']
['Air Quality']
UPGRADE TO CONTINUE READING