Compliance Just Got Easier: Stay ahead of regulatory changes with instant notifications on updates that matter.
['Air Programs']
['Chlorofluorocarbons', 'Criteria Air Pollutants', 'Air Quality']
05/13/2025

Copyright 2026 J. J. Keller & Associate, Inc. For re-use options please contact copyright@jjkeller.com or call 800-558-5011.
Authority: 42 U.S.C. 7401, et seq.
§50.1 Definitions.
(a) As used in this part, all terms not defined herein shall have the meaning given them by the Act.
(b) Act means the Clean Air Act, as amended (42 U.S.C. 1857-18571, as amended by Pub. L. 91-604).
(c) Agency means the Environmental Protection Agency.
(d) Administrator means the Administrator of the Environmental Protection Agency.
(e) Ambient air means that portion of the atmosphere, external to buildings, to which the general public has access.
(f) Reference method means a method of sampling and analyzing the ambient air for an air pollutant that is specified as a reference method in an appendix to this part, or a method that has been designated as a reference method in accordance with §53.11 of this chapter; it does not include a method for which a reference method designation has been cancelled in accordance with §53.11 or §53.16 of this chapter.
(g) Equivalent method means a method of sampling and analyzing the ambient air for an air pollutant that has been designated as an equivalent method in accordance with §53.11 of this chapter; it does not include a method for which an equivalent method designation has been cancelled in accordance with §53.11 or §53.16 of this chapter.
(h) Traceable means that a local standard has been compared and certified either directly or via not more than one intermediate standard, to a primary standard such as a National Bureau of Standards Standard Reference Material (NBS SRM), or a USEPA/NBS-approved Certified Reference Material (CRM).
(i) Indian country is as defined in 18 U.S.C. 1151.
(j) Exceptional event means an event(s) and its resulting emissions that affect air quality in such a way that there exists a clear causal relationship between the specific event(s) and the monitored exceedance(s) or violation(s), is not reasonably controllable or preventable, is an event(s) caused by human activity that is unlikely to recur at a particular location or a natural event(s), and is determined by the Administrator in accordance with 40 CFR 50.14 to be an exceptional event. It does not include air pollution relating to source noncompliance. Stagnation of air masses and meteorological inversions do not directly cause pollutant emissions and are not exceptional events. Meteorological events involving high temperatures or lack of precipitation (i.e., severe, extreme or exceptional drought) also do not directly cause pollutant emissions and are not considered exceptional events. However, conditions involving high temperatures or lack of precipitation may promote occurrences of particular types of exceptional events, such as wildfires or high wind events, which do directly cause emissions.
(k) Natural event means an event and its resulting emissions, which may recur at the same location, in which human activity plays little or no direct causal role. For purposes of the definition of a natural event, anthropogenic sources that are reasonably controlled shall be considered to not play a direct role in causing emissions.
(l) Exceedance with respect to a national ambient air quality standard means one occurrence of a measured or modeled concentration that exceeds the specified concentration level of such standard for the averaging period specified by the standard.
(m) Prescribed fire is any fire intentionally ignited by management actions in accordance with applicable laws, policies, and regulations to meet specific land or resource management objectives.
(n) Wildfire is any fire started by an unplanned ignition caused by lightning; volcanoes; other acts of nature; unauthorized activity; or accidental, human-caused actions, or a prescribed fire that has developed into a wildfire. A wildfire that predominantly occurs on wildland is a natural event.
(o) Wildland means an area in which human activity and development are essentially non-existent, except for roads, railroads, power lines, and similar transportation facilities. Structures, if any, are widely scattered.
(p) High wind dust event is an event that includes the high-speed wind and the dust that the wind entrains and transports to a monitoring site.
(q) High wind threshold is the minimum wind speed capable of causing particulate matter emissions from natural undisturbed lands in the area affected by a high wind dust event.
(r) Federal land manager means, consistent with the definition in 40 CFR 51.301, the Secretary of the department with authority over the Federal Class I area (or the Secretary's designee) or, with respect to Roosevelt-Campobello International Park, the Chairman of the Roosevelt-Campobello International Park Commission.
[36 FR 22384, Nov. 25, 1971, as amended at 41 FR 11253, Mar. 17, 1976; 48 FR 2529, Jan. 20, 1983; 63 FR 7274, Feb. 12, 1998; 72 FR 13580, Mar. 22, 2007; 81 FR 68276, Oct. 3, 2016]
§50.2 Scope.
(a) National primary and secondary ambient air quality standards under section 109 of the Act are set forth in this part.
(b) National primary ambient air quality standards define levels of air quality which the Administrator judges are necessary, with an adequate margin of safety, to protect the public health. National secondary ambient air quality standards define levels of air quality which the Administrator judges necessary to protect the public welfare from any known or anticipated adverse effects of a pollutant. Such standards are subject to revision, and additional primary and secondary standards may be promulgated as the Administrator deems necessary to protect the public health and welfare.
(c) The promulgation of national primary and secondary ambient air quality standards shall not be considered in any manner to allow significant deterioration of existing air quality in any portion of any State or Indian country.
(d) The proposal, promulgation, or revision of national primary and secondary ambient air quality standards shall not prohibit any State or Indian country from establishing ambient air quality standards for that State or area under a tribal CAA program or any portion thereof which are more stringent than the national standards.
[36 FR 22384, Nov. 25, 1971, as amended at 63 FR 7274, Feb. 12, 1998]
§50.3 Reference conditions.
All measurements of air quality that are expressed as mass per unit volume (e.g., micrograms per cubic meter) other than for particulate matter (PM2.5) standards contained in §§50.7, 50.13, and 50.18, and lead standards contained in §50.16 shall be corrected to a reference temperature of 25 (deg) C and a reference pressure of 760 millimeters of mercury (1,013.2 millibars). Measurements of PM2.5 for purposes of comparison to the standards contained in §§50.7, 50.13, and 50.18, and of lead for purposes of comparison to the standards contained in §50.16 shall be reported based on actual ambient air volume measured at the actual ambient temperature and pressure at the monitoring site during the measurement period.
[78 FR 3277, Jan. 15, 2013]
§50.4 National primary ambient air quality standards for sulfur oxides (sulfur dioxide).
(a) The level of the annual standard is 0.030 parts per million (ppm), not to be exceeded in a calendar year. The annual arithmetic mean shall be rounded to three decimal places (fractional parts equal to or greater than 0.0005 ppm shall be rounded up).
(b) The level of the 24-hour standard is 0.14 parts per million (ppm), not to be exceeded more than once per calendar year. The 24-hour averages shall be determined from successive nonoverlapping 24-hour blocks starting at midnight each calendar day and shall be rounded to two decimal places (fractional parts equal to or greater than 0.005 ppm shall be rounded up).
(c) Sulfur oxides shall be measured in the ambient air as sulfur dioxide by the reference method described in appendix A to this part or by an equivalent method designated in accordance with §53.11 of this chapter.
(d) To demonstrate attainment, the annual arithmetic mean and the second-highest 24-hour averages must be based upon hourly data that are at least 75 percent complete in each calendar quarter. A 24-hour block average shall be considered valid if at least 75 percent of the hourly averages for the 24-hour period are available. In the event that only 18, 19, 20, 21, 22, or 23 hourly averages are available, the 24-hour block average shall be computed as the sum of the available hourly averages using 18, 19, etc. as the divisor. If fewer than 18 hourly averages are available, but the 24-hour average would exceed the level of the standard when zeros are substituted for the missing values, subject to the rounding rule of paragraph (b) of this section, then this shall be considered a valid 24-hour average. In this case, the 24-hour block average shall be computed as the sum of the available hourly averages divided by 24.
(e) The standards set forth in this section will remain applicable to all areas notwithstanding the promulgation of SO2 national ambient air quality standards (NAAQS) in §50.17. The SO2 NAAQS set forth in this section will no longer apply to an area one year after the effective date of the designation of that area, pursuant to section 107 of the Clean Air Act, for the SO2 NAAQS set forth in §50.17; except that for areas designated nonattainment for the SO2 NAAQS set forth in this section as of the effective date of §50.17, and areas not meeting the requirements of a SIP call with respect to requirements for the SO2 NAAQS set forth in this section, the SO2 NAAQS set forth in this section will apply until that area submits, pursuant to section 191 of the Clean Air Act, and EPA approves, an implementation plan providing for attainment of the SO2 NAAQS set forth in §50.17.
[61 FR 25579, May 22, 1996, as amended at 75 FR 35592, June 22, 2010]
§50.5 National secondary ambient air quality standard for sulfur oxides (sulfur dioxide).
(a) The level of the 3-hour standard is 0.5 parts per million (ppm), not to be exceeded more than once per calendar year. The 3-hour averages shall be determined from successive nonoverlapping 3-hour blocks starting at midnight each calendar day and shall be rounded to 1 decimal place (fractional parts equal to or greater than 0.05 ppm shall be rounded up).
(b) Sulfur oxides shall be measured in the ambient air as sulfur dioxide by the reference method described in appendix A of this part or by an equivalent method designated in accordance with §53.11 of this chapter.
(c) To demonstrate attainment, the second-highest 3-hour average must be based upon hourly data that are at least 75 percent complete in each calendar quarter. A 3-hour block average shall be considered valid only if all three hourly averages for the 3-hour period are available. If only one or two hourly averages are available, but the 3-hour average would exceed the level of the standard when zeros are substituted for the missing values, subject to the rounding rule of paragraph (a) of this section, then this shall be considered a valid 3-hour average. In all cases, the 3-hour block average shall be computed as the sum of the hourly averages divided by 3.
[61 FR 25580, May 22, 1996]
§50.6 National primary and secondary ambient air quality standards for PM 10.
(a) The level of the national primary and secondary 24-hour ambient air quality standards for particulate matter is 150 micrograms per cubic meter (µg/m 3), 24-hour average concentration. The standards are attained when the expected number of days per calendar year with a 24-hour average concentration above 150 µg/m 3, as determined in accordance with appendix K to this part, is equal to or less than one.
(b) [Reserved]
(c) For the purpose of determining attainment of the primary and secondary standards, particulate matter shall be measured in the ambient air as PM10 (particles with an aerodynamic diameter less than or equal to a nominal 10 micrometers) by:
(1) A reference method based on appendix J and designated in accordance with §53.11 of this chapter, or
(2) An equivalent method designated in accordance with §53.11 of this chapter.
[52 FR 24663, July 1, 1987, as amended at 62 FR 38711, July 18, 1997; 65 FR 80779, Dec. 22, 2000; 71 FR 61224, Oct. 17, 2006]
§50.7 National primary and secondary ambient air quality standards for PM2.5.
(a) The national primary and secondary ambient air quality standards for particulate matter are 15.0 micrograms per cubic meter (µg/m 3) annual arithmetic mean concentration, and 65 µg/m 3 24-hour average concentration measured in the ambient air as PM2.5 (particles with an aerodynamic diameter less than or equal to a nominal 2.5 micrometers) by either:
(1) A reference method based on appendix L of this part and designated in accordance with §53.11 of this chapter; or
(2) An equivalent method designated in accordance with §53.11 of this chapter.
(b) The annual primary and secondary PM2.5 standards are met when the annual arithmetic mean concentration, as determined in accordance with appendix N of this part, is less than or equal to 15.0 micrograms per cubic meter.
(c) The 24-hour primary and secondary PM2.5 standards are met when the 98 th percentile 24-hour concentration, as determined in accordance with appendix N of this part, is less than or equal to 65 micrograms per cubic meter.
[62 FR 38711, July 18, 1997, as amended at 69 FR 45595, July 30, 2004]
§50.8 National primary ambient air quality standards for carbon monoxide.
(a) The national primary ambient air quality standards for carbon monoxide are:
(1) 9 parts per million (10 milligrams per cubic meter) for an 8-hour average concentration not to be exceeded more than once per year and
(2) 35 parts per million (40 milligrams per cubic meter) for a 1-hour average concentration not to be exceeded more than once per year.
(b) The levels of carbon monoxide in the ambient air shall be measured by:
(1) A reference method based on appendix C and designated in accordance with §53.11 of this chapter, or
(2) An equivalent method designated in accordance with §53.11 of this chapter.
(c) An 8-hour average shall be considered valid if at least 75 percent of the hourly average for the 8-hour period are available. In the event that only six (or seven) hourly averages are available, the 8-hour average shall be computed on the basis of the hours available using six (or seven) as the divisor.
(d) When summarizing data for comparision with the standards, averages shall be stated to one decimal place. Comparison of the data with the levels of the standards in parts per million shall be made in terms of integers with fractional parts of 0.5 or greater rounding up.
[50 FR 37501, Sept. 13, 1985]
§50.9 National 1-hour primary and secondary ambient air quality standards for ozone.
(a) The level of the national 1-hour primary and secondary ambient air quality standards for ozone measured by a reference method based on appendix D to this part and designated in accordance with §53.11 of this chapter, is 0.12 parts per million (235 µg/m 3). The standard is attained when the expected number of days per calendar year with maximum hourly average concentrations above 0.12 parts per million (235 µg/m 3) is equal to or less than 1, as determined by appendix H to this part.
(b) The 1-hour standards set forth in this section will remain applicable to all areas notwithstanding the promulgation of 8-hour ozone standards under §50.10. The 1-hour NAAQS set forth in paragraph (a) of this section will no longer apply to an area one year after the effective date of the designation of that area for the 8-hour ozone NAAQS pursuant to section 107 of the Clean Air Act. Area designations and classifications with respect to the 1-hour standards are codified in 40 CFR part 81.
[62 FR 38894, July 18, 1997, as amended at 65 FR 45200, July 20, 2000; 68 FR 38163, June 26, 2003, 69 FR 23996, Apr. 30, 2004; 77 FR 28441, May 14, 2012]
§50.10 National 8-hour primary and secondary ambient air quality standards for ozone.
(a) The level of the national 8-hour primary and secondary ambient air quality standards for ozone, measured by a reference method based on appendix D to this part and designated in accordance with §53.11 of this chapter, is 0.08 parts per million (ppm), daily maximum 8-hour average.
(b) The 8-hour primary and secondary ozone ambient air quality standards are met at an ambient air quality monitoring site when the average of the annual fourth-highest daily maximum 8-hour average ozone concentration is less than or equal to 0.08 ppm, as determined in accordance with appendix I to this part.
(c) Until the effective date of the final Implementation of the 2008 National Ambient Air Quality Standards for Ozone: State Implementation Plan Requirements Rule (final SIP Requirements Rule) to be codified at 40 CFR 51.1100 et seq., the 1997 ozone NAAQS set forth in this section will continue in effect, notwithstanding the promulgation of the 2008 ozone NAAQS under §50.15. The 1997 ozone NAAQS set forth in this section will no longer apply upon the effective date of the final SIP Requirements Rule. For purposes of the anti-backsliding requirements of §51.1105, §51.165 and Appendix S to part 51, the area designations and classifications with respect to the revoked 1997 ozone NAAQS are codified in 40 CFR part 81.
[62 FR 38894, July 18, 1997, as amended at 77 FR 30170, May 21, 2012; 80 FR 12312, Mar. 6, 2015]
§50.11 National primary and secondary ambient air quality standards for oxides of nitrogen (with nitrogen dioxide as the indicator).
(a) The level of the national primary annual ambient air quality standard for oxides of nitrogen is 53 parts per billion (ppb, which is 1 part in 1,000,000,000), annual average concentration, measured in the ambient air as nitrogen dioxide.
(b) The level of the national primary 1-hour ambient air quality standard for oxides of nitrogen is 100 ppb, 1-hour average concentration, measured in the ambient air as nitrogen dioxide.
(c) The level of the national secondary ambient air quality standard for nitrogen dioxide is 0.053 parts per million (100 micrograms per cubic meter), annual arithmetic mean concentration.
(d) The levels of the standards shall be measured by:
(1) A reference method based on appendix F to this part; or
(2) By a Federal equivalent method (FEM) designated in accordance with §53.11 of this chapter.
(e) The annual primary standard is met when the annual average concentration in a calendar year is less than or equal to 53 ppb, as determined in accordance with appendix S of this part for the annual standard.
(f) The 1-hour primary standard is met when the three-year average of the annual 98th percentile of the daily maximum 1-hour average concentration is less than or equal to 100 ppb, as determined in accordance with appendix S of this part for the 1-hour standard.
(g) The secondary standard is attained when the annual arithmetic mean concentration in a calendar year is less than or equal to 0.053 ppm, rounded to three decimal places (fractional parts equal to or greater than 0.0005 ppm must be rounded up). To demonstrate attainment, an annual mean must be based upon hourly data that are at least 75 percent complete or upon data derived from manual methods that are at least 75 percent complete for the scheduled sampling days in each calendar quarter.
[75 FR 6531, Feb. 9, 2010]
§50.12 National primary and secondary ambient air quality standards for lead.
(a) National primary and secondary ambient air quality standards for lead and its compounds, measured as elemental lead by a reference method based on appendix G to this part, or by an equivalent method, are: 1.5 micrograms per cubic meter, maximum arithmetic mean averaged over a calendar quarter.
(b) The standards set forth in this section will remain applicable to all areas notwithstanding the promulgation of lead national ambient air quality standards (NAAQS) in §50.16. The lead NAAQS set forth in this section will no longer apply to an area one year after the effective date of the designation of that area, pursuant to section 107 of the Clean Air Act, for the lead NAAQS set forth in §50.16; except that for areas designated nonattainment for the lead NAAQS set forth in this section as of the effective date of §50.16, the lead NAAQS set forth in this section will apply until that area submits, pursuant to section 191 of the Clean Air Act, and EPA approves, an implementation plan providing for attainment and/or maintenance of the lead NAAQS set forth in §50.16.
(Secs. 109, 301(a) Clean Air Act as amended (42 U.S.C. 7409, 7601(a)))
[43 FR 46258, Oct. 5, 1978, as amended at 73 FR 67051, Nov. 12, 2008]
§50.13 National primary and secondary ambient air quality standards for PM2.5.
(a) The national primary and secondary ambient air quality standards for particulate matter are 15.0 micrograms per cubic meter (µg/m 3) annual arithmetic mean concentration, and 35 µg/m 3 24-hour average concentration measured in the ambient air as PM2.5 (particles with an aerodynamic diameter less than or equal to a nominal 2.5 micrometers) by either:
(1) A reference method based on appendix L of this part and designated in accordance with §53.11 of this chapter; or
(2) An equivalent method designated in accordance with §53.11 of this chapter.
(b) The annual primary and secondary PM2.5 standards are met when the annual arithmetic mean concentration, as determined in accordance with appendix N of this part, is less than or equal to 15.0 µg/m 3.
(c) The 24-hour primary and secondary PM2.5 standards are met when the 98th percentile 24-hour concentration, as determined in accordance with appendix N of this part, is less than or equal to 35 µg/m 3.
(d) Until the effective date of the final Fine Particulate Matter National Ambient Air Quality Standards: State Implementation Plan Requirements rule to be codified at 40 CFR 51.1000 through 51.1016, the 1997 annual PM2.5 NAAQS set forth in this section will continue in effect, notwithstanding the promulgation of the 2012 primary annual PM2.5 NAAQS under §50.18. The 1997 primary annual PM2.5 NAAQS set forth in this section will no longer apply upon the effective date of the final Fine Particulate Matter National Ambient Air Quality Standards: State Implementation Plan Requirements rule; except that for areas designated nonattainment for the 1997 annual PM2.5 NAAQS set forth in this section as of the effective date of the final Fine Particulate Matter National Ambient Air Quality Standards: State Implementation Plan Requirements rule, the requirements applicable to the 1997 primary annual PM2.5 NAAQS set forth in this section will apply until the effective date of an area's redesignation to attainment for the 1997 annual PM2.5 NAAQS pursuant to the requirements of section 107 of the Clean Air Act. The 1997 secondary annual PM2.5 NAAQS and the 1997 24-hour PM2.5 NAAQS shall remain in effect. The area designations and classifications with respect to the 1997 annual and 24-hour PM2.5 NAAQS remain codified in 40 CFR §53.11 in order to provide information on where the 1997 primary annual PM2.5 NAAQS has been revoked and to facilitate the implementation of the 1997 secondary annual PM2.5 NAAQS and the 1997 24-hour PM2.5 NAAQS.
[71 FR 61224, Oct. 17, 2006, as amended at 81 FR 58149, Aug. 24, 2016]
§50.14 Treatment of air quality monitoring data influenced by exceptional events.
(a) Requirements - (1) Scope. (i) This section applies to the treatment of data showing exceedances or violations of any national ambient air quality standard for purposes of the following types of regulatory determinations by the Administrator:
(A) An action to designate an area, pursuant to Clean Air Act section 107(d)(1), or redesignate an area, pursuant to Clean Air Act section 107(d)(3), for a particular national ambient air quality standard;
(B) The assignment or re-assignment of a classification category to a nonattainment area where such classification is based on a comparison of pollutant design values, calculated according to the specific data handling procedures in 40 CFR §53.11 for each national ambient air quality standard, to the level of the relevant national ambient air quality standard;
(C) A determination regarding whether a nonattainment area has attained the level of the appropriate national ambient air quality standard by its specified deadline;
(D) A determination that an area has data for the specific NAAQS, which qualify the area for an attainment date extension under the CAA provisions for the applicable pollutant;
(E) A determination under Clean Air Act section 110(k)(5), if based on an area violating a national ambient air quality standard, that the state implementation plan is inadequate under the requirements of Clean Air Act section 110; and
(F) Other actions on a case-by-case basis as determined by the Administrator.
(ii) A State, federal land manager or other federal agency may request the Administrator to exclude data showing exceedances or violations of any national ambient air quality standard that are directly due to an exceptional event from use in determinations identified in paragraph (a)(1)(i) of this section by demonstrating to the Administrator's satisfaction that such event caused a specific air pollution concentration at a particular air quality monitoring location.
(A) For a federal land manager or other federal agency to be eligible to initiate such a request for data exclusion, the federal land manager or other federal agency must:
(1) Either operate a regulatory monitor that has been affected by an exceptional event or manage land on which an exceptional event occurred that influenced a monitored concentration at a regulatory monitor; and
(2) Initiate such a request only after the State in which the affected monitor is located concurs with the federal land manager's or other federal agency's submittal.
(B) With regard to such a request, all provisions in this section that are expressed as requirements applying to a State shall, except as noted, be requirements applying to the federal land manager or other federal agency.
(C) Provided all provisions in this section are met, the Administrator shall allow a State to submit demonstrations for any regulatory monitor within its jurisdictional bounds, including those operated by federal land managers, other federal agencies and delegated local agencies.
(D) Where multiple agencies within a state submit demonstrations for events that meet the requirements of the Exceptional Events Rule, a State submittal shall have primacy for any regulatory monitor within its jurisdictional bounds.
(2) A demonstration to justify data exclusion may include any reliable and accurate data, but must specifically address the elements in paragraphs (c)(3)(iv) and (v) of this section.
(b) Determinations by the Administrator - (1) Generally. The Administrator shall exclude data from use in determinations of exceedances and violations identified in paragraph (a)(1)(i) of this section where a State demonstrates to the Administrator's satisfaction that an exceptional event caused a specific air pollution concentration at a particular air quality monitoring location and otherwise satisfies the requirements of this section.
(2) Fireworks displays. The Administrator shall exclude data from use in determinations of exceedances and violations where a State demonstrates to the Administrator's satisfaction that emissions from fireworks displays caused a specific air pollution concentration in excess of one or more national ambient air quality standards at a particular air quality monitoring location and otherwise satisfies the requirements of this section. Such data will be treated in the same manner as exceptional events under this rule, provided a State demonstrates that such use of fireworks is significantly integral to traditional national, ethnic, or other cultural events including, but not limited to, July Fourth celebrations that satisfy the requirements of this section.
(3) Prescribed fires. (i) The Administrator shall exclude data from use in determinations of exceedances and violations, where a State demonstrates to the Administrator's satisfaction that emissions from prescribed fires caused a specific air pollution concentration in excess of one or more national ambient air quality standards at a particular air quality monitoring location and otherwise satisfies the requirements of this section.
(ii) In addressing the requirements set forth in paragraph (c)(3)(iv)(D) of this section regarding the not reasonably controllable or preventable criterion:
(A) With respect to the requirement that a prescribed fire be not reasonably controllable, the State must either certify to the Administrator that it has adopted and is implementing a smoke management program or the State must demonstrate that the burn manager employed appropriate basic smoke management practices identified in Table 1 to §50.14. Where a burn manager employs appropriate basic smoke management practices, the State may rely on a statement or other documentation provided by the burn manager that he or she employed those practices. If an exceedance or violation of a NAAQS occurs when a prescribed fire is employing an appropriate basic smoke management practices approach, the State and the burn manager must undertake a review of the subject fire, including a review of the basic smoke management practices applied during the subject fire to ensure the protection of air quality and public health and progress towards restoring and/or maintaining a sustainable and resilient wildland ecosystem. If the prescribed fire becomes the subject of an exceptional events demonstration, documentation of the post-burn review must accompany the demonstration.
(B) If the State anticipates satisfying the requirements of paragraph (c)(3)(iv)(D) of this section by employing the appropriate basic smoke management practices identified in Table 1 to §50.14, then:
(1) The State, federal land managers, and other entities as appropriate, must periodically collaborate with burn managers operating within the jurisdiction of the State to discuss and document the process by which air agencies and land managers will work together to protect public health and manage air quality impacts during the conduct of prescribed fires on wildland. Such discussions must include outreach and education regarding general expectations for the selection and application of appropriate basic smoke management practices and goals for advancing strategies and increasing adoption and communication of the benefits of appropriate basic smoke management practices;
(2) The State, federal land managers and burn managers shall have an initial implementation period, defined as being 2 years from September 30, 2016, to implement the collaboration and outreach effort identified in paragraph (b)(3)(ii)(B)(1) of this section; and
(3) Except as provided in paragraph (b)(3)(ii)(B)(2) of this section, the Administrator shall not place a concurrence flag in the appropriate field for the data record in the AQS database, as specified in paragraph (c)(2)(ii) of this section, if the data are associated with a prescribed fire on wildland unless the requirements of paragraph (b)(3)(ii)(B)(1) of this section have been met and associated documentation accompanies any applicable exceptional events demonstration. The Administrator may nonconcur or defer action on such a demonstration.
(C) With respect to the requirement that a prescribed fire be not reasonably preventable, the State may rely upon and reference a multi-year land or resource management plan for a wildland area with a stated objective to establish, restore and/or maintain a sustainable and resilient wildland ecosystem and/or to preserve endangered or threatened species through a program of prescribed fire provided that the Administrator determines that there is no compelling evidence to the contrary in the record and the use of prescribed fire in the area has not exceeded the frequency indicated in that plan.
(iii) Provided the Administrator determines that there is no compelling evidence to the contrary in the record, in addressing the requirements set forth in paragraph (c)(3)(iv)(E) of this section regarding the human activity unlikely to recur at a particular location criterion for demonstrations involving prescribed fires on wildland, the State must describe the actual frequency with which a burn was conducted, but may rely upon and reference an assessment of the natural fire return interval or the prescribed fire frequency needed to establish, restore and/or maintain a sustainable and resilient wildland ecosystem contained in a multi-year land or resource management plan with a stated objective to establish, restore and/or maintain a sustainable and resilient wildland ecosystem and/or to preserve endangered or threatened species through a program of prescribed fire.
| Basic Smoke Management Practice b | Benefit achieved with the BSMP | When the BSMP is applied - before/during/after the burn |
|---|---|---|
| Evaluate Smoke Dispersion Conditions | Minimize smoke impacts | Before, During, After. |
| Monitor Effects on Air Quality | Be aware of where the smoke is going and degree it impacts air quality | Before, During, After. |
| Record-Keeping/Maintain a Burn/Smoke Journal | Retain information about the weather, burn and smoke. If air quality problems occur, documentation helps analyze and address air regulatory issues. | Before, During, After. |
| Communication - Public Notification | Notify neighbors and those potentially impacted by smoke, especially sensitive receptors | Before, During. |
| Consider Emission Reduction Techniques | Reducing emissions through mechanisms such as reducing fuel loading can reduce downwind impacts | Before, During, After. |
| Share the Airshed - Coordination of Area Burning | Coordinate multiple burns in the area to manage exposure of the public to smoke | Before, During, After. |
| a The EPA believes that elements of these BSMP could also be practical and beneficial to apply to wildfires for areas likely to experience recurring wildfires. b The listing of BSMP in this table is not intended to be all-inclusive. Not all BSMP are appropriate for all burns. Goals for applicability should retain flexibility to allow for onsite variation and site-specific conditions that can be variable on the day of the burn. Burn managers can consider other appropriate BSMP as they become available due to technological advancement or programmatic refinement. | ||
(4) Wildfires. The Administrator shall exclude data from use in determinations of exceedances and violations where a State demonstrates to the Administrator's satisfaction that emissions from wildfires caused a specific air pollution concentration in excess of one or more national ambient air quality standard at a particular air quality monitoring location and otherwise satisfies the requirements of this section. Provided the Administrator determines that there is no compelling evidence to the contrary in the record, the Administrator will determine every wildfire occurring predominantly on wildland to have met the requirements identified in paragraph (c)(3)(iv)(D) of this section regarding the not reasonably controllable or preventable criterion.
(5) High wind dust events. (i) The Administrator shall exclude data from use in determinations of exceedances and violations, where a State demonstrates to the Administrator's satisfaction that emissions from a high wind dust event caused a specific air pollution concentration in excess of one or more national ambient air quality standards at a particular air quality monitoring location and otherwise satisfies the requirements of this section provided that such emissions are from high wind dust events.
(ii) The Administrator will consider high wind dust events to be natural events in cases where windblown dust is entirely from natural undisturbed lands in the area or where all anthropogenic sources are reasonably controlled as determined in accordance with paragraph (b)(8) of this section.
(iii) The Administrator will accept a high wind threshold of a sustained wind of 25 mph for areas in the States of Arizona, California, Colorado, Kansas, Nebraska, Nevada, New Mexico, North Dakota, Oklahoma, South Dakota, Texas, Utah, and Wyoming provided this value is not contradicted by evidence in the record at the time the State submits a demonstration. In lieu of this threshold, States can identify and use an Administrator-approved alternate area-specific high wind threshold that is more representative of local or regional conditions, if appropriate.
(iv) In addressing the requirements set forth in paragraph (c)(3)(iv)(D) of this section regarding the not reasonably preventable criterion, the State shall not be required to provide a case-specific justification for a high wind dust event.
(v) With respect to the not reasonably controllable criterion of paragraph (c)(3)(iv)(D) of this section, dust controls on an anthropogenic source shall be considered reasonable in any case in which the controls render the anthropogenic source as resistant to high winds as natural undisturbed lands in the area affected by the high wind dust event. The Administrator may determine lesser controls reasonable on a case-by-case basis.
(vi) For large-scale and high-energy high wind dust events, the Administrator will generally consider a demonstration documenting the nature and extent of the event to be sufficient with respect to the not reasonably controllable criterion of paragraph (c)(3)(iv)(D) of this section provided the State provides evidence showing that the event satisfies the following:
(A) The event is associated with a dust storm and is the focus of a Dust Storm Warning.
(B) The event has sustained winds that are greater than or equal to 40 miles per hour.
(C) The event has reduced visibility equal to or less than 0.5 miles.
(6) Stratospheric Intrusions. Where a State demonstrates to the Administrator's satisfaction that emissions from stratospheric intrusions caused a specific air pollution concentration in excess of one or more national ambient air quality standard at a particular air quality monitoring location and otherwise satisfies the requirements of this section, the Administrator will determine stratospheric intrusions to have met the requirements identified in paragraph (c)(3)(iv)(D) of this section regarding the not reasonably controllable or preventable criterion and shall exclude data from use in determinations of exceedances and violations.
(7) Determinations with respect to event aggregation, multiple national ambient air quality standards for the same pollutant, and exclusion of 24-hour values for particulate matter.
(i) Where a State demonstrates to the Administrator's satisfaction that for national ambient air quality standards with averaging or cumulative periods less than or equal to 24 hours the aggregate effect of events occurring on the same day has caused an exceedance or violation, the Administrator shall determine such collective data to satisfy the requirements in paragraph (c)(3)(iv)(B) of this section regarding the clear causal relationship criterion. Where a State demonstrates to the Administrator's satisfaction that for national ambient air quality standards with averaging or cumulative periods longer than 24 hours the aggregate effect of events occurring on different days has caused an exceedance or violation, the Administrator shall determine such collective data to satisfy the requirements in paragraph (c)(3)(iv)(B) of this section regarding the clear causal relationship criterion.
(ii) The Administrator shall accept as part of a demonstration for the clear causal relationship in paragraph (c)(3)(iv)(B) of this section with respect to a 24-hour NAAQS, a State's comparison of a 24-hour concentration of any national ambient air quality standard pollutant to the level of a national ambient air quality standard for the same pollutant with a longer averaging period. The Administrator shall also accept as part of a demonstration for the clear causal relationship in paragraph (c)(3)(iv)(B) of this section with respect to a NAAQS with a longer averaging period, a State's comparison of a 24-hour concentration of any national ambient air quality standard pollutant to the level of the national ambient air quality standard for the same pollutant with a longer averaging period, without the State having to demonstrate that the event caused the annual average concentration of the pollutant to exceed the level of the NAAQS with the longer averaging period.
(iii) Where a State operates a continuous analyzer that has been designated as a Federal Equivalent Method monitor as defined in 40 CFR 50.1(g) that complies with the monitoring requirements of 40 CFR part 58, Appendix C, and the State believes that collected data have been influenced by an event, in following the process outlined in paragraph (c)(2) of this section, the State shall create an initial event description and flag the associated event-influenced data that have been submitted to the AQS database for the affected monitor. Where a State demonstrates to the Administrator's satisfaction that such data satisfy the requirements in paragraph (c)(3)(iv)(B) of this section regarding the clear causal relationship criterion and otherwise satisfy the requirements of this section, the Administrator shall agree to exclude all data within the affected calendar day(s).
(8) Determinations with respect to the not reasonably controllable or preventable criterion. (i) The not reasonably controllable or preventable criterion has two prongs that the State must demonstrate: prevention and control.
(ii) The Administrator shall determine that an event is not reasonably preventable if the State shows that reasonable measures to prevent the event were applied at the time of the event.
(iii) The Administrator shall determine that an event is not reasonably controllable if the State shows that reasonable measures to control the impact of the event on air quality were applied at the time of the event.
(iv) The Administrator shall assess the reasonableness of available controls for anthropogenic sources based on information available as of the date of the event.
(v) Except where a State, tribal or federal air agency is obligated to revise its state implementation plan, tribal implementation plan, or federal implementation plan, the Administrator shall consider enforceable control measures implemented in accordance with a state implementation plan, tribal implementation plan, or federal implementation plan, approved by the EPA within 5 years of the date of the event, that address the event-related pollutant and all sources necessary to fulfill the requirements of the Clean Air Act for the state implementation plan, tribal implementation plan, or federal implementation plan to be reasonable controls with respect to all anthropogenic sources that have or may have contributed to the monitored exceedance or violation.
(vi) Where a State, tribal or federal air agency is obligated to revise its state implementation plan, tribal implementation plan, or federal implementation plan, the deference to enforceable control measures identified in paragraph (b)(8)(v) of this section shall remain only until the due date of the required state implementation plan, tribal implementation plan, or federal implementation plan revisions. However, where an air agency is obligated to revise the enforceable control measures identified in paragraph (b)(8)(v) of this section in its implementation plan as a result of an action pursuant to Clean Air Act section 110(k)(5), the deference, if any, to those enforceable control measures shall be determined on a case-by-case basis.
(vii) The Administrator shall not require a State to provide case-specific justification to support the not reasonably controllable or preventable criterion for emissions-generating activity that occurs outside of the State's jurisdictional boundaries within which the concentration at issue was monitored. In the case of a tribe treated as a state under 40 CFR 49.2 with respect to exceptional events requirements, the tribe's jurisdictional boundaries for purposes of requiring or directly implementing emission controls apply. In the case of a federal land manager or other federal agency submitting a demonstration under the requirements of this section, the jurisdictional boundaries that apply are those of the State or the tribe depending on which has jurisdiction over the area where the event has occurred.
(viii) In addition to the provisions that apply to specific event types identified in paragraphs (b)(3)(ii) and (b)(5)(i) through (iii) of this section in addressing the requirements set forth in paragraph (c)(3)(iv)(D) of this section regarding the not reasonably controllable or preventable criterion, the State must include the following components:
(A) Identification of the natural and anthropogenic sources of emissions causing and contributing to the monitored exceedance or violation, including the contribution from local sources.
(B) Identification of the relevant state implementation plan, tribal implementation plan, or federal implementation plan or other enforceable control measures in place for the sources identified in paragraph (b)(8)(vii)(A) of this section and the implementation status of these controls.
(C) Evidence of effective implementation and enforcement of the measures identified in paragraph (b)(8)(vii)(B) of this section.
(D) The provisions in this paragraph shall not apply if the provisions in paragraph (b)(4), (b)(5)(vi), or (b)(6) of this section apply.
(9) Mitigation plans. (i) Except as provided for in paragraph (b)(9)(ii) of this section, where a State is subject to the requirements of 40 CFR 51.930(b), the Administrator shall not place a concurrence flag in the appropriate field for the data record in the AQS database, as specified in paragraph (c)(2)(ii) of this section, if the data are of the type and pollutant that are the focus of the mitigation plan until the State fulfills its obligations under the requirements of 40 CFR 51.930(b). The Administrator may nonconcur or defer action on such a demonstration.
(ii) The prohibition on placing a concurrence flag in the appropriate field for the data record in the AQS database by the Administrator stated in paragraph (b)(9(i) of this section does not apply to data that are included in an exceptional events demonstration that is:
(A) submitted in accordance with paragraph (c)(3) of this section that is also of the type and pollutant that is the focus of the mitigation plan, and
(B) submitted within the 2-year period allowed for mitigation plan development as specified in 40 CFR 51.930(b)(3).
(c) Schedules and procedures - (1) Public notification. (>(i) In accordance with the mitigation requirement at 40 CFR 51.930(a)(1), all States and, where applicable, their political subdivisions must notify the public promptly whenever an event occurs or is reasonably anticipated to occur which may result in the exceedance of an applicable air quality standard.
(ii) [Reserved]
(2) Initial notification of potential exceptional event. (i) A State shall notify the Administrator of its intent to request exclusion of one or more measured exceedances of an applicable national ambient air quality standard as being due to an exceptional event by creating an initial event description and flagging the associated data that have been submitted to the AQS database and by engaging in the Initial Notification of Potential Exceptional Event process as follows:
(A) The State and the appropriate EPA Regional office shall engage in regular communications to identify those data that have been potentially influenced by an exceptional event, to determine whether the identified data may affect a regulatory determination and to discuss whether the State should develop and submit an exceptional events demonstration according to the requirements in this section;
(B) For data that may affect an anticipated regulatory determination or where circumstances otherwise compel the Administrator to prioritize the resulting demonstration, the Administrator shall respond to a State's Initial Notification of Potential Exceptional Event with a due date for demonstration submittal that considers the nature of the event and the anticipated timing of the associated regulatory decision;
(C) The Administrator may waive the Initial Notification of Potential Exceptional Event process on a case-by-case basis.
(ii) The data shall not be excluded from determinations with respect to exceedances or violations of the national ambient air quality standards unless and until, following the State's submittal of its demonstration pursuant to paragraph (c)(3) of this section and the Administrator's review, the Administrator notifies the State of its concurrence by placing a concurrence flag in the appropriate field for the data record in the AQS database.
(iii) [Reserved]
(iv) [Reserved]
(v) [Reserved]
(vi) Table 2 to §50.14 identifies the submission process for data that will or may influence the initial designation of areas for any new or revised national ambient air quality standard.
| Exceptional events/Regulatory action | Condition | Exceptional events deadline schedule d |
|---|---|---|
| (A) Initial Notification deadline for data years 1, 2 and 3. a | If state and tribal initial designation recommendations for a new/revised national ambient air quality standard are due August through January, | then the Initial Notification deadline will be the July 1 prior to the recommendation deadline. |
| (B) Initial Notification deadline for data years 1, 2 and 3. a | If state and tribal recommendations for a new/revised national ambient air quality standard are due February through July, | then the Initial Notification deadline will be the January 1 prior to the recommendation deadline. |
| (C) Exceptional events demonstration submittal deadline for data years 1, 2 and 3 a | None | no later than the later of November 29, 2016 or the date that state and tribal recommendations are due to the Administrator. |
| (D) Initial Notification and exceptional events demonstration submittal deadline for data year 4 b and, where applicable, data year 5. c | None | by the last day of the month that is 1 year and 7 months after promulgation of a new/revised national ambient air quality standard, unless either paragraph (E) or paragraph (F) applies. |
| (E) Initial Notification and exceptional events demonstration submittal deadline for data year 4 b and, where applicable, data year 5. c | If the Administrator follows a 3-year designation schedule | the deadline is 2 years and 7 months after promulgation of a new/revised national ambient air quality standard. |
| (F) Initial Notification and exceptional events demonstration submittal deadline for data year 4 b and, where applicable, data year 5. c | If the Administrator notifies the state/tribe that it intends to complete the initial area designations process according to a schedule between 2 and 3 years, | the deadline is 5 months prior to the date specified for final designations decisions in such Administrator notification. |
| a Where data years 1, 2, and 3 are those years expected to be considered in state and tribal recommendations. b Where data year 4 is the additional year of data that the Administrator may consider when making final area designations for a new/revised national ambient air quality standard under the standard designations schedule. c Where data year 5 is the additional year of data that the Administrator may consider when making final area designations for a new/revised national ambient air quality standard under an extended designations schedule. d The date by which air agencies must certify their ambient air quality monitoring data in AQS is annually on May 1 of the year following the year of data collection as specified in 40 CFR 58.15(a)(2). In some cases, however, air agencies may choose to certify a prior year's data in advance of May 1 of the following year, particularly if the Administrator has indicated intent to promulgate final designations in the first 8 months of the calendar year. Exceptional events demonstration deadlines for “early certified” data will follow the deadlines for “year 4” and “year 5” data. | ||
(3) Submission of demonstrations. (i) Except as provided under paragraph (c)(2)(vi) of this section, a State that has flagged data as being due to an exceptional event and is requesting exclusion of the affected measurement data shall, after notice and opportunity for public comment, submit a demonstration to justify data exclusion to the Administrator according to the schedule established under paragraph (c)(2)(i)(B).
(ii) [Reserved]
(iii) [Reserved]
(iv) The demonstration to justify data exclusion must include:
(A) A narrative conceptual model that describes the event(s) causing the exceedance or violation and a discussion of how emissions from the event(s) led to the exceedance or violation at the affected monitor(s);
(B) A demonstration that the event affected air quality in such a way that there exists a clear causal relationship between the specific event and the monitored exceedance or violation;
(C) Analyses comparing the claimed event-influenced concentration(s) to concentrations at the same monitoring site at other times to support the requirement at paragraph (c)(3)(iv)(B) of this section. The Administrator shall not require a State to prove a specific percentile point in the distribution of data;
(D) A demonstration that the event was both not reasonably controllable and not reasonably preventable; and
(E) A demonstration that the event was a human activity that is unlikely to recur at a particular location or was a natural event.
(v) With the submission of the demonstration containing the elements in paragraph (c)(3)(iv) of this section, the State must:
(A) Document that the State followed the public comment process and that the comment period was open for a minimum of 30 days, which could be concurrent with the beginning of the Administrator's initial review period of the associated demonstration provided the State can meet all requirements in this paragraph;
(B) Submit the public comments it received along with its demonstration to the Administrator; and
(C) Address in the submission to the Administrator those comments disputing or contradicting factual evidence provided in the demonstration.
(vi) Where the State has submitted a demonstration according to the requirements of this section after September 30, 2016 and the Administrator has reviewed such demonstration and requested additional evidence to support one of the elements in paragraph (c)(3)(iv) of this section, the State shall have 12 months from the date of the Administrator's request to submit such evidence. At the conclusion of this time, if the State has not submitted the requested additional evidence, the Administrator will notify the State in writing that it considers the demonstration to be inactive and will not pursue additional review of the demonstration. After a 12-month period of inactivity by the State, if a State desires to pursue the inactive demonstration, it must reinitiate its request to exclude associated data by following the process beginning with paragraph (c)(2)(i) of this section.
[81 FR 68277, Oct. 3, 2016]
§50.15 National primary and secondary ambient air quality standards for ozone.
(a) The level of the national 8-hour primary and secondary ambient air quality standards for ozone (O3) is 0.075 parts per million (ppm), daily maximum 8-hour average, measured by a reference method based on appendix D to this part and designated in accordance with §53.11 of this chapter or an equivalent method designated in accordance with §53.11 of this chapter.
(b) The 8-hour primary and secondary O3 ambient air quality standards are met at an ambient air quality monitoring site when the 3-year average of the annual fourth-highest daily maximum 8-hour average O3 concentration is less than or equal to 0.075 ppm, as determined in accordance with appendix P to this part.
[73 FR 16511, Mar. 27, 2008]
§50.16 National primary and secondary ambient air quality standards for lead.
(a) The national primary and secondary ambient air quality standards for lead (Pb) and its compounds are 0.15 micrograms per cubic meter, arithmetic mean concentration over a 3-month period, measured in the ambient air as Pb either by:
(1) A reference method based on appendix G of this part and designated in accordance with §53.11 of this chapter or;
(2) An equivalent method designated in accordance with §53.11 of this chapter.
(b) The national primary and secondary ambient air quality standards for Pb are met when the maximum arithmetic 3-month mean concentration for a 3-year period, as determined in accordance with appendix R of this part, is less than or equal to 0.15 micrograms per cubic meter.
[73 FR 67052, Nov. 12, 2008]
§50.17 National primary ambient air quality standards for sulfur oxides (sulfur dioxide).
(a) The level of the national primary 1-hour annual ambient air quality standard for oxides of sulfur is 75 parts per billion (ppb, which is 1 part in 1,000,000,000), measured in the ambient air as sulfur dioxide (SO2).
(b) The 1-hour primary standard is met at an ambient air quality monitoring site when the three-year average of the annual (99th percentile) of the daily maximum 1-hour average concentrations is less than or equal to 75 ppb, as determined in accordance with appendix T of this part.
(c) The level of the standard shall be measured by a reference method based on appendix A or A-1 of this part, or by a Federal Equivalent Method (FEM) designated in accordance with §53.11 of this chapter.
[75 FR 35592, June 22, 2010]
§50.18 National primary ambient air quality standards for PM2.5.
(a) The national primary ambient air quality standards for PM2.5 are 12.0 micrograms per cubic meter (µg/m 3) annual arithmetic mean concentration and 35 µg/m 3 24-hour average concentration measured in the ambient air as PM2.5 (particles with an aerodynamic diameter less than or equal to a nominal 2.5 micrometers) by either:
(1) A reference method based on appendix L to this part and designated in accordance with §53.11 of this chapter; or
(2) An equivalent method designated in accordance with §53.11 of this chapter.
(b) The primary annual PM2.5 standard is met when the annual arithmetic mean concentration, as determined in accordance with appendix N of this part, is less than or equal to 12.0 µg/m 3.
(c) The primary 24-hour PM2.5 standard is met when the 98th percentile 24-hour concentration, as determined in accordance with appendix N of this part, is less than or equal to 35 µg/m 3.
[78 FR 3277, Jan. 15, 2013]
§50.19 National primary and secondary ambient air quality standards for ozone.
(a) The level of the national 8-hour primary ambient air quality standard for ozone (O3) is 0.070 parts per million (ppm), daily maximum 8-hour average, measured by a reference method based on appendix D to this part and designated in accordance with §53.11 of this chapter or an equivalent method designated in accordance with §53.11 of this chapter.
(b) The 8-hour primary O3 ambient air quality standard is met at an ambient air quality monitoring site when the 3-year average of the annual fourth-highest daily maximum 8-hour average O3 concentration is less than or equal to 0.070 ppm, as determined in accordance with appendix U to this part.
(c) The level of the national secondary ambient air quality standard for O3 is 0.070 ppm, daily maximum 8-hour average, measured by a reference method based on appendix D to this part and designated in accordance with §53.11 of this chapter or an equivalent method designated in accordance with §53.11 of this chapter.
(d) The 8-hour secondary O3 ambient air quality standard is met at an ambient air quality monitoring site when the 3-year average of the annual fourth-highest daily maximum 8-hour average O3 concentration is less than or equal to 0.070 ppm, as determined in accordance with appendix U to this part.
[80 FR 65452, Oct. 26, 2015]
§50.20 National primary ambient air quality standards for PM2.5.
(a) The national primary ambient air quality standards for PM 2.5 are 9.0 micrograms per cubic meter (µg/m 3 ) annual arithmetic mean concentration and 35 µg/m 3 24-hour average concentration measured in the ambient air as PM 2.5 (particles with an aerodynamic diameter less than or equal to a nominal 2.5 micrometers) by either:
(1) A reference method based on appendix L to this part and designated in accordance with part 53 of this chapter; or
(2) An equivalent method designated in accordance with part 53 of this chapter.
(b) The primary annual PM 2.5 standard is met when the annual arithmetic mean concentration, as determined in accordance with appendix N to this part, is less than or equal to 9.0 µg/m 3 .
(c) The primary 24-hour PM 2.5 standard is met when the 98th percentile 24-hour concentration, as determined in accordance with appendix N to this part, is less than or equal to 35 µg/m 3 .
[89 FR 16380, March 6, 2024]
Appendix A-1 to Part 50 - Reference Measurement Principle and Calibration Procedure for the Measurement of Sulfur Dioxide in the Atmosphere (Ultraviolet Fluorescence Method)
1.0 Applicability
1.1 This ultraviolet fluorescence (UVF) method provides a measurement of the concentration of sulfur dioxide (SO2) in ambient air for determining compliance with the national primary and secondary ambient air quality standards for sulfur oxides (sulfur dioxide) as specified in §50.4, §50.5, and §50.17 of this chapter. The method is applicable to the measurement of ambient SO2 concentrations using continuous (real-time) sampling. Additional quality assurance procedures and guidance are provided in part 58, appendix A, of this chapter and in Reference 3.
2.0 Principle
2.1 This reference method is based on automated measurement of the intensity of the characteristic fluorescence released by SO2 in an ambient air sample contained in a measurement cell of an analyzer when the air sample is irradiated by ultraviolet (UV) light passed through the cell. The fluorescent light released by the SO2 is also in the ultraviolet region, but at longer wavelengths than the excitation light. Typically, optimum instrumental measurement of SO2 concentrations is obtained with an excitation wavelength in a band between approximately 190 to 230 nm, and measurement of the SO2 fluorescence in a broad band around 320 nm, but these wavelengths are not necessarily constraints of this reference method. Generally, the measurement system (analyzer) also requires means to reduce the effects of aromatic hydrocarbon species, and possibly other compounds, in the air sample to control measurement interferences from these compounds, which may be present in the ambient air. References 1 and 2 describe UVF method.
2.2 The measurement system is calibrated by referencing the instrumental fluorescence measurements to SO2 standard concentrations traceable to a National Institute of Standards and Technology (NIST) primary standard for SO2 (see Calibration Procedure below).
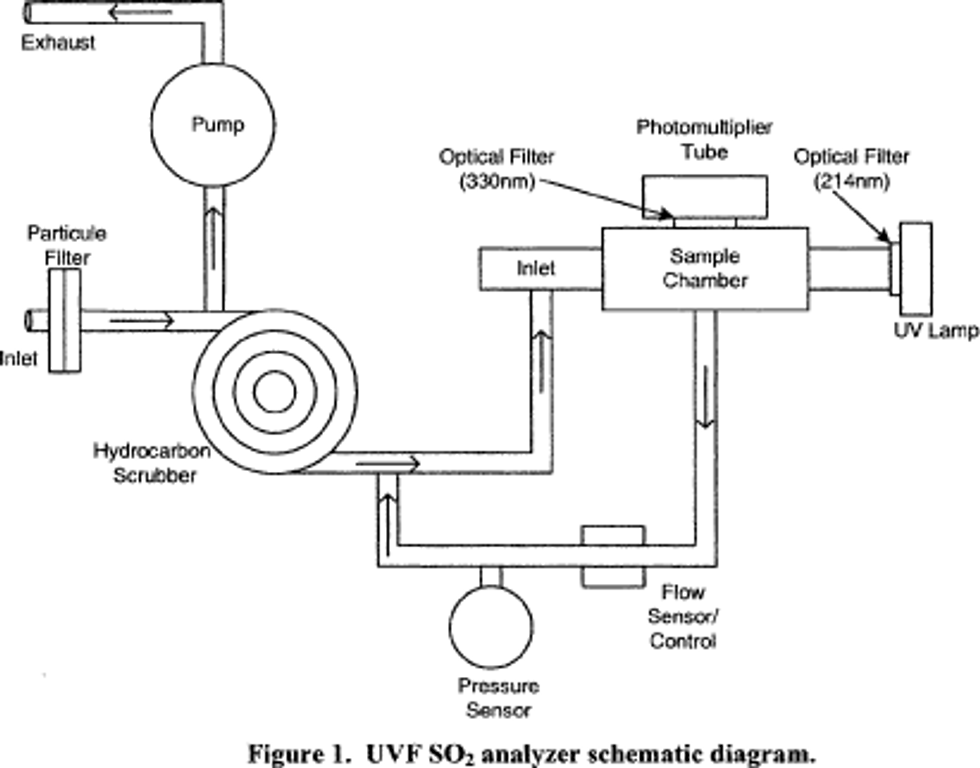
2.3 An analyzer implementing this measurement principle is shown schematically in Figure 1. Designs should include a measurement cell, a UV light source of appropriate wavelength, a UV detector system with appropriate wave length sensitivity, a pump and flow control system for sampling the ambient air and moving it into the measurement cell, sample air conditioning components as necessary to minimize measurement interferences, suitable control and measurement processing capability, and other apparatus as may be necessary. The analyzer must be designed to provide accurate, repeatable, and continuous measurements of SO2 concentrations in ambient air, with measurement performance as specified in Subpart B of Part 53 of this chapter.
2.4 Sampling considerations: The use of a particle filter on the sample inlet line of a UVF SO2 analyzer is required to prevent interference, malfunction, or damage due to particles in the sampled air.
3.0 Interferences
3.1 The effects of the principal potential interferences may need to be mitigated to meet the interference equivalent requirements of §53.11 of this chapter. Aromatic hydrocarbons such as xylene and naphthalene can fluoresce and act as strong positive interferences. These gases can be removed by using a permeation type scrubber (hydrocarbon “kicker”). Nitrogen oxide (NO) in high concentrations can also fluoresce and cause positive interference. Optical filtering can be employed to improve the rejection of interference from high NO. Ozone can absorb UV light given off by the SO2 molecule and cause a measurement offset. This effect can be reduced by minimizing the measurement path length between the area where SO2 fluorescence occurs and the photomultiplier tube detector (e.g., <5 cm). A hydrocarbon scrubber, optical filter and appropriate distancing of the measurement path length may be required method components to reduce interference.
4.0 Calibration Procedure
Atmospheres containing accurately known concentrations of sulfur dioxide are prepared using a compressed gas transfer standard diluted with accurately metered clean air flow rates.
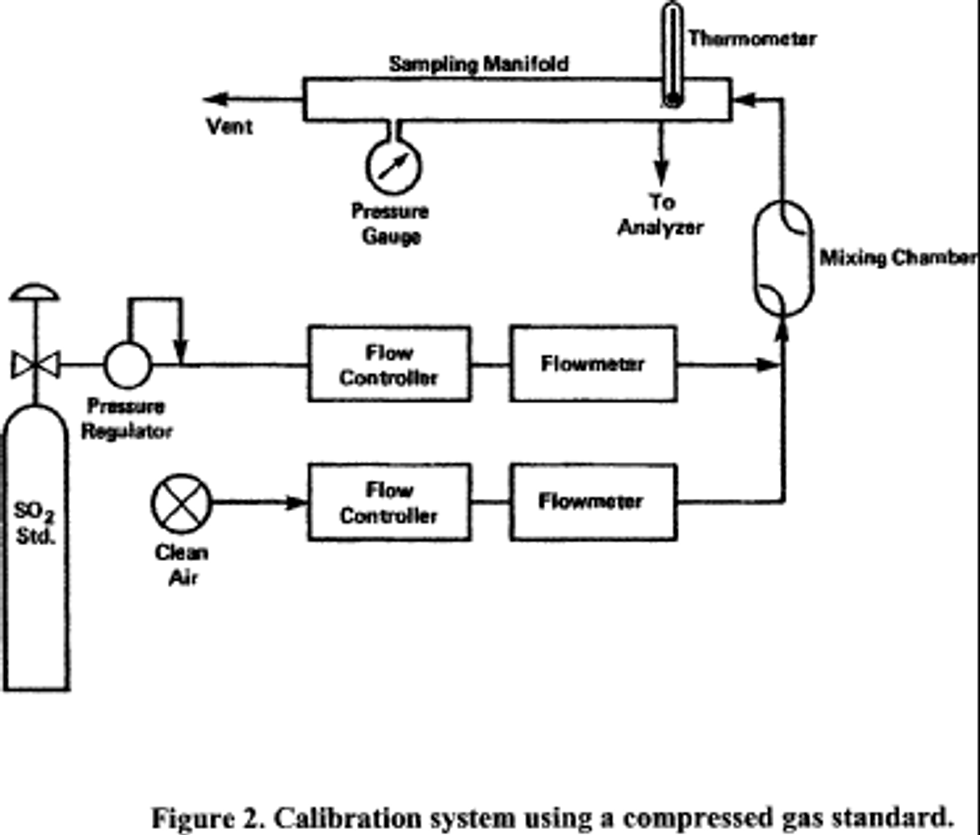
4.1 Apparatus: Figure 2 shows a typical generic system suitable for diluting a SO2 gas cylinder concentration standard with clean air through a mixing chamber to produce the desired calibration concentration standards. A valve may be used to conveniently divert the SO2 from the sampling manifold to provide clean zero air at the output manifold for zero adjustment. The system may be made up using common laboratory components, or it may be a commercially manufactured system. In either case, the principle components are as follows:
4.1.1 SO2 standard gas flow control and measurement devices (or a combined device) capable of regulating and maintaining the standard gas flow rate constant to within ±2 percent and measuring the gas flow rate accurate to within ±2, properly calibrated to a NIST-traceable standard.
4.1.2 Dilution air flow control and measurement devices (or a combined device) capable of regulating and maintaining the air flow rate constant to within ±2 percent and measuring the air flow rate accurate to within ±2, properly calibrated to a NIST-traceable standard.
4.1.3 Mixing chamber, of an inert material such as glass and of proper design to provide thorough mixing of pollutant gas and diluent air streams.
4.1.4 Sampling manifold, constructed of glass, polytetrafluoroethylene (PTFE Teflon TM), or other suitably inert material and of sufficient diameter to insure a minimum pressure drop at the analyzer connection, with a vent designed to insure a minimum over-pressure (relative to ambient air pressure) at the analyzer connection and to prevent ambient air from entering the manifold.
4.1.5 Standard gas pressure regulator, of clean stainless steel with a stainless steel diaphragm, suitable for use with a high pressure SO2 gas cylinder.
4.1.6 Reagents
4.1.6.1 SO2 gas concentration transfer standard having a certified SO2 concentration of not less than 10 ppm, in N2, traceable to a NIST Standard Reference Material (SRM).
4.1.6.2 Clean zero air, free of contaminants that could cause a detectable response or a change in sensitivity of the analyzer. Since ultraviolet fluorescence analyzers may be sensitive to aromatic hydrocarbons and O2-to-N2 ratios, it is important that the clean zero air contains less than 0.1 ppm aromatic hydrocarbons and O2 and N2 percentages approximately the same as in ambient air. A procedure for generating zero air is given in reference 1.
4.2 Procedure
4.2.1 Obtain a suitable calibration apparatus, such as the one shown schematically in Figure 1, and verify that all materials in contact with the pollutant are of glass, Teflon TM, or other suitably inert material and completely clean.
4.2.2 Purge the SO2 standard gas lines and pressure regulator to remove any residual air.
4.2.3 Ensure that there are no leaks in the system and that the flow measuring devices are properly and accurately calibrated under the conditions of use against a reliable volume or flow rate standard such as a soap-bubble meter or a wet-test meter traceable to a NIST standard. All volumetric flow rates should be corrected to the same reference temperature and pressure by using the formula below:

Where:
Fc = corrected flow rate (L/min at 25°C and 760 mm Hg),
Fm = measured flow rate, (at temperature, Tm and pressure, Pm),
Pm = measured pressure in mm Hg, (absolute), and
Tm = measured temperature in degrees Celsius.
4.2.4 Allow the SO2 analyzer under calibration to sample zero air until a stable response is obtained, then make the proper zero adjustment.
4.2.5 Adjust the airflow to provide an SO2 concentration of approximately 80 percent of the upper measurement range limit of the SO2 instrument and verify that the total air flow of the calibration system exceeds the demand of all analyzers sampling from the output manifold (with the excess vented).
4.2.6 Calculate the actual SO2 calibration concentration standard as:
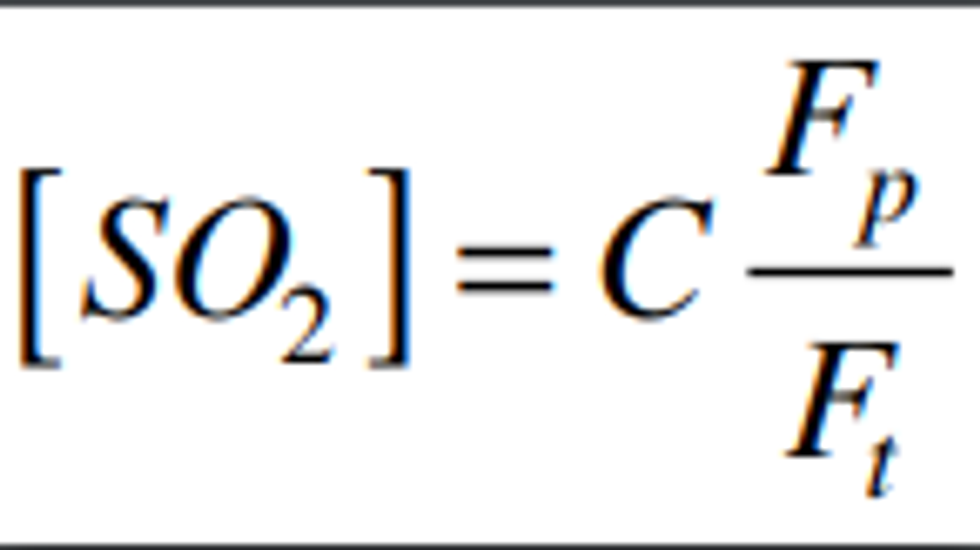
Where:
C = the concentration of the SO2 gas standard
Fp = the flow rate of SO2 gas standard
Ft = the total air flow rate of pollutant and diluent gases
4.2.7 When the analyzer response has stabilized, adjust the SO2 span control to obtain the desired response equivalent to the calculated standard concentration. If substantial adjustment of the span control is needed, it may be necessary to re-check the zero and span adjustments by repeating steps 4.2.4 through 4.2.7 until no further adjustments are needed.
4.2.8 Adjust the flow rate(s) to provide several other SO2 calibration concentrations over the analyzer's measurement range. At least five different concentrations evenly spaced throughout the analyzer's range are suggested.
4.2.9 Plot the analyzer response (vertical or Y-axis) versus SO2 concentration (horizontal or X-axis). Compute the linear regression slope and intercept and plot the regression line to verify that no point deviates from this line by more than 2 percent of the maximum concentration tested.
Note:
Additional information on calibration and pollutant standards is provided in Section 12 of Reference 3.
5.0 Frequency of Calibration
The frequency of calibration, as well as the number of points necessary to establish the calibration curve and the frequency of other performance checking will vary by analyzer; however, the minimum frequency, acceptance criteria, and subsequent actions are specified in Reference 3, Appendix D: Measurement Quality Objectives and Validation Template for SO2 (page 9 of 30). The user's quality control program should provide guidelines for initial establishment of these variables and for subsequent alteration as operational experience is accumulated. Manufacturers of analyzers should include in their instruction/operation manuals information and guidance as to these variables and on other matters of operation, calibration, routine maintenance, and quality control.
6.0 References for SO2 Method
1. H. Okabe, P. L. Splitstone, and J. J. Ball, “Ambient and Source SO2 Detector Based on a Fluorescence Method”, Journal of the Air Control Pollution Association, vol. 23, p. 514-516 (1973).
2. F. P. Schwarz, H. Okabe, and J. K. Whittaker, “Fluorescence Detection of Sulfur Dioxide in Air at the Parts per Billion Level,” Analytical Chemistry, vol. 46, pp. 1024-1028 (1974).
3. QA Handbook for Air Pollution Measurement Systems - Volume II. Ambient Air Quality Monitoring Programs. U.S.
[75 FR 35593, June 22, 2010]
Appendix A-2 to Part 50 - Reference Method for the Determination of Sulfur Dioxide in the Atmosphere (Pararosaniline Method)
1.0 Applicability.
1.1 This method provides a measurement of the concentration of sulfur dioxide (SO2) in ambient air for determining compliance with the primary and secondary national ambient air quality standards for sulfur oxides (sulfur dioxide) as specified in §50.4 and §50.5 of this chapter. The method is applicable to the measurement of ambient SO2 concentrations using sampling periods ranging from 30 minutes to 24 hours. Additional quality assurance procedures and guidance are provided in part 58, appendixes A and B, of this chapter and in references 1 and 2.
2.0 Principle.
2.1 A measured volume of air is bubbled through a solution of 0.04 M potassium tetrachloromercurate (TCM). The SO2 present in the air stream reacts with the TCM solution to form a stable monochlorosulfonatomercurate(3) complex. Once formed, this complex resists air oxidation(4, 5) and is stable in the presence of strong oxidants such as ozone and oxides of nitrogen. During subsequent analysis, the complex is reacted with acid-bleached pararosaniline dye and formaldehyde to form an intensely colored pararosaniline methyl sulfonic acid.
(6) The optical density of this species is determined spectrophotometrically at 548 nm and is directly related to the amount of SO2 collected. The total volume of air sampled, corrected to EPA reference conditions (25°C, 760 mm Hg [101 kPa]), is determined from the measured flow rate and the sampling time. The concentration of SO2 in the ambient air is computed and expressed in micrograms per standard cubic meter (µg/std m 3).
3.0 Range.
3.1 The lower limit of detection of SO2 in 10 mL of TCM is 0.75 µg (based on collaborative test results).(7) This represents a concentration of 25 µg SO2/m 3 (0.01 ppm) in an air sample of 30 standard liters (short-term sampling) and a concentration of 13 µg SO2/m 3 (0.005 ppm) in an air sample of 288 standard liters (long-term sampling). Concentrations less than 25 µg SO2/m 3 can be measured by sampling larger volumes of ambient air; however, the collection efficiency falls off rapidly at low concentrations.(8, 9) Beer's law is adhered to up to 34 µg of SO2 in 25 mL of final solution. This upper limit of the analysis range represents a concentration of 1,130 µg SO2/m 3 (0.43 ppm) in an air sample of 30 standard liters and a concentration of 590 µg SO2/m 3 (0.23 ppm) in an air sample of 288 standard liters. Higher concentrations can be measured by collecting a smaller volume of air, by increasing the volume of absorbing solution, or by diluting a suitable portion of the collected sample with absorbing solution prior to analysis.
4.0 Interferences.
4.1 The effects of the principal potential interferences have been minimized or eliminated in the following manner: Nitrogen oxides by the addition of sulfamic acid,(10, 11) heavy metals by the addition of ethylenediamine tetracetic acid disodium salt (EDTA) and phosphoric acid,(10, 12) and ozone by time delay.(10) Up to 60 µg Fe (III), 22 µg V (V), 10 µg Cu (II), 10 µg Mn (II), and 10 µg Cr (III) in 10 mL absorbing reagent can be tolerated in the procedure.(10) No significant interference has been encountered with 2.3 µg NH3.(13)
5.0 Precision and Accuracy.
5.1 The precision of the analysis is 4.6 percent (at the 95 percent confidence level) based on the analysis of standard sulfite samples.(10)
5.2 Collaborative test results (14) based on the analysis of synthetic test atmospheres (SO2 in scrubbed air) using the 24-hour sampling procedure and the sulfite-TCM calibration procedure show that:
The replication error varies linearly with concentration from ±2.5 µg/m 3 at concentrations of 100 µg/m 3 to ±7 µg/m 3 at concentrations of 400 µg/m 3.
The day-to-day variability within an individual laboratory (repeatability) varies linearly with concentration from ±18.1 µg/m 3 at levels of 100 µg/m 3 to ±50.9 µg/m 3 at levels of 400 µg/m 3.
The day-to-day variability between two or more laboratories (reproducibility) varies linearly with concentration from ±36.9 µg/m 3 at levels of 100 µg/m 3 to ±103.5 µ g/m 3 at levels of 400 µg/m 3.
The method has a concentration-dependent bias, which becomes significant at the 95 percent confidence level at the high concentration level. Observed values tend to be lower than the expected SO2 concentration level.
6.0 Stability.
6.1 By sampling in a controlled temperature environment of 15° ±10°C, greater than 98.9 percent of the SO2-TCM complex is retained at the completion of sampling. (15) If kept at 5°C following the completion of sampling, the collected sample has been found to be stable for up to 30 days. (10) The presence of EDTA enhances the stability of SO2 in the TCM solution and the rate of decay is independent of the concentration of SO2. (16)
7.0 Apparatus.
7.1 Sampling.
7.1.1 Sample probe: A sample probe meeting the requirements of section 7 of 40 CFR part 58, appendix E (Teflon ® or glass with residence time less than 20 sec.) is used to transport ambient air to the sampling train location. The end of the probe should be designed or oriented to preclude the sampling of precipitation, large particles, etc. A suitable probe can be constructed from Teflon ® tubing connected to an inverted funnel.
7.1.2 Absorber - short-term sampling: An all glass midget impinger having a solution capacity of 30 mL and a stem clearance of 4 ±1 mm from the bottom of the vessel is used for sampling periods of 30 minutes and 1 hour (or any period considerably less than 24 hours). Such an impinger is shown in Figure 1. These impingers are commercially available from distributors such as Ace Glass, Incorporated.
7.1.3 Absorber - 24-hour sampling: A polypropylene tube 32 mm in diameter and 164 mm long (available from Bel Art Products, Pequammock, NJ) is used as the absorber. The cap of the absorber must be a polypropylene cap with two ports (rubber stoppers are unacceptable because the absorbing reagent can react with the stopper to yield erroneously high SO2 concentrations). A glass impinger stem, 6 mm in diameter and 158 mm long, is inserted into one port of the absorber cap. The tip of the stem is tapered to a small diameter orifice (0.4 ±0.1 mm) such that a No. 79 jeweler's drill bit will pass through the opening but a No. 78 drill bit will not. Clearance from the bottom of the absorber to the tip of the stem must be 6 ±2 mm. Glass stems can be fabricated by any reputable glass blower or can be obtained from a scientific supply firm. Upon receipt, the orifice test should be performed to verify the orifice size. The 50 mL volume level should be permanently marked on the absorber. The assembled absorber is shown in Figure 2.
7.1.4 Moisture trap: A moisture trap constructed of a glass trap as shown in Figure 1 or a polypropylene tube as shown in Figure 2 is placed between the absorber tube and flow control device to prevent entrained liquid from reaching the flow control device. The tube is packed with indicating silica gel as shown in Figure 2. Glass wool may be substituted for silica gel when collecting short-term samples (1 hour or less) as shown in Figure 1, or for long term (24 hour) samples if flow changes are not routinely encountered.
7.1.5 Cap seals: The absorber and moisture trap caps must seal securely to prevent leaks during use. Heat-shrink material as shown in Figure 2 can be used to retain the cap seals if there is any chance of the caps coming loose during sampling, shipment, or storage.
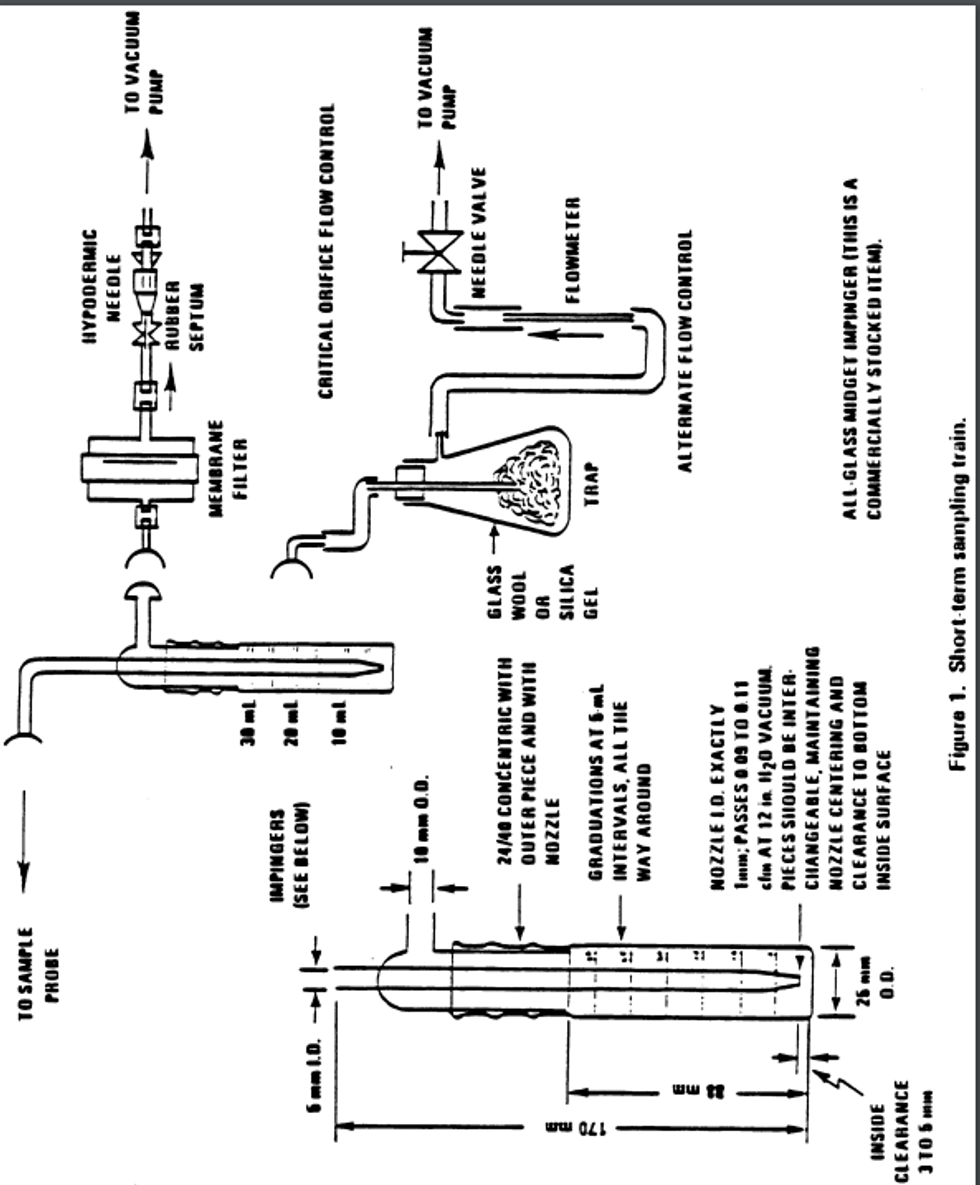
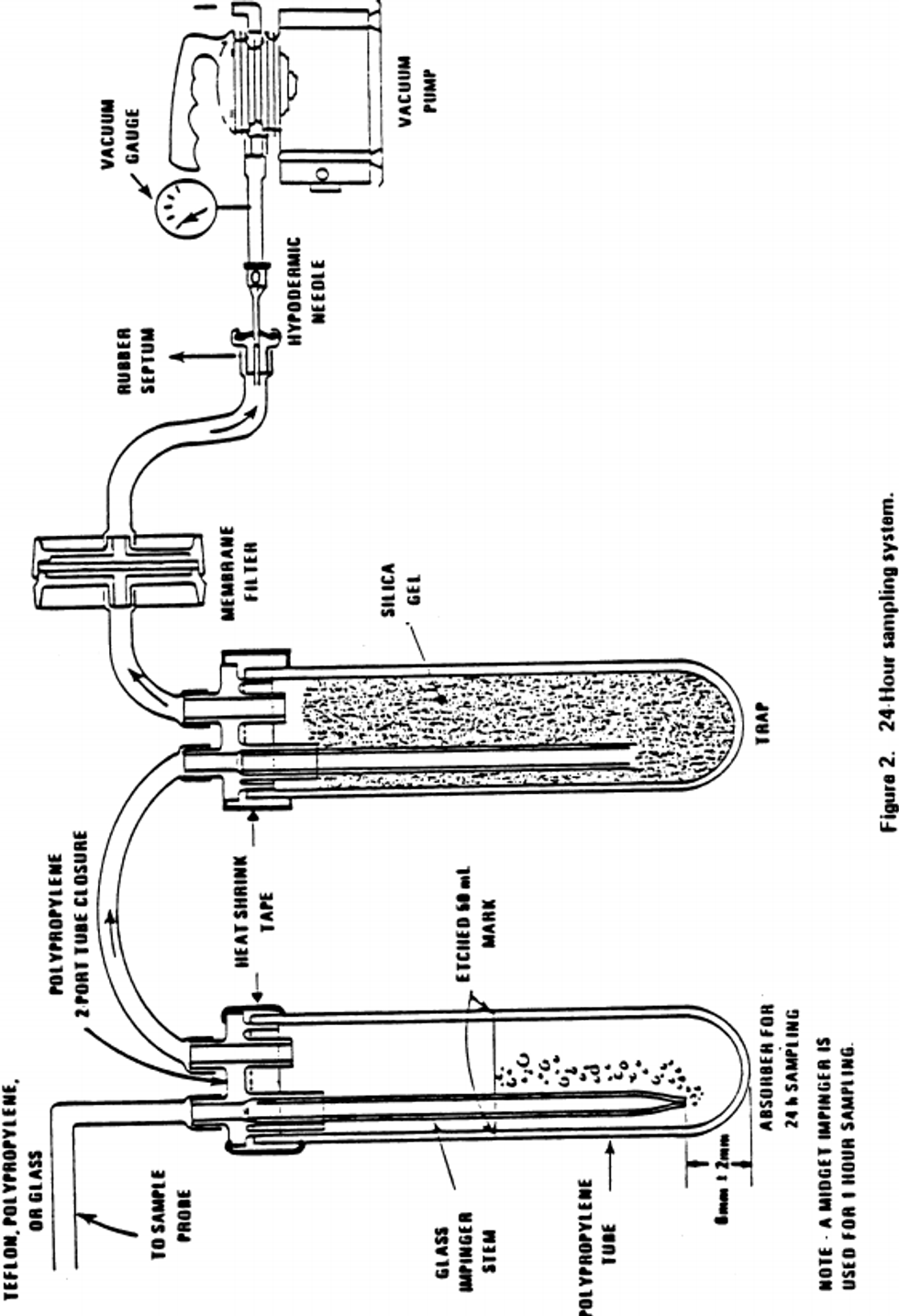
7.1.6 Flow control device: A calibrated rotameter and needle valve combination capable of maintaining and measuring air flow to within ±2 percent is suitable for short-term sampling but may not be used for long-term sampling. A critical orifice can be used for regulating flow rate for both long-term and short-term sampling. A 22-gauge hypodermic needle 25 mm long may be used as a critical orifice to yield a flow rate of approximately 1 L/min for a 30-minute sampling period. When sampling for 1 hour, a 23-gauge hypodermic needle 16 mm in length will provide a flow rate of approximately 0.5 L/min. Flow control for a 24-hour sample may be provided by a 27-gauge hypodermic needle critical orifice that is 9.5 mm in length. The flow rate should be in the range of 0.18 to 0.22 L/min.
7.1.7 Flow measurement device: Device calibrated as specified in 9.4.1 and used to measure sample flow rate at the monitoring site.
7.1.8 Membrane particle filter: A membrane filter of 0.8 to 2 µm porosity is used to protect the flow controller from particles during long-term sampling. This item is optional for short-term sampling.
7.1.9 Vacuum pump: A vacuum pump equipped with a vacuum gauge and capable of maintaining at least 70 kPa (0.7 atm) vacuum differential across the flow control device at the specified flow rate is required for sampling.
7.1.10 Temperature control device: The temperature of the absorbing solution during sampling must be maintained at 15° ±10°C. As soon as possible following sampling and until analysis, the temperature of the collected sample must be maintained at 5° ±5°C. Where an extended period of time may elapse before the collected sample can be moved to the lower storage temperature, a collection temperature near the lower limit of the 15 ±10°C range should be used to minimize losses during this period. Thermoelectric coolers specifically designed for this temperature control are available commercially and normally operate in the range of 5° to 15°C. Small refrigerators can be modified to provide the required temperature control; however, inlet lines must be insulated from the lower temperatures to prevent condensation when sampling under humid conditions. A small heating pad may be necessary when sampling at low temperatures (<7°C) to prevent the absorbing solution from freezing.(17)
7.1.11 Sampling train container: The absorbing solution must be shielded from light during and after sampling. Most commercially available sampler trains are enclosed in a light-proof box.
7.1.12 Timer: A timer is recommended to initiate and to stop sampling for the 24-hour period. The timer is not a required piece of equipment; however, without the timer a technician would be required to start and stop the sampling manually. An elapsed time meter is also recommended to determine the duration of the sampling period.
7.2 Shipping.
7.2.1 Shipping container: A shipping container that can maintain a temperature of 5° ±5°C is used for transporting the sample from the collection site to the analytical laboratory. Ice coolers or refrigerated shipping containers have been found to be satisfactory. The use of eutectic cold packs instead of ice will give a more stable temperature control. Such equipment is available from Cole-Parmer Company, 7425 North Oak Park Avenue, Chicago, IL 60648.
7.3 Analysis.
7.3.1 Spectrophotometer: A spectrophotometer suitable for measurement of absorbances at 548 nm with an effective spectral bandwidth of less than 15 nm is required for analysis. If the spectrophotometer reads out in transmittance, convert to absorbance as follows:
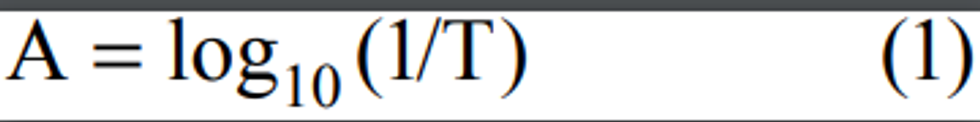
where:
A = absorbance, and
T = transmittance (0<≥T<1).
A standard wavelength filter traceable to the National Bureau of Standards is used to verify the wavelength calibration according to the procedure enclosed with the filter. The wavelength calibration must be verified upon initial receipt of the instrument and after each 160 hours of normal use or every 6 months, whichever occurs first.
7.3.2 Spectrophotometer cells: A set of 1-cm path length cells suitable for use in the visible region is used during analysis. If the cells are unmatched, a matching correction factor must be determined according to Section 10.1.
7.3.3 Temperature control device: The color development step during analysis must be conducted in an environment that is in the range of 20° to 30°C and controlled to ±1°C. Both calibration and sample analysis must be performed under identical conditions (within 1°C). Adequate temperature control may be obtained by means of constant temperature baths, water baths with manual temperature control, or temperature controlled rooms.
7.3.4 Glassware: Class A volumetric glassware of various capacities is required for preparing and standardizing reagents and standards and for dispensing solutions during analysis. These included pipets, volumetric flasks, and burets.
7.3.5 TCM waste receptacle: A glass waste receptacle is required for the storage of spent TCM solution. This vessel should be stoppered and stored in a hood at all times.
8.0 Reagents.
8.1 Sampling.
8.1.1 Distilled water: Purity of distilled water must be verified by the following procedure:(18)
Place 0.20 mL of potassium permanganate solution (0.316 g/L), 500 mL of distilled water, and 1mL of concentrated sulfuric acid in a chemically resistant glass bottle, stopper the bottle, and allow to stand.
If the permanganate color (pink) does not disappear completely after a period of 1 hour at room temperature, the water is suitable for use.
If the permanganate color does disappear, the water can be purified by redistilling with one crystal each of barium hydroxide and potassium permanganate in an all glass still.
8.1.2 Absorbing reagent (0.04 M potassium tetrachloromercurate [TCM]): Dissolve 10.86 g mercuric chloride, 0.066 g EDTA, and 6.0 g potassium chloride in distilled water and dilute to volume with distilled water in a 1,000-mL volumetric flask. (Caution: Mercuric chloride is highly poisonous. If spilled on skin, flush with water immediately.) The pH of this reagent should be between 3.0 and 5.0 (10) Check the pH of the absorbing solution by using pH indicating paper or a pH meter. If the pH of the solution is not between 3.0 and 5.0, dispose of the solution according to one of the disposal techniques described in Section 13.0. The absorbing reagent is normally stable for 6 months. If a precipitate forms, dispose of the reagent according to one of the procedures described in Section 13.0.
8.2 Analysis.
8.2.1 Sulfamic acid (0.6%): Dissolve 0.6 g sulfamic acid in 100 mL distilled water. Perpare fresh daily.
8.2.2 Formaldehyde (0.2%): Dilute 5 mL formaldehyde solution (36 to 38 percent) to 1,000 mL with distilled water. Prepare fresh daily.
8.2.3 Stock iodine solution (0.1 N): Place 12.7 g resublimed iodine in a 250-mL beaker and add 40 g potassium iodide and 25 mL water. Stir until dissolved, transfer to a 1,000 mL volumetric flask and dilute to volume with distilled water.
8.2.4 Iodine solution (0.01 N): Prepare approximately 0.01 N iodine solution by diluting 50 mL of stock iodine solution (Section 8.2.3) to 500 mL with distilled water.
8.2.5 Starch indicator solution: Triturate 0.4 g soluble starch and 0.002 g mercuric iodide (preservative) with enough distilled water to form a paste. Add the paste slowly to 200 mL of boiling distilled water and continue boiling until clear. Cool and transfer the solution to a glass stopperd bottle.
8.2.6 1 N hydrochloric acid: Slowly and while stirring, add 86 mL of concentrated hydrochloric acid to 500 mL of distilled water. Allow to cool and dilute to 1,000 mL with distilled water.
8.2.7 Potassium iodate solution: Accurately weigh to the nearest 0.1 mg, 1.5 g (record weight) of primary standard grade potassium iodate that has been previously dried at 180°C for at least 3 hours and cooled in a dessicator. Dissolve, then dilute to volume in a 500-mL volumetric flask with distilled water.
8.2.8 Stock sodium thiosulfate solution (0.1 N): Prepare a stock solution by dissolving 25 g sodium thiosulfate (Na2 S2 O3 ÷ 5H2 O) in 1,000 mL freshly boiled, cooled, distilled water and adding 0.1 g sodium carbonate to the solution. Allow the solution to stand at least 1 day before standardizing. To standardize, accurately pipet 50 mL of potassium iodate solution (Section 8.2.7) into a 500-mL iodine flask and add 2.0 g of potassium iodide and 10 mL of 1 N HCl. Stopper the flask and allow to stand for 5 minutes. Titrate the solution with stock sodium thiosulfate solution (Section 8.2.8) to a pale yellow color. Add 5 mL of starch solution (Section 8.2.5) and titrate until the blue color just disappears. Calculate the normality (Ns) of the stock sodium thiosulfate solution as follows:

where:
M = volume of thiosulfate required in mL, and
W = weight of potassium iodate in g (recorded weight in Section 8.2.7).

8.2.9 Working sodium thiosulfate titrant (0.01 N): Accurately pipet 100 mL of stock sodium thiosulfate solution (Section 8.2.8) into a 1,000-mL volumetric flask and dilute to volume with freshly boiled, cooled, distilled water. Calculate the normality of the working sodium thiosulfate titrant (NT) as follows:
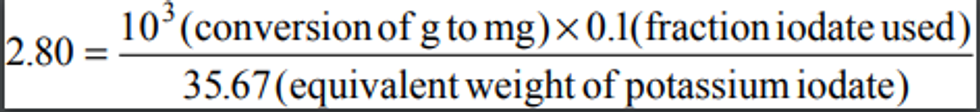
8.2.10 Standardized sulfite solution for the preparation of working sulfite-TCM solution: Dissolve 0.30 g sodium metabisulfite (Na2 S2 O5) or 0.40 g sodium sulfite (Na2 SO3) in 500 mL of recently boiled, cooled, distilled water. (Sulfite solution is unstable; it is therefore important to use water of the highest purity to minimize this instability.) This solution contains the equivalent of 320 to 400 µg SO2/mL. The actual concentration of the solution is determined by adding excess iodine and back-titrating with standard sodium thiosulfate solution. To back-titrate, pipet 50 mL of the 0.01 N iodine solution (Section 8.2.4) into each of two 500-mL iodine flasks (A and B). To flask A (blank) add 25 mL distilled water, and to flask B (sample) pipet 25 mL sulfite solution. Stopper the flasks and allow to stand for 5 minutes. Prepare the working sulfite-TCM solution (Section 8.2.11) immediately prior to adding the iodine solution to the flasks. Using a buret containing standardized 0.01 N thiosulfate titrant (Section 8.2.9), titrate the solution in each flask to a pale yellow color. Then add 5 mL starch solution (Section 8.2.5) and continue the titration until the blue color just disappears.
8.2.11 Working sulfite-TCM solution: Accurately pipet 5 mL of the standard sulfite solution (Section 8.2.10) into a 250-mL volumetric flask and dilute to volume with 0.04 M TCM. Calculate the concentration of sulfur dioxide in the working solution as follows:
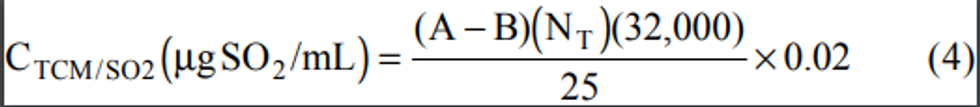
where:
A = volume of thiosulfate titrant required for the blank, mL;
B = volume of thiosulfate titrant required for the sample, mL;
NT = normality of the thiosulfate titrant, from equation (3);
32,000 = milliequivalent weight of SO2, µg;
25 = volume of standard sulfite solution, mL; and
0.02 = dilution factor.
This solution is stable for 30 days if kept at 5°C. (16) If not kept at 5°C, prepare fresh daily.
8.2.12 Purified pararosaniline (PRA) stock solution (0.2% nominal):
8.2.12.1 Dye specifications -
The dye must have a maximum absorbance at a wavelength of 540 nm when assayed in a buffered solution of 0.1 M sodium acetate-acetic acid;
• The absorbance of the reagent blank, which is temperature sensitive (0.015 absorbance unit/°C), must not exceed 0.170 at 22°C with a 1-cm optical path length when the blank is prepared according to the specified procedure;
The calibration curve (Section 10.0) must have a slope equal to 0.030 ±0.002 absorbance unit/µg SO2 with a 1-cm optical path length when the dye is pure and the sulfite solution is properly standardized.
8.2.12.2 Preparation of stock PRA solution - A specially purified (99 to 100 percent pure) solution of pararosaniline, which meets the above specifications, is commercially available in the required 0.20 percent concentration (Harleco Co.). Alternatively, the dye may be purified, a stock solution prepared, and then assayed according to the procedure as described below.(10)
8.2.12.3 Purification procedure for PRA -
1. Place 100 mL each of 1-butanol and 1 N HCl in a large separatory funnel (250-mL) and allow to equilibrate. Note: Certain batches of 1-butanol contain oxidants that create an SO2 demand. Before using, check by placing 20 mL of 1-butanol and 5 mL of 20 percent potassium iodide (KI) solution in a 50-mL separatory funnel and shake thoroughly. If a yellow color appears in the alcohol phase, redistill the 1-butanol from silver oxide and collect the middle fraction or purchase a new supply of 1-butanol.
2. Weigh 100 mg of pararosaniline hydrochloride dye (PRA) in a small beaker. Add 50 mL of the equilibrated acid (drain in acid from the bottom of the separatory funnel in 1.) to the beaker and let stand for several minutes. Discard the remaining acid phase in the separatory funnel.
3. To a 125-mL separatory funnel, add 50 mL of the equilibrated 1-butanol (draw the 1-butanol from the top of the separatory funnel in 1.). Transfer the acid solution (from 2.) containing the dye to the funnel and shake carefully to extract. The violet impurity will transfer to the organic phase.
4. Transfer the lower aqueous phase into another separatory funnel, add 20 mL of equilibrated 1-butanol, and extract again.
5. Repeat the extraction procedure with three more 10-mL portions of equilibrated 1-butanol.
6. After the final extraction, filter the acid phase through a cotton plug into a 50-mL volumetric flask and bring to volume with 1 N HCl. This stock reagent will be a yellowish red.
7. To check the purity of the PRA, perform the assay and adjustment of concentration (Section 8.2.12.4) and prepare a reagent blank (Section 11.2); the absorbance of this reagent blank at 540 nm should be less than 0.170 at 22°C. If the absorbance is greater than 0.170 under these conditions, further extractions should be performed.
8.2.12.4 PRA assay procedure - The concentration of pararosaniline hydrochloride (PRA) need be assayed only once after purification. It is also recommended that commercial solutions of pararosaniline be assayed when first purchased. The assay procedure is as follows:(10)
1. Prepare 1 M acetate-acetic acid buffer stock solution with a pH of 4.79 by dissolving 13.61 g of sodium acetate trihydrate in distilled water in a 100-mL volumetric flask. Add 5.70 mL of glacial acetic acid and dilute to volume with distilled water.
2. Pipet 1 mL of the stock PRA solution obtained from the purification process or from a commercial source into a 100-mL volumetric flask and dilute to volume with distilled water.
3. Transfer a 5-mL aliquot of the diluted PRA solution from 2. into a 50-mL volumetric flask. Add 5mL of 1 M acetate-acetic acid buffer solution from 1. and dilute the mixture to volume with distilled water. Let the mixture stand for 1 hour.
4. Measure the absorbance of the above solution at 540 nm with a spectrophotometer against a distilled water reference. Compute the percentage of nominal concentration of PRA by
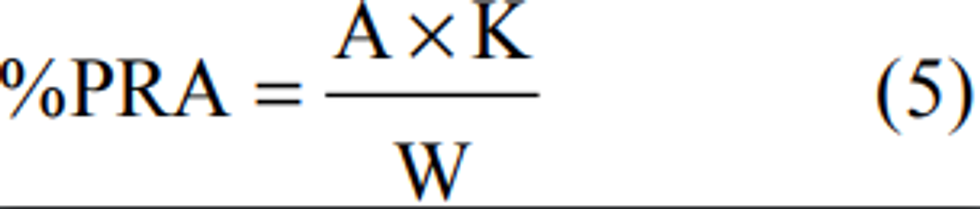
where:
A = measured absorbance of the final mixture (absorbance units);
W = weight in grams of the PRA dye used in the assay to prepare 50 mL of stock solution (for example, 0.100 g of dye was used to prepare 50 mL of solution in the purification procedure; when obtained from commercial sources, use the stated concentration to compute W; for 98% PRA, W = .098 g.); and
K = 21.3 for spectrophotometers having a spectral bandwidth of less than 15 nm and a path length of 1 cm.
8.2.13 Pararosaniline reagent: To a 250-mL volumetric flask, add 20 mL of stock PRA solution. Add an additional 0.2 mL of stock solution for each percentage that the stock assays below 100 percent. Then add 25 mL of 3 M phosphoric acid and dilute to volume with distilled water. The reagent is stable for at least 9 months. Store away from heat and light.
9.0 Sampling Procedure.
9.1 General Considerations. Procedures are described for short-term sampling (30-minute and 1-hour) and for long-term sampling (24-hour). Different combinations of absorbing reagent volume, sampling rate, and sampling time can be selected to meet special needs. For combinations other than those specifically described, the conditions must be adjusted so that linearity is maintained between absorbance and concentration over the dynamic range. Absorbing reagent volumes less than 10 mL are not recommended. The collection efficiency is above 98 percent for the conditions described; however, the efficiency may be substantially lower when sampling concentrations below 25 µγSO2/m 3.(8,9)
9.2 30-Minute and 1-Hour Sampling. Place 10 mL of TCM absorbing reagent in a midget impinger and seal the impinger with a thin film of silicon stopcock grease (around the ground glass joint). Insert the sealed impinger into the sampling train as shown in Figure 1, making sure that all connections between the various components are leak tight. Greaseless ball joint fittings, heat shrinkable Teflon ® tubing, or Teflon ® tube fittings may be used to attain leakfree conditions for portions of the sampling train that come into contact with air containing SO2. Shield the absorbing reagent from direct sunlight by covering the impinger with aluminum foil or by enclosing the sampling train in a light-proof box. Determine the flow rate according to Section 9.4.2. Collect the sample at 1 ±0.10 L/min for 30-minute sampling or 0.500 ±0.05 L/min for 1-hour sampling. Record the exact sampling time in minutes, as the sample volume will later be determined using the sampling flow rate and the sampling time. Record the atmospheric pressure and temperature.
9.3 24-Hour Sampling. Place 50 mL of TCM absorbing solution in a large absorber, close the cap, and, if needed, apply the heat shrink material as shown in Figure 3. Verify that the reagent level is at the 50 mL mark on the absorber. Insert the sealed absorber into the sampling train as shown in Figure 2. At this time verify that the absorber temperature is controlled to 15 ±10°C. During sampling, the absorber temperature must be controlled to prevent decomposition of the collected complex. From the onset of sampling until analysis, the absorbing solution must be protected from direct sunlight. Determine the flow rate according to Section 9.4.2. Collect the sample for 24 hours from midnight to midnight at a flow rate of 0.200 ±0.020 L/min. A start/stop timer is helpful for initiating and stopping sampling and an elapsed time meter will be useful for determining the sampling time.
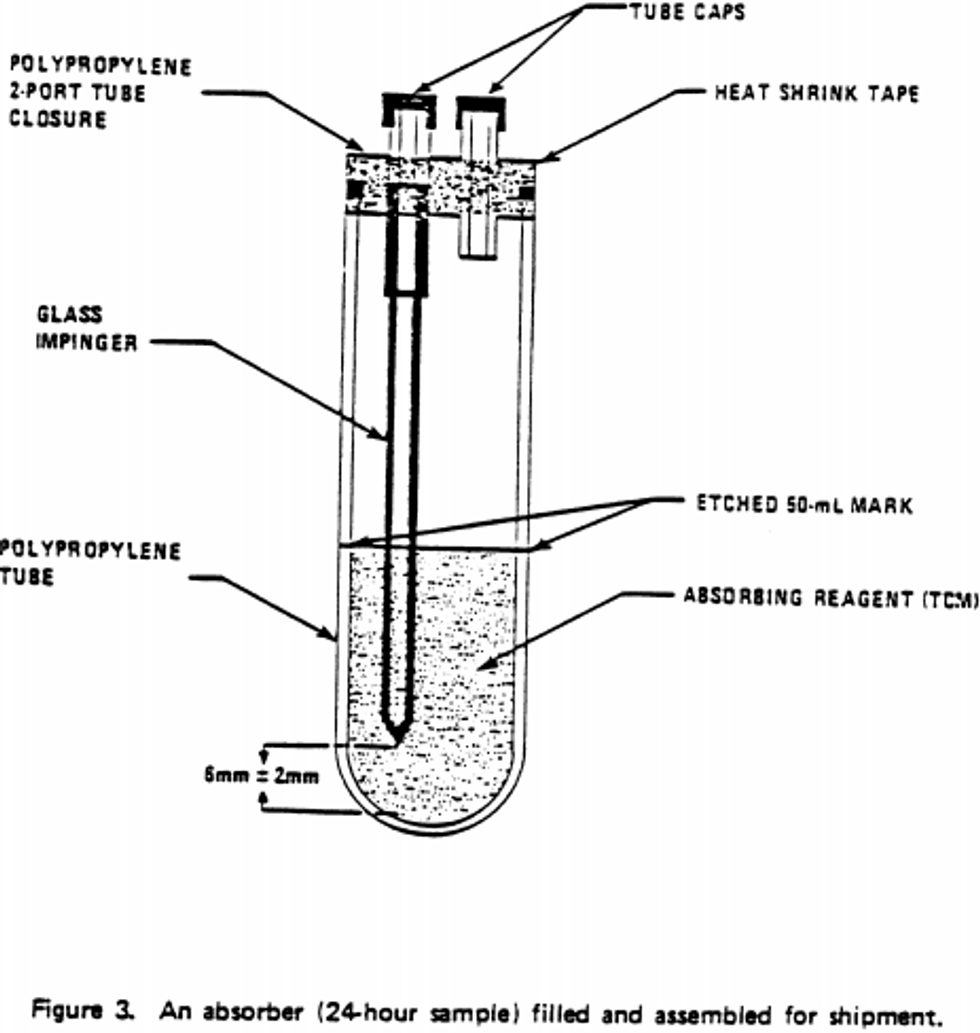
9.4 Flow Measurement.
9.4.1 Calibration: Flow measuring devices used for the on-site flow measurements required in 9.4.2 must be calibrated against a reliable flow or volume standard such as an NBS traceable bubble flowmeter or calibrated wet test meter. Rotameters or critical orifices used in the sampling train may be calibrated, if desired, as a quality control check, but such calibration shall not replace the on-site flow measurements required by 9.4.2. In-line rotameters, if they are to be calibrated, should be calibrated in situ, with the appropriate volume of solution in the absorber.
9.4.2 Determination of flow rate at sampling site: For short-term samples, the standard flow rate is determined at the sampling site at the initiation and completion of sample collection with a calibrated flow measuring device connected to the inlet of the absorber. For 24-hour samples, the standard flow rate is determined at the time the absorber is placed in the sampling train and again when the absorber is removed from the train for shipment to the analytical laboratory with a calibrated flow measuring device connected to the inlet of the sampling train. The flow rate determination must be made with all components of the sampling system in operation (e.g., the absorber temperature controller and any sample box heaters must also be operating). Equation 6 may be used to determine the standard flow rate when a calibrated positive displacement meter is used as the flow measuring device. Other types of calibrated flow measuring devices may also be used to determine the flow rate at the sampling site provided that the user applies any appropriate corrections to devices for which output is dependent on temperature or pressure.
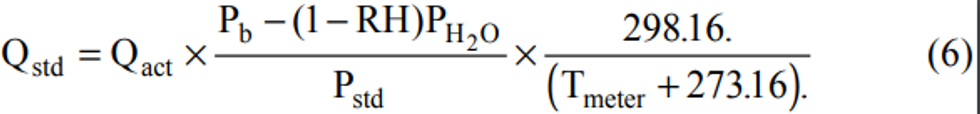
where:
Qstd = flow rate at standard conditions, std L/min (25°C and 760 mm Hg);
Qact = flow rate at monitoring site conditions, L/min;
Pb = barometric pressure at monitoring site conditions, mm Hg or kPa;
RH = fractional relative humidity of the air being measured;
PH2O = vapor pressure of water at the temperature of the air in the flow or volume standard, in the same units as Pb, (for wet volume standards only, i.e., bubble flowmeter or wet test meter; for dry standards, i.e., dry test meter, PH2O = 0);
Pstd = standard barometric pressure, in the same units as Pb (760 mm Hg or 101 kPa); and
Tmeter = temperature of the air in the flow or volume standard,°C (e.g., bubble flowmeter).
If a barometer is not available, the following equation may be used to determine the barometric pressure:

where:
H = sampling site elevation above sea level in meters.
If the initial flow rate (Qi) differs from the flow rate of the critical orifice or the flow rate indicated by the flowmeter in the sampling train (Qc) by more than 5 percent as determined by equation (8), check for leaks and redetermine Qi.
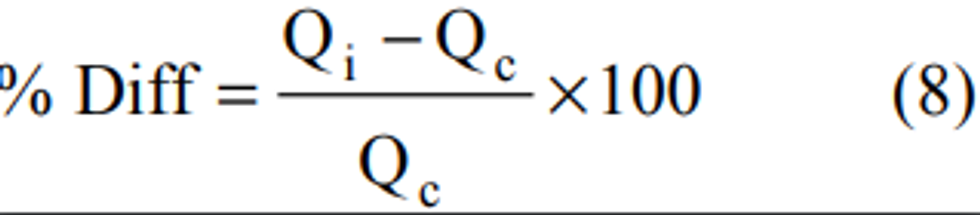
Invalidate the sample if the difference between the initial (Qi) and final (Qf) flow rates is more than 5 percent as determined by equation (9):
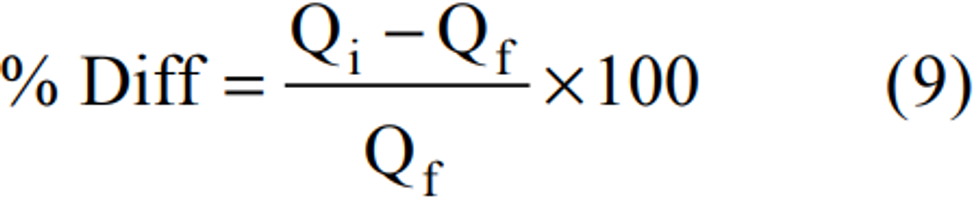
9.5 Sample Storage and Shipment. Remove the impinger or absorber from the sampling train and stopper immediately. Verify that the temperature of the absorber is not above 25°C. Mark the level of the solution with a temporary (e.g., grease pencil) mark. If the sample will not be analyzed within 12 hours of sampling, it must be stored at 5° ±5°C until analysis. Analysis must occur within 30 days. If the sample is transported or shipped for a period exceeding 12 hours, it is recommended that thermal coolers using eutectic ice packs, refrigerated shipping containers, etc., be used for periods up to 48 hours. (17) Measure the temperature of the absorber solution when the shipment is received. Invalidate the sample if the temperature is above 10°C. Store the sample at 5° ±5°C until it is analyzed.
10.0 Analytical Calibration.
10.1 Spectrophotometer Cell Matching. If unmatched spectrophotometer cells are used, an absorbance correction factor must be determined as follows:
1. Fill all cells with distilled water and designate the one that has the lowest absorbance at 548 nm as the reference. (This reference cell should be marked as such and continually used for this purpose throughout all future analyses.)
2. Zero the spectrophotometer with the reference cell.
3. Determine the absorbance of the remaining cells (Ac) in relation to the reference cell and record these values for future use. Mark all cells in a manner that adequately identifies the correction.
The corrected absorbance during future analyses using each cell is determining as follows:
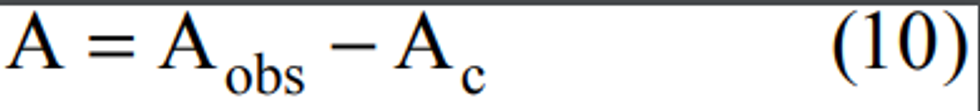
where:
A = corrected absorbance,
Aobs = uncorrected absorbance, and
Ac = cell correction.
10.2 Static Calibration Procedure (Option 1). Prepare a dilute working sulfite-TCM solution by diluting 10 mL of the working sulfite-TCM solution (Section 8.2.11) to 100 mL with TCM absorbing reagent. Following the table below, accurately pipet the indicated volumes of the sulfite-TCM solutions into a series of 25-mL volumetric flasks. Add TCM absorbing reagent as indicated to bring the volume in each flask to 10 mL.
| Sulfite-TCM solution | Volume of sulfite-TCM solution | Volume of TCM, mL | Total µg SO2 (approx.* |
|---|---|---|---|
| *Based on working sulfite-TCM solution concentration of 7.2 µg SO2/mL; the actual total µg SO2 must be calculated using equation 11 below. | |||
| Working | 4.0 | 6.0 | 28.8 |
| Working | 3.0 | 7.0 | 21.6 |
| Working | 2.0 | 8.0 | 14.4 |
| Dilute working | 10.0 | 0.0 | 7.2 |
| Dilute working | 5.0 | 5.0 | 3.6 |
| 0.0 | 10.0 | 0.0 | |
To each volumetric flask, add 1 mL 0.6% sulfamic acid (Section 8.2.1), accurately pipet 2 mL 0.2% formaldehyde solution (Section 8.2.2), then add 5 mL pararosaniline solution (Section 8.2.13). Start a laboratory timer that has been set for 30 minutes. Bring all flasks to volume with recently boiled and cooled distilled water and mix thoroughly. The color must be developed (during the 30-minute period) in a temperature environment in the range of 20° to 30°C, which is controlled to ±1°C. For increased precision, a constant temperature bath is recommended during the color development step. After 30 minutes, determine the corrected absorbance of each standard at 548 nm against a distilled water reference (Section 10.1). Denote this absorbance as (A). Distilled water is used in the reference cell rather than the reagant blank because of the temperature sensitivity of the reagent blank. Calculate the total micrograms SO2 in each solution:
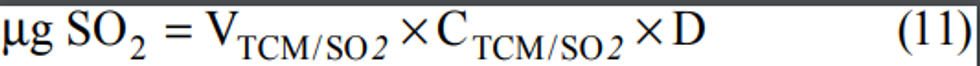
where:
VTCM/SO2 = volume of sulfite-TCM solution used, mL;
CTCM/SO2 = concentration of sulfur dioxide in the working sulfite-TCM, µg SO2/mL (from equation 4); and
D = dilution factor (D = 1 for the working sulfite-TCM solution; D = 0.1 for the diluted working sulfite-TCM solution).
A calibration equation is determined using the method of linear least squares (Section 12.1). The total micrograms SO2 contained in each solution is the x variable, and the corrected absorbance (eq. 10) associated with each solution is the y variable. For the calibration to be valid, the slope must be in the range of 0.030 ±0.002 absorbance unit/µg SO2, the intercept as determined by the least squares method must be equal to or less than 0.170 absorbance unit when the color is developed at 22°C (add 0.015 to this 0.170 specification for each°C above 22°C) and the correlation coefficient must be greater than 0.998. If these criteria are not met, it may be the result of an impure dye and/or an improperly standardized sulfite-TCM solution. A calibration factor (Bs) is determined by calculating the reciprocal of the slope and is subsequently used for calculating the sample concentration (Section 12.3).
10.3 Dynamic Calibration Procedures (Option 2). Atmospheres containing accurately known concentrations of sulfur dioxide are prepared using permeation devices. In the systems for generating these atmospheres, the permeation device emits gaseous SO2 at a known, low, constant rate, provided the temperature of the device is held constant (±0.1°C) and the device has been accurately calibrated at the temperature of use. The SO2 permeating from the device is carried by a low flow of dry carrier gas to a mixing chamber where it is diluted with SO2-free air to the desired concentration and supplied to a vented manifold. A typical system is shown schematically in Figure 4 and this system and other similar systems have been described in detail by O'Keeffe and Ortman; (19) Scaringelli, Frey, and Saltzman, (20) and Scaringelli, O'Keeffe, Rosenberg, and Bell. (21) Permeation devices may be prepared or purchased and in both cases must be traceable either to a National Bureau of Standards (NBS) Standard Reference Material (SRM 1625, SRM 1626, SRM 1627) or to an NBS/EPA-approved commercially available Certified Reference Material (CRM). CRM's are described in Reference 22, and a list of CRM sources is available from the address shown for Reference 22. A recommended protocol for certifying a permeation device to an NBS SRM or CRM is given in Section 2.0.7 of Reference 2. Device permeation rates of 0.2 to 0.4 µg/min, inert gas flows of about 50 mL/min, and dilution air flow rates from 1.1 to 15 L/min conveniently yield standard atmospheres in the range of 25 to 600 µg SO2/m 3 (0.010 to 0.230 ppm).
10.3.1 Calibration Option 2A (30-minute and 1-hour samples): Generate a series of six standard atmospheres of SO2 (e.g., 0, 50, 100, 200, 350, 500, 750 µg/m 3) by adjusting the dilution flow rates appropriately. The concentration of SO2 in each atmosphere is calculated as follows:
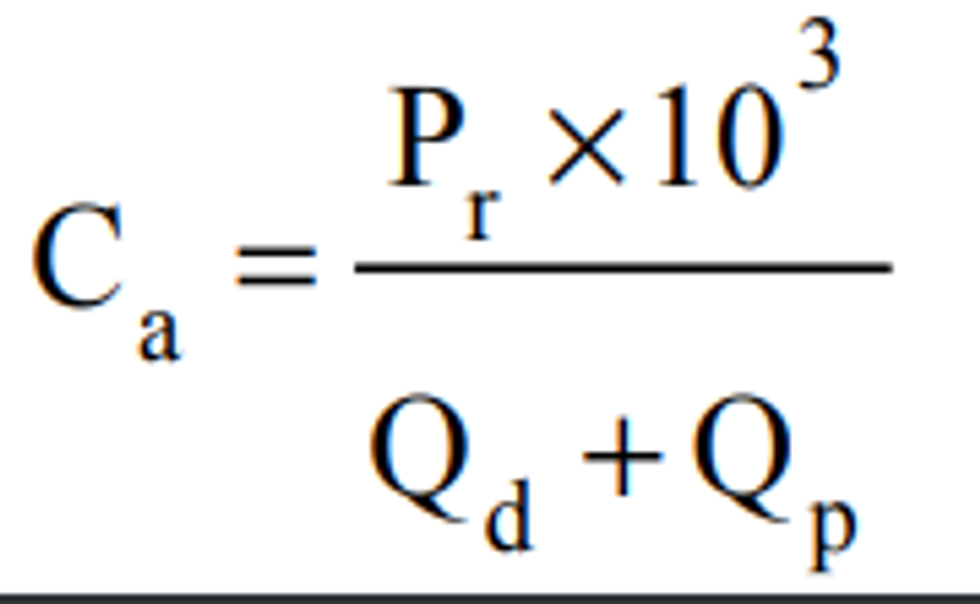
where:
Ca = concentration of SO2 at standard conditions, µg/m 3;
Pr = permeation rate, µg/min;
Qd = flow rate of dilution air, std L/min; and
Qp = flow rate of carrier gas across permeation device, std L/min.
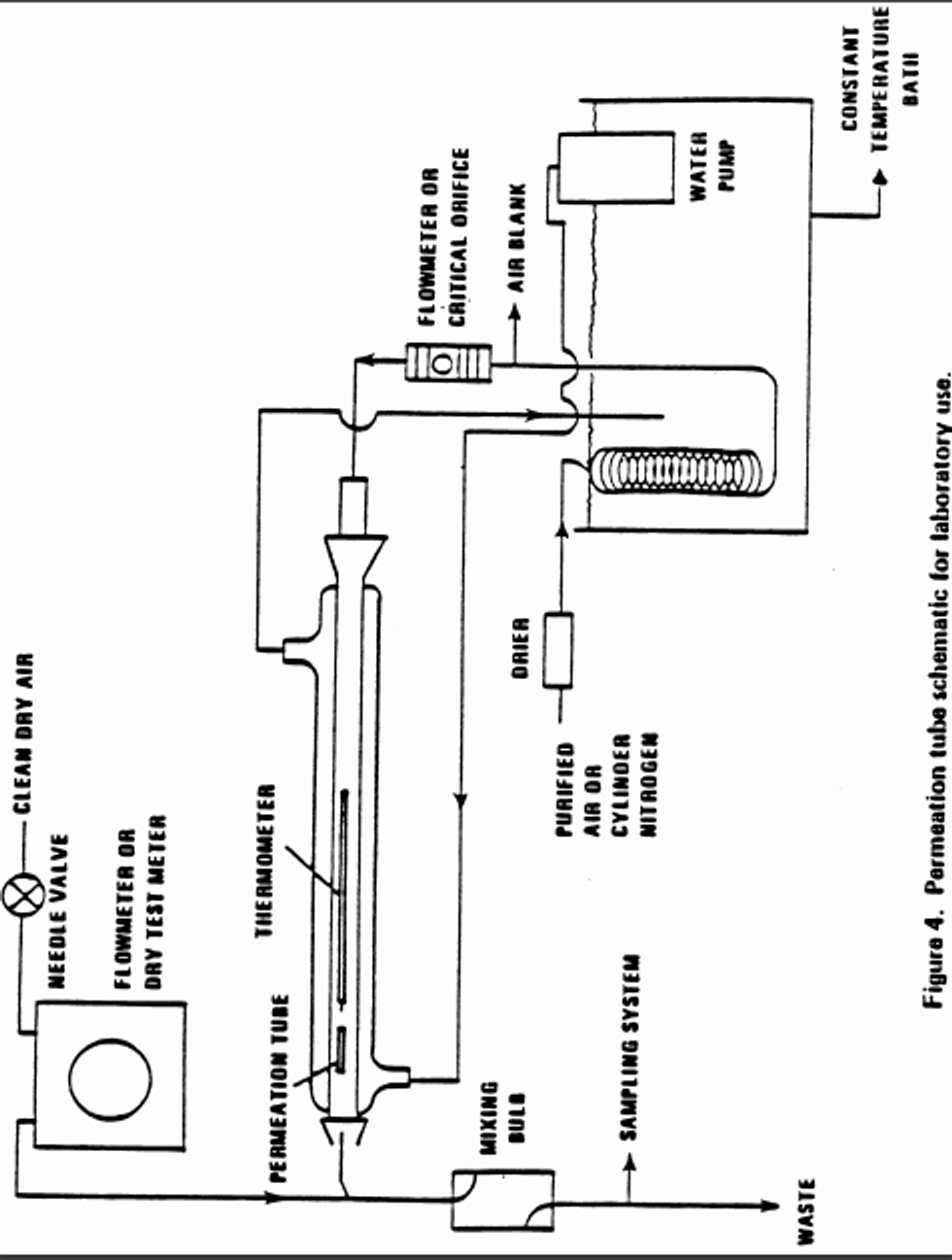
Be sure that the total flow rate of the standard exceeds the flow demand of the sample train, with the excess flow vented at atmospheric pressure. Sample each atmosphere using similar apparatus as shown in Figure 1 and under the same conditions as field sampling (i.e., use same absorbing reagent volume and sample same volume of air at an equivalent flow rate). Due to the length of the sampling periods required, this method is not recommended for 24-hour sampling. At the completion of sampling, quantitatively transfer the contents of each impinger to one of a series of 25-mL volumetric flasks (if 10 mL of absorbing solution was used) using small amounts of distilled water for rinse (<5mL). If >10 mL of absorbing solution was used, bring the absorber solution in each impinger to orginal volume with distilled H2 O and pipet 10-mL portions from each impinger into a series of 25-mL volumetric flasks. If the color development steps are not to be started within 12 hours of sampling, store the solutions at 5° ±5°C. Calculate the total micrograms SO2 in each solution as follows:
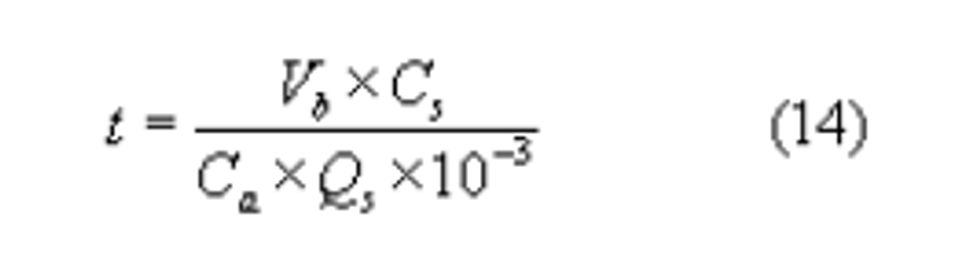
where:
Ca = concentration of SO2 in the standard atmosphere, µg/m 3;
Os = sampling flow rate, std L/min;
t = sampling time, min;
Va = volume of absorbing solution used for color development (10 mL); and
Vb = volume of absorbing solution used for sampling, mL.
Add the remaining reagents for color development in the same manner as in Section 10.2 for static solutions. Calculate a calibration equation and a calibration factor (Bg) according to Section 10.2, adhering to all the specified criteria.
10.3.2 Calibration Option 2B (24-hour samples): Generate a standard atmosphere containing approximately 1,050 µg SO2/m 3 and calculate the exact concentration according to equation 12. Set up a series of six absorbers according to Figure 2 and connect to a common manifold for sampling the standard atmosphere. Be sure that the total flow rate of the standard exceeds the flow demand at the sample manifold, with the excess flow vented at atmospheric pressure. The absorbers are then allowed to sample the atmosphere for varying time periods to yield solutions containing 0, 0.2, 0.6, 1.0, 1.4, 1.8, and 2.2 µg SO2/mL solution. The sampling times required to attain these solution concentrations are calculated as follows:

where:
t = sampling time, min;
Vb = volume of absorbing solution used for sampling (50 mL);
Cs = desired concentration of SO2 in the absorbing solution, µg/mL;
Ca = concentration of the standard atmosphere calculated according to equation 12, µg/m 3; and
Qs = sampling flow rate, std L/min.
At the completion of sampling, bring the absorber solutions to original volume with distilled water. Pipet a 10-mL portion from each absorber into one of a series of 25-mL volumetric flasks. If the color development steps are not to be started within 12 hours of sampling, store the solutions at 5° ±5°C. Add the remaining reagents for color development in the same manner as in Section 10.2 for static solutions. Calculate the total µg SO2 in each standard as follows:
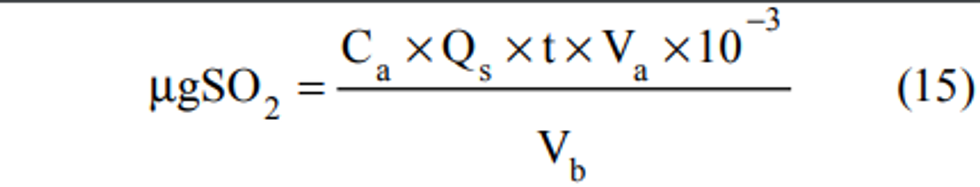
where:
Va = volume of absorbing solution used for color development (10 mL).
All other parameters are defined in equation 14.
Calculate a calibration equation and a calibration factor (Bt) according to Section 10.2 adhering to all the specified criteria.
11.0 Sample Preparation and Analysis.
11.1 Sample Preparation. Remove the samples from the shipping container. If the shipment period exceeded 12 hours from the completion of sampling, verify that the temperature is below 10°C. Also, compare the solution level to the temporary level mark on the absorber. If either the temperature is above 10°C or there was significant loss (more than 10 mL) of the sample during shipping, make an appropriate notation in the record and invalidate the sample. Prepare the samples for analysis as follows:
1. For 30-minute or 1-hour samples: Quantitatively transfer the entire 10 mL amount of absorbing solution to a 25-mL volumetric flask and rinse with a small amount (<5 mL) of distilled water.
2. For 24-hour samples: If the volume of the sample is less than the original 50-mL volume (permanent mark on the absorber), adjust the volume back to the original volume with distilled water to compensate for water lost to evaporation during sampling. If the final volume is greater than the original volume, the volume must be measured using a graduated cylinder. To analyze, pipet 10 mL of the solution into a 25-mL volumetric flask.
11.2 Sample Analysis. For each set of determinations, prepare a reagent blank by adding 10 mL TCM absorbing solution to a 25-mL volumetric flask, and two control standards containing approximately 5 and 15 µg SO2, respectively. The control standards are prepared according to Section 10.2 or 10.3. The analysis is carried out as follows:
1. Allow the sample to stand 20 minutes after the completion of sampling to allow any ozone to decompose (if applicable).
2. To each 25-mL volumetric flask containing reagent blank, sample, or control standard, add 1 mL of 0.6% sulfamic acid (Section 8.2.1) and allow to react for 10 min.
3. Accurately pipet 2 mL of 0.2% formaldehyde solution (Section 8.2.2) and then 5 mL of pararosaniline solution (Section 8.2.13) into each flask. Start a laboratory timer set at 30 minutes.
4. Bring each flask to volume with recently boiled and cooled distilled water and mix thoroughly.
5. During the 30 minutes, the solutions must be in a temperature controlled environment in the range of 20° to 30°C maintained to ±1°C. This temperature must also be within 1°C of that used during calibration.
6. After 30 minutes and before 60 minutes, determine the corrected absorbances (equation 10) of each solution at 548 nm using 1-cm optical path length cells against a distilled water reference (Section 10.1). (Distilled water is used as a reference instead of the reagent blank because of the sensitivity of the reagent blank to temperature.)
7. Do not allow the colored solution to stand in the cells because a film may be deposited. Clean the cells with isopropyl alcohol after use.
8. The reagent blank must be within 0.03 absorbance units of the intercept of the calibration equation determined in Section 10.
11.3 Absorbance range. If the absorbance of the sample solution ranges between 1.0 and 2.0, the sample can be diluted 1:1 with a portion of the reagent blank and the absorbance redetermined within 5 minutes. Solutions with higher absorbances can be diluted up to sixfold with the reagent blank in order to obtain scale readings of less than 1.0 absorbance unit. However, it is recommended that a smaller portion (<10 mL) of the original sample be reanalyzed (if possible) if the sample requires a dilution greater than 1:1.
11.4 Reagent disposal. All reagents containing mercury compounds must be stored and disposed of using one of the procedures contained in Section 13. Until disposal, the discarded solutions can be stored in closed glass containers and should be left in a fume hood.
12.0 Calculations.
12.1 Calibration Slope, Intercept, and Correlation Coefficient. The method of least squares is used to calculate a calibration equation in the form of:

where:
y = corrected absorbance,
m = slope, absorbance unit/µg SO2,
x = micrograms of SO2,
b = y intercept (absorbance units).
The slope (m), intercept (b), and correlation coefficient (r) are calculated as follows:
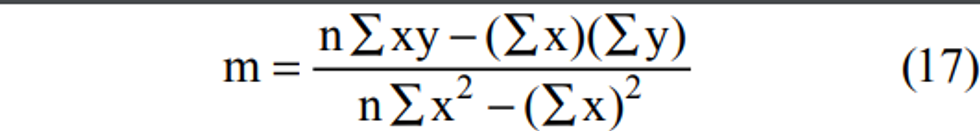
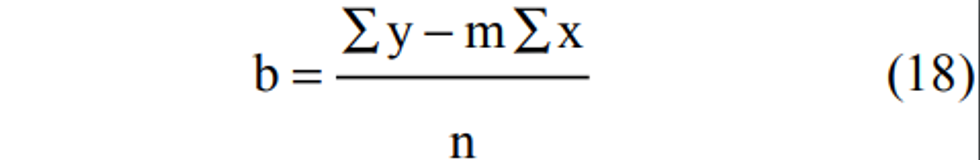
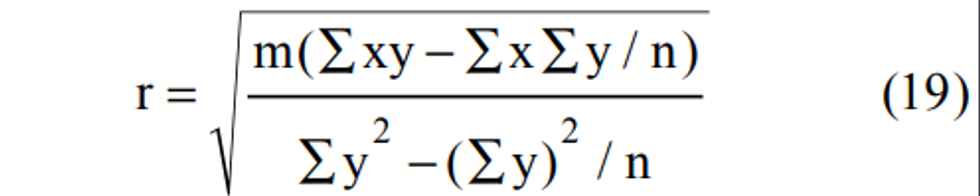
where n is the number of calibration points.
A data form (Figure 5) is supplied for easily organizing calibration data when the slope, intercept, and correlation coefficient are calculated by hand.
12.2 Total Sample Volume. Determine the sampling volume at standard conditions as follows:
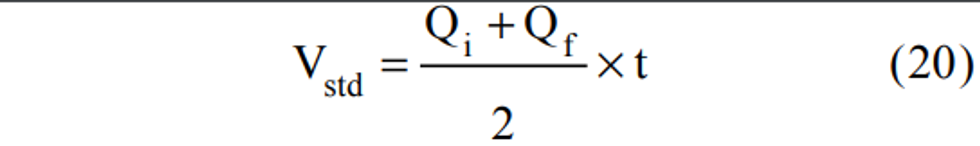
where:
Vstd = sampling volume in std L,
Qi = standard flow rate determined at the initiation of sampling in std L/min,
Qf = standard flow rate determined at the completion of sampling is std L/min, and
t = total sampling time, min.
12.3 Sulfur Dioxide Concentration. Calculate and report the concentration of each sample as follows:
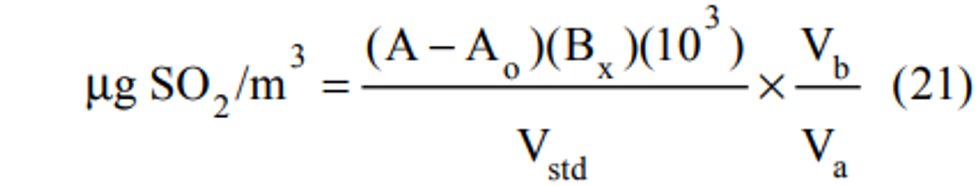
where:
A = corrected absorbance of the sample solution, from equation (10);
Ao = corrected absorbance of the reagent blank, using equation (10);
BX = calibration factor equal to Bs, Bg, or Bt depending on the calibration procedure used, the reciprocal of the slope of the calibration equation;
Va = volume of absorber solution analyzed, mL;
Vb = total volume of solution in absorber (see 11.1-2), mL; and
Vstd = standard air volume sampled, std L (from Section 12.2).
| Calibration point no. | Micro- grams So2 | Absor- bance units | |||
|---|---|---|---|---|---|
| (x) | (y) | x 2 | xy | y 2 | |
| 1 | |||||
| 2 | |||||
| 3 | |||||
| 4 | |||||
| 5 | |||||
| 6 |
Σ x=___ Σ y=___ Σ x 2=___ Σxy___ Σy 2___
n=___ (number of pairs of coordinates.)
Figure 5. Data form for hand calculations.
12.4 Control Standards. Calculate the analyzed micrograms of SO2 in each control standard as follows:
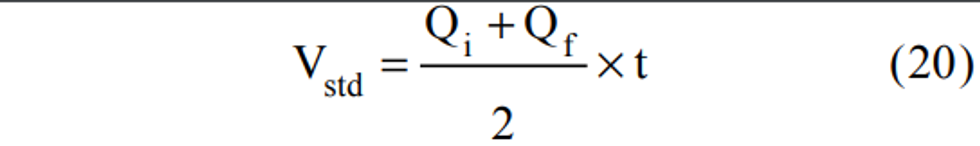
where:
Cq = analyzed µg SO2 in each control standard,
A = corrected absorbance of the control standard, and
Ao = corrected absorbance of the reagent blank.
The difference between the true and analyzed values of the control standards must not be greater than 1 µg. If the difference is greater than 1 µg, the source of the discrepancy must be identified and corrected.
12.5 Conversion of µg/m 3 to ppm (v/v). If desired, the concentration of sulfur dioxide at reference conditions can be converted to ppm SO2 (v/v) as follows:
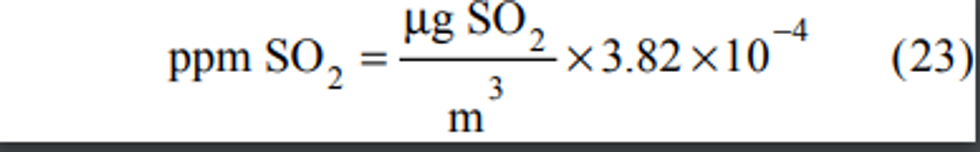
13.0 The TCM absorbing solution and any reagents containing mercury compounds must be treated and disposed of by one of the methods discussed below. Both methods remove greater than 99.99 percent of the mercury.
13.1 Disposal of Mercury-Containing Solutions.
13.2 Method for Forming an Amalgam.
1. Place the waste solution in an uncapped vessel in a hood.
2. For each liter of waste solution, add approximately 10 g of sodium carbonate until neutralization has occurred (NaOH may have to be used).
3. Following neutralization, add 10 g of granular zinc or magnesium.
4. Stir the solution in a hood for 24 hours. Caution must be exercised as hydrogen gas is evolved by this treatment process.
5. After 24 hours, allow the solution to stand without stirring to allow the mercury amalgam (solid black material) to settle to the bottom of the waste receptacle.
6. Upon settling, decant and discard the supernatant liquid.
7. Quantitatively transfer the solid material to a container and allow to dry.
8. The solid material can be sent to a mercury reclaiming plant. It must not be discarded.
13.3 Method Using Aluminum Foil Strips.
1. Place the waste solution in an uncapped vessel in a hood.
2. For each liter of waste solution, add approximately 10 g of aluminum foil strips. If all the aluminum is consumed and no gas is evolved, add an additional 10 g of foil. Repeat until the foil is no longer consumed and allow the gas to evolve for 24 hours.
3. Decant the supernatant liquid and discard.
4. Transfer the elemental mercury that has settled to the bottom of the vessel to a storage container.
5. The mercury can be sent to a mercury reclaiming plant. It must not be discarded.
14.0 References for SO2Method.
1. Quality Assurance Handbook for Air Pollution Measurement Systems, Volume I, Principles. EPA-600/9-76-005, U.S. Environmental Protection Agency, Research Triangle Park, NC 27711, 1976.
2. Quality Assurance Handbook for Air Pollution Measurement Systems, Volume II, Ambient Air Specific Methods. EPA-600/4-77-027a, U.S. Environmental Protection Agency, Research Triangle Park, NC 27711, 1977.
3. Dasqupta, P. K., and K. B. DeCesare. Stability of Sulfur Dioxide in Formaldehyde and Its Anomalous Behavior in Tetrachloromercurate (II). Submitted for publication in Atmospheric Environment, 1982.
4. West, P. W., and G. C. Gaeke. Fixation of Sulfur Dioxide as Disulfitomercurate (II) and Subsequent Colorimetric Estimation. Anal. Chem., 28:1816, 1956.
5. Ephraim, F. Inorganic Chemistry. P. C. L. Thorne and E. R. Roberts, Eds., 5th Edition, Interscience, 1948, p. 562.
6. Lyles, G. R., F. B. Dowling, and V. J. Blanchard. Quantitative Determination of Formaldehyde in the Parts Per Hundred Million Concentration Level. J. Air. Poll. Cont. Assoc., Vol. 15(106), 1965.
7. McKee, H. C., R. E. Childers, and O. Saenz, Jr. Collaborative Study of Reference Method for Determination of Sulfur Dioxide in the Atmosphere (Pararosaniline Method). EPA-APTD-0903, U.S. Environmental Protection Agency, Research Triangle Park, NC 27711, September 1971.
8. Urone, P., J. B. Evans, and C. M. Noyes. Tracer Techniques in Sulfur - Air Pollution Studies Apparatus and Studies of Sulfur Dioxide Colorimetric and Conductometric Methods. Anal. Chem., 37: 1104, 1965.
9. Bostrom, C. E. The Absorption of Sulfur Dioxide at Low Concentrations (pphm) Studied by an Isotopic Tracer Method. Intern. J. Air Water Poll., 9:333, 1965.
10. Scaringelli, F. P., B. E. Saltzman, and S. A. Frey. Spectrophotometric Determination of Atmospheric Sulfur Dioxide. Anal. Chem., 39: 1709, 1967.
11. Pate, J. B., B. E. Ammons, G. A. Swanson, and J. P. Lodge, Jr. Nitrite Interference in Spectrophotometric Determination of Atmospheric Sulfur Dioxide. Anal. Chem., 37:942, 1965.
12. Zurlo, N., and A. M. Griffini. Measurement of the Sulfur Dioxide Content of the Air in the Presence of Oxides of Nitrogen and Heavy Metals. Medicina Lavoro, 53:330, 1962.
13. Rehme, K. A., and F. P. Scaringelli. Effect of Ammonia on the Spectrophotometric Determination of Atmospheric Concentrations of Sulfur Dioxide. Anal. Chem., 47:2474, 1975.
14. McCoy, R. A., D. E. Camann, and H. C. McKee. Collaborative Study of Reference Method for Determination of Sulfur Dioxide in the Atmosphere (Pararosaniline Method) (24-Hour Sampling). EPA-650/4-74-027, U.S. Environmental Protection Agency, Research Triangle Park, NC 27711, December 1973.
15. Fuerst, R. G. Improved Temperature Stability of Sulfur Dioxide Samples Collected by the Federal Reference Method. EPA-600/4-78-018, U.S. Environmental Protection Agency, Research Triangle Park, NC 27711, April 1978.
16. Scaringelli, F. P., L. Elfers, D. Norris, and S. Hochheiser. Enhanced Stability of Sulfur Dioxide in Solution. Anal. Chem., 42:1818, 1970.
17. Martin, B. E. Sulfur Dioxide Bubbler Temperature Study. EPA-600/4-77-040, U.S. Environmental Protection Agency, Research Triangle Park, NC 27711, August 1977.
18. American Society for Testing and Materials. ASTM Standards, Water; Atmospheric Analysis. Part 23. Philadelphia, PA, October 1968, p. 226.
19. O'Keeffe, A. E., and G. C. Ortman. Primary Standards for Trace Gas Analysis. Anal. Chem., 38:760, 1966.
20. Scaringelli, F. P., S. A. Frey, and B. E. Saltzman. Evaluation of Teflon Permeation Tubes for Use with Sulfur Dioxide. Amer. Ind. Hygiene Assoc. J., 28:260, 1967.
21. Scaringelli, F. P., A. E. O'Keeffe, E. Rosenberg, and J. P. Bell, Preparation of Known Concentrations of Gases and Vapors With Permeation Devices Calibrated Gravimetrically. Anal. Chem., 42:871, 1970.
22. A Procedure for Establishing Traceability of Gas Mixtures to Certain National Bureau of Standards Standard Reference Materials. EPA-600/7-81-010, U.S. Environmental Protection Agency, Environmental Monitoring Systems Laboratory (MD-77), Research Triangle Park, NC 27711, January 1981.
[47 FR 54899, Dec. 6, 1982; 48 FR 17355, Apr. 22, 1983. Redesignated at 75 FR 35595, June 22, 2010]
Appendix B to Part 50 - Reference Method for the Determination of Suspended Particulate Matter in the Atmosphere (High-Volume Method)
1.0 Applicability.
1.1 This method provides a measurement of the mass concentration of total suspended particulate matter (TSP) in ambient air for determining compliance with the primary and secondary national ambient air quality standards for particulate matter as specified in §50.6 and §50.7 of this chapter. The measurement process is nondestructive, and the size of the sample collected is usually adequate for subsequent chemical analysis. Quality assurance procedures and guidance are provided in part 58, appendixes A and B, of this chapter and in References 1 and 2.
2.0 Principle.
2.1 An air sampler, properly located at the measurement site, draws a measured quantity of ambient air into a covered housing and through a filter during a 24-hr (nominal) sampling period. The sampler flow rate and the geometry of the shelter favor the collection of particles up to 25-50 µm (aerodynamic diameter), depending on wind speed and direction.(3) The filters used are specified to have a minimum collection efficiency of 99 percent for 0.3 µm (DOP) particles (see Section 7.1.4).
2.2 The filter is weighed (after moisture equilibration) before and after use to determine the net weight (mass) gain. The total volume of air sampled, corrected to EPA standard conditions (25°C, 760 mm Hg [101 kPa]), is determined from the measured flow rate and the sampling time. The concentration of total suspended particulate matter in the ambient air is computed as the mass of collected particles divided by the volume of air sampled, corrected to standard conditions, and is expressed in micrograms per standard cubic meter (µg/std m 3). For samples collected at temperatures and pressures significantly different than standard conditions, these corrected concentrations may differ substantially from actual concentrations (micrograms per actual cubic meter), particularly at high elevations. The actual particulate matter concentration can be calculated from the corrected concentration using the actual temperature and pressure during the sampling period.
3.0 Range.
3.1 The approximate concentration range of the method is 2 to 750 µg/std m 3. The upper limit is determined by the point at which the sampler can no longer maintain the specified flow rate due to the increased pressure drop of the loaded filter. This point is affected by particle size distribution, moisture content of the collected particles, and variability from filter to filter, among other things. The lower limit is determined by the sensitivity of the balance (see Section 7.10) and by inherent sources of error (see Section 6).
3.2 At wind speeds between 1.3 and 4.5 m/sec (3 and 10 mph), the high-volume air sampler has been found to collect particles up to 25 to 50 µm, depending on wind speed and direction.(3) For the filter specified in Section 7.1, there is effectively no lower limit on the particle size collected.
4.0 Precision.
4.1 Based upon collaborative testing, the relative standard deviation (coefficient of variation) for single analyst precision (repeatability) of the method is 3.0 percent. The corresponding value for interlaboratory precision (reproducibility) is 3.7 percent.(4)
5.0 Accuracy.
5.1 The absolute accuracy of the method is undefined because of the complex nature of atmospheric particulate matter and the difficulty in determining the “true” particulate matter concentration. This method provides a measure of particulate matter concentration suitable for the purpose specified under Section 1.0, Applicability.
6.0 Inherent Sources of Error.
6.1 Airflow variation. The weight of material collected on the filter represents the (integrated) sum of the product of the instantaneous flow rate times the instantaneous particle concentration. Therefore, dividing this weight by the average flow rate over the sampling period yields the true particulate matter concentration only when the flow rate is constant over the period. The error resulting from a nonconstant flow rate depends on the magnitude of the instantaneous changes in the flow rate and in the particulate matter concentration. Normally, such errors are not large, but they can be greatly reduced by equipping the sampler with an automatic flow controlling mechanism that maintains constant flow during the sampling period. Use of a contant flow controller is recommended.*
*At elevated altitudes, the effectiveness of automatic flow controllers may be reduced because of a reduction in the maximum sampler flow.
6.2 Air volume measurement. If the flow rate changes substantially or nonuniformly during the sampling period, appreciable error in the estimated air volume may result from using the average of the presampling and postsampling flow rates. Greater air volume measurement accuracy may be achieved by (1) equipping the sampler with a flow controlling mechanism that maintains constant air flow during the sampling period,* (2) using a calibrated, continuous flow rate recording device to record the actual flow rate during the samping period and integrating the flow rate over the period, or (3) any other means that will accurately measure the total air volume sampled during the sampling period. Use of a continuous flow recorder is recommended, particularly if the sampler is not equipped with a constant flow controller.
6.3 Loss of volatiles. Volatile particles collected on the filter may be lost during subsequent sampling or during shipment and/or storage of the filter prior to the postsampling weighing.(5) Although such losses are largely unavoidable, the filter should be reweighed as soon after sampling as practical.
6.4 Artifact particulate matter. Artifact particulate matter can be formed on the surface of alkaline glass fiber filters by oxidation of acid gases in the sample air, resulting in a higher than true TSP determination.(6 7) This effect usually occurs early in the sample period and is a function of the filter pH and the presence of acid gases. It is generally believed to account for only a small percentage of the filter weight gain, but the effect may become more significant where relatively small particulate weights are collected.
6.5 Humidity. Glass fiber filters are comparatively insensitive to changes in relative humidity, but collected particulate matter can be hygroscopic.(8) The moisture conditioning procedure minimizes but may not completely eliminate error due to moisture.
6.6 Filter handling. Careful handling of the filter between the presampling and postsampling weighings is necessary to avoid errors due to loss of fibers or particles from the filter. A filter paper cartridge or cassette used to protect the filter can minimize handling errors. (See Reference 2, Section 2).
6.7 Nonsampled particulate matter. Particulate matter may be deposited on the filter by wind during periods when the sampler is inoperative. (9) It is recommended that errors from this source be minimized by an automatic mechanical device that keeps the filter covered during nonsampling periods, or by timely installation and retrieval of filters to minimize the nonsampling periods prior to and following operation.
6.8 Timing errors. Samplers are normally controlled by clock timers set to start and stop the sampler at midnight. Errors in the nominal 1,440-min sampling period may result from a power interruption during the sampling period or from a discrepancy between the start or stop time recorded on the filter information record and the actual start or stop time of the sampler. Such discrepancies may be caused by (1) poor resolution of the timer set-points, (2) timer error due to power interruption, (3) missetting of the timer, or (4) timer malfunction. In general, digital electronic timers have much better set-point resolution than mechanical timers, but require a battery backup system to maintain continuity of operation after a power interruption. A continuous flow recorder or elapsed time meter provides an indication of the sampler run-time, as well as indication of any power interruption during the sampling period and is therefore recommended.
6.9 Recirculation of sampler exhaust. Under stagnant wind conditions, sampler exhaust air can be resampled. This effect does not appear to affect the TSP measurement substantially, but may result in increased carbon and copper in the collected sample. (10) This problem can be reduced by ducting the exhaust air well away, preferably downwind, from the sampler.
7.0 Apparatus.
(See References 1 and 2 for quality assurance information.)
Note:
Samplers purchased prior to the effective date of this amendment are not subject to specifications preceded by (†).
7.1 Filter. (Filters supplied by the Environmental Protection Agency can be assumed to meet the following criteria. Additional specifications are required if the sample is to be analyzed chemically.)
7.1.1 Size: 20.3 ±0.2 × 25.4 ±0.2 cm (nominal 8 × 10 in).
7.1.2 Nominal exposed area: 406.5 cm 2 (63 in 2).
7.1.3. Material: Glass fiber or other relatively inert, nonhygroscopic material. (8)
7.1.4 Collection efficiency: 99 percent minimum as measured by the DOP test (ASTM-2986) for particles of 0.3 µm diameter.
7.1.5 Recommended pressure drop range: 42-54 mm Hg (5.6-7.2 kPa) at a flow rate of 1.5 std m 3/min through the nominal exposed area.
7.1.6 pH: 6 to 10. (11)
7.1.7 Integrity: 2.4 mg maximum weight loss. (11)
7.1.8 Pinholes: None.
7.1.9 Tear strength: 500 g minimum for 20 mm wide strip cut from filter in weakest dimension. (See ASTM Test D828-60).
7.1.10 Brittleness: No cracks or material separations after single lengthwise crease.
7.2 Sampler. The air sampler shall provide means for drawing the air sample, via reduced pressure, through the filter at a uniform face velocity.
7.2.1 The sampler shall have suitable means to:
a. Hold and seal the filter to the sampler housing.
b. Allow the filter to be changed conveniently.
c. Preclude leaks that would cause error in the measurement of the air volume passing through the filter.
d. (†) Manually adjust the flow rate to accommodate variations in filter pressure drop and site line voltage and altitude. The adjustment may be accomplished by an automatic flow controller or by a manual flow adjustment device. Any manual adjustment device must be designed with positive detents or other means to avoid unintentional changes in the setting.
(†) See note at beginning of Section 7 of this appendix.
7.2.2 Minimum sample flow rate, heavily loaded filter: 1.1 m 3/min (39 ft 3/min).‡
‡ These specifications are in actual air volume units; to convert to EPA standard air volume units, multiply the specifications by (Pb/Pstd)(298/T) where Pb and T are the barometric pressure in mm Hg (or kPa) and the temperature in K at the sampler, and Pstd is 760 mm Hg (or 101 kPa).
7.2.3 Maximum sample flow rate, clean filter: 1.7 m 3/min (60 ft 3/min).‡
7.2.4 Blower Motor: The motor must be capable of continuous operation for 24-hr periods.
7.3 Sampler shelter.
7.3.1 The sampler shelter shall:
a. Maintain the filter in a horizontal position at least 1 m above the sampler supporting surface so that sample air is drawn downward through the filter.
b. Be rectangular in shape with a gabled roof, similar to the design shown in Figure 1.
c. Cover and protect the filter and sampler from precipitation and other weather.
d. Discharge exhaust air at least 40 cm from the sample air inlet.
e. Be designed to minimize the collection of dust from the supporting surface by incorporating a baffle between the exhaust outlet and the supporting surface.
7.3.2 The sampler cover or roof shall overhang the sampler housing somewhat, as shown in Figure 1, and shall be mounted so as to form an air inlet gap between the cover and the sampler housing walls. † This sample air inlet should be approximately uniform on all sides of the sampler. † The area of the sample air inlet must be sized to provide an effective particle capture air velocity of between 20 and 35 cm/sec at the recommended operational flow rate. The capture velocity is the sample air flow rate divided by the inlet area measured in a horizontal plane at the lower edge of the cover. † Ideally, the inlet area and operational flow rate should be selected to obtain a capture air velocity of 25 ±2 cm/sec.
7.4 Flow rate measurement devices.
7.4.1 The sampler shall incorporate a flow rate measurement device capable of indicating the total sampler flow rate. Two common types of flow indicators covered in the calibration procedure are (1) an electronic mass flowmeter and (2) an orifice or orifices located in the sample air stream together with a suitable pressure indicator such as a manometer, or aneroid pressure gauge. A pressure recorder may be used with an orifice to provide a continuous record of the flow. Other types of flow indicators (including rotameters) having comparable precision and accuracy are also acceptable.
7.4.2 † The flow rate measurement device must be capable of being calibrated and read in units corresponding to a flow rate which is readable to the nearest 0.02 std m 3/min over the range 1.0 to 1.8 std m 3/min.
7.5 Thermometer, to indicate the approximate air temperature at the flow rate measurement orifice, when temperature corrections are used.
7.5.1 Range: −40° to + 50°C (223-323 K).
7.5.2 Resolution: 2°C (2 K).
7.6 Barometer, to indicate barometric pressure at the flow rate measurement orifice, when pressure corrections are used.
7.6.1 Range: 500 to 800 mm Hg (66-106 kPa).
7.6.2 Resolution: ±5 mm Hg (0.67 kPa).
7.7 Timing/control device.
7.7.1 The timing device must be capable of starting and stopping the sampler to obtain an elapsed run-time of 24 hr ±1 hr (1,440 ±60 min).
7.7.2 Accuracy of time setting: ±30 min, or better. (See Section 6.8).
7.8 Flow rate transfer standard, traceable to a primary standard. (See Section 9.2.)
7.8.1 Approximate range: 1.0 to 1.8 m 3/min.
7.8.2 Resolution: 0.02 m 3/min.
7.8.3 Reproducibility: ±2 percent (2 times coefficient of variation) over normal ranges of ambient temperature and pressure for the stated flow rate range. (See Reference 2, Section 2.)
7.8.4 Maximum pressure drop at 1.7 std m3/min; 50 cm H2 O (5 kPa).
7.8.5 The flow rate transfer standard must connect without leaks to the inlet of the sampler and measure the flow rate of the total air sample.
7.8.6 The flow rate transfer standard must include a means to vary the sampler flow rate over the range of 1.0 to 1.8 m 3/min (35-64 ft 3/min) by introducing various levels of flow resistance between the sampler and the transfer standard inlet.
7.8.7 The conventional type of flow transfer standard consists of: An orifice unit with adapter that connects to the inlet of the sampler, a manometer or other device to measure orifice pressure drop, a means to vary the flow through the sampler unit, a thermometer to measure the ambient temperature, and a barometer to measure ambient pressure. Two such devices are shown in Figures 2a and 2b. Figure 2a shows multiple fixed resistance plates, which necessitate disassembly of the unit each time the flow resistance is changed. A preferable design, illustrated in Figure 2b, has a variable flow restriction that can be adjusted externally without disassembly of the unit. Use of a conventional, orifice-type transfer standard is assumed in the calibration procedure (Section 9). However, the use of other types of transfer standards meeting the above specifications, such as the one shown in Figure 2c, may be approved; see the note following Section 9.1.
7.9 Filter conditioning environment
7.9.1 Controlled temperature: between 15° and 30°C with less than ±3°C variation during equilibration period.
7.9.2 Controlled humidity: Less than 50 percent relative humidity, constant within ±5 percent.
7.10 Analytical balance.
7.10.1 Sensitivity: 0.1 mg.
7.10.2 Weighing chamber designed to accept an unfolded 20.3 × 25.4 cm (8 × 10 in) filter.
7.11 Area light source, similar to X-ray film viewer, to backlight filters for visual inspection.
7.12 Numbering device, capable of printing identification numbers on the filters before they are placed in the filter conditioning environment, if not numbered by the supplier.
8.0 Procedure.
(See References 1 and 2 for quality assurance information.)
8.1 Number each filter, if not already numbered, near its edge with a unique identification number.
8.2 Backlight each filter and inspect for pinholes, particles, and other imperfections; filters with visible imperfections must not be used.
8.3 Equilibrate each filter in the conditioning environment for at least 24-hr.
8.4 Following equilibration, weigh each filter to the nearest milligram and record this tare weight (Wi) with the filter identification number.
8.5 Do not bend or fold the filter before collection of the sample.
8.6 Open the shelter and install a numbered, preweighed filter in the sampler, following the sampler manufacturer's instructions. During inclement weather, precautions must be taken while changing filters to prevent damage to the clean filter and loss of sample from or damage to the exposed filter. Filter cassettes that can be loaded and unloaded in the laboratory may be used to minimize this problem (See Section 6.6).
8.7 Close the shelter and run the sampler for at least 5 min to establish run-temperature conditions.
8.8 Record the flow indicator reading and, if needed, the barometric pressure (P 33) and the ambient temperature (T 33) see NOTE following step 8.12). Stop the sampler. Determine the sampler flow rate (see Section 10.1); if it is outside the acceptable range (1.1 to 1.7 m 3/min [39-60 ft 3/min]), use a different filter, or adjust the sampler flow rate. Warning: Substantial flow adjustments may affect the calibration of the orifice-type flow indicators and may necessitate recalibration.
8.9 Record the sampler identification information (filter number, site location or identification number, sample date, and starting time).
8.10 Set the timer to start and stop the sampler such that the sampler runs 24-hrs, from midnight to midnight (local time).
8.11 As soon as practical following the sampling period, run the sampler for at least 5 min to again establish run-temperature conditions.
8.12 Record the flow indicator reading and, if needed, the barometric pressure (P 33) and the ambient temperature (T 33).
Note:
No onsite pressure or temperature measurements are necessary if the sampler flow indicator does not require pressure or temperature corrections (e.g., a mass flowmeter) or if average barometric pressure and seasonal average temperature for the site are incorporated into the sampler calibration (see step 9.3.9). For individual pressure and temperature corrections, the ambient pressure and temperature can be obtained by onsite measurements or from a nearby weather station. Barometric pressure readings obtained from airports must be station pressure, not corrected to sea level, and may need to be corrected for differences in elevation between the sampler site and the airport. For samplers having flow recorders but not constant flow controllers, the average temperature and pressure at the site during the sampling period should be estimated from weather bureau or other available data.
8.13 Stop the sampler and carefully remove the filter, following the sampler manufacturer's instructions. Touch only the outer edges of the filter. See the precautions in step 8.6.
8.14 Fold the filter in half lengthwise so that only surfaces with collected particulate matter are in contact and place it in the filter holder (glassine envelope or manila folder).
8.15 Record the ending time or elapsed time on the filter information record, either from the stop set-point time, from an elapsed time indicator, or from a continuous flow record. The sample period must be 1,440 ±60 min. for a valid sample.
8.16 Record on the filter information record any other factors, such as meteorological conditions, construction activity, fires or dust storms, etc., that might be pertinent to the measurement. If the sample is known to be defective, void it at this time.
8.17 Equilibrate the exposed filter in the conditioning environment for at least 24-hrs.
8.18 Immediately after equilibration, reweigh the filter to the nearest milligram and record the gross weight with the filter identification number. See Section 10 for TSP concentration calculations.
9.0 Calibration.
9.1 Calibration of the high volume sampler's flow indicating or control device is necessary to establish traceability of the field measurement to a primary standard via a flow rate transfer standard. Figure 3a illustrates the certification of the flow rate transfer standard and Figure 3b illustrates its use in calibrating a sampler flow indicator. Determination of the corrected flow rate from the sampler flow indicator, illustrated in Figure 3c, is addressed in Section 10.1
Note:
The following calibration procedure applies to a conventional orifice-type flow transfer standard and an orifice-type flow indicator in the sampler (the most common types). For samplers using a pressure recorder having a square-root scale, 3 other acceptable calibration procedures are provided in Reference 12. Other types of transfer standards may be used if the manufacturer or user provides an appropriately modified calibration procedure that has been approved by EPA under Section 2.8 of appendix C to §53.11 of this chapter.
9.2 Certification of the flow rate transfer standard.
9.2.1 Equipment required: Positive displacement standard volume meter traceable to the National Bureau of Standards (such as a Roots meter or equivalent), stop-watch, manometer, thermometer, and barometer.
9.2.2 Connect the flow rate transfer standard to the inlet of the standard volume meter. Connect the manometer to measure the pressure at the inlet of the standard volume meter. Connect the orifice manometer to the pressure tap on the transfer standard. Connect a high-volume air pump (such as a high-volume sampler blower) to the outlet side of the standard volume meter. See Figure 3a.
9.2.3 Check for leaks by temporarily clamping both manometer lines (to avoid fluid loss) and blocking the orifice with a large-diameter rubber stopper, wide cellophane tape, or other suitable means. Start the high-volume air pump and note any change in the standard volume meter reading. The reading should remain constant. If the reading changes, locate any leaks by listening for a whistling sound and/or retightening all connections, making sure that all gaskets are properly installed.
9.2.4 After satisfactorily completing the leak check as described above, unclamp both manometer lines and zero both manometers.
9.2.5 Achieve the appropriate flow rate through the system, either by means of the variable flow resistance in the transfer standard or by varying the voltage to the air pump. (Use of resistance plates as shown in Figure 1a is discouraged because the above leak check must be repeated each time a new resistance plate is installed.) At least five different but constant flow rates, evenly distributed, with at least three in the specified flow rate interval (1.1 to 1.7 m 3/min [39-60 ft 3/min]), are required.
9.2.6 Measure and record the certification data on a form similar to the one illustrated in Figure 4 according to the following steps.
9.2.7 Observe the barometric pressure and record as P1 (item 8 in Figure 4).
9.2.8 Read the ambient temperature in the vicinity of the standard volume meter and record it as T1 (item 9 in Figure 4).
9.2.9 Start the blower motor, adjust the flow, and allow the system to run for at least 1 min for a constant motor speed to be attained.
9.2.10 Observe the standard volume meter reading and simultaneously start a stopwatch. Record the initial meter reading (Vi) in column 1 of Figure 4.
9.2.11 Maintain this constant flow rate until at least 3 m 3 of air have passed through the standard volume meter. Record the standard volume meter inlet pressure manometer reading as ΔP (column 5 in Figure 4), and the orifice manometer reading as ΔH (column 7 in Figure 4). Be sure to indicate the correct units of measurement.
9.2.12 After at least 3 m 3 of air have passed through the system, observe the standard volume meter reading while simultaneously stopping the stopwatch. Record the final meter reading (Vf) in column 2 and the elapsed time (t) in column 3 of Figure 4.
9.2.13 Calculate the volume measured by the standard volume meter at meter conditions of temperature and pressures as Vm = Vf−Vi. Record in column 4 of Figure 4.
9.2.14 Correct this volume to standard volume (std m 3) as follows:
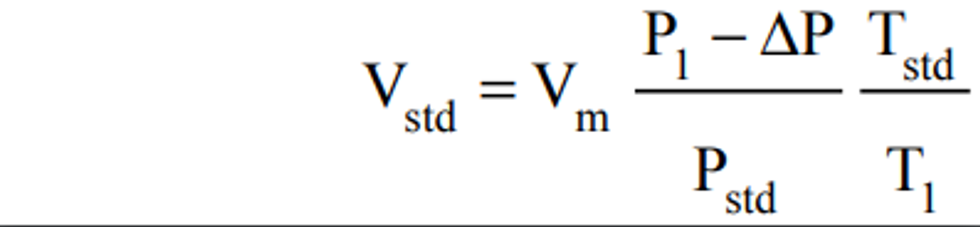
where:
Vstd = standard volume, std m 3;
Vm = actual volume measured by the standard volume meter;
P1 = barometric pressure during calibration, mm Hg or kPa;
ΔP = differential pressure at inlet to volume meter, mm Hg or kPa;
Pstd = 760 mm Hg or 101 kPa;
Tstd = 298 K;
T1 = ambient temperature during calibration, K.
Calculate the standard flow rate (std m 3/min) as follows:
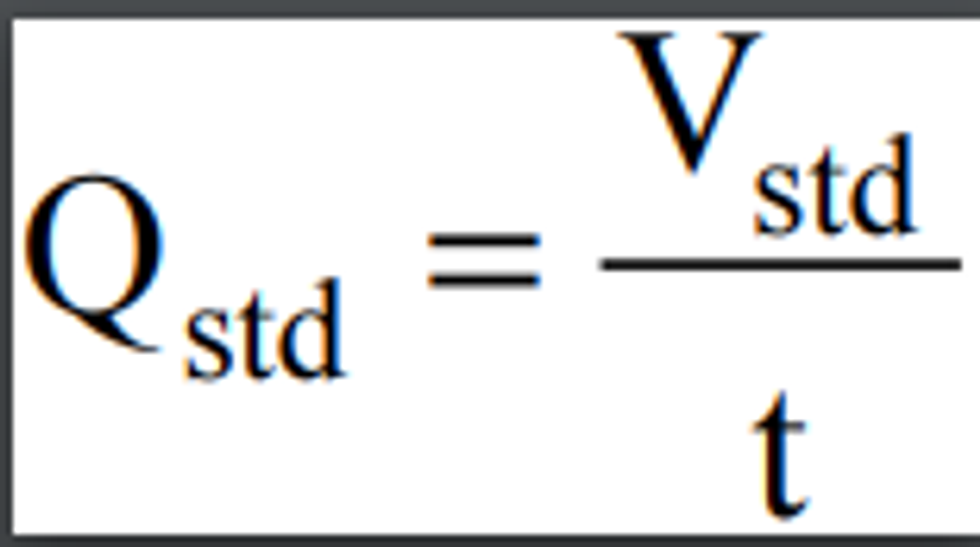
where:
Qstd = standard volumetric flow rate, std m 3/min
t = elapsed time, minutes.
Record Qstd to the nearest 0.01 std m 3/min in column 6 of Figure 4.
9.2.15 Repeat steps 9.2.9 through 9.2.14 for at least four additional constant flow rates, evenly spaced over the approximate range of 1.0 to 1.8 std m 3/min (35-64 ft 3/min).
9.2.16 For each flow, compute
√ΔΔH (P1/Pstd)(298/T1)
(column 7a of Figure 4) and plot these value against Qstd as shown in Figure 3a. Be sure to use consistent units (mm Hg or kPa) for barometric pressure. Draw the orifice transfer standard certification curve or calculate the linear least squares slope (m) and intercept (b) of the certification curve:
√ΔΔH (P1/Pstd)(298/T1)
= mQstd + b. See Figures 3 and 4. A certification graph should be readable to 0.02 std m 3/min.
9.2.17 Recalibrate the transfer standard annually or as required by applicable quality control procedures. (See Reference 2.)
9.3 Calibration of sampler flow indicator.
Note:
For samplers equipped with a flow controlling device, the flow controller must be disabled to allow flow changes during calibration of the sampler's flow indicator, or the alternate calibration of the flow controller given in 9.4 may be used. For samplers using an orifice-type flow indicator downstream of the motor, do not vary the flow rate by adjusting the voltage or power supplied to the sampler.
9.3.1 A form similar to the one illustrated in Figure 5 should be used to record the calibration data.
9.3.2 Connect the transfer standard to the inlet of the sampler. Connect the orifice manometer to the orifice pressure tap, as illustrated in Figure 3b. Make sure there are no leaks between the orifice unit and the sampler.
9.3.3 Operate the sampler for at least 5 minutes to establish thermal equilibrium prior to the calibration.
9.3.4 Measure and record the ambient temperature, T2, and the barometric pressure, P2, during calibration.
9.3.5 Adjust the variable resistance or, if applicable, insert the appropriate resistance plate (or no plate) to achieve the desired flow rate.
9.3.6 Let the sampler run for at least 2 min to re-establish the run-temperature conditions. Read and record the pressure drop across the orifice (ΔH) and the sampler flow rate indication (I) in the appropriate columns of Figure 5.
9.3.7 Calculate √ΔΔH(P2/Pstd)(298/T2) and determine the flow rate at standard conditions (Qstd) either graphically from the certification curve or by calculating Qstd from the least square slope and intercept of the transfer standard's transposed certification curve: Qstd = 1/m √ΔH(P2/Pstd)(298/T2)−b. Record the value of Qstd on Figure 5.
9.3.8 Repeat steps 9.3.5, 9.3.6, and 9.3.7 for several additional flow rates distributed over a range that includes 1.1 to 1.7 std m 3/min.
9.3.9 Determine the calibration curve by plotting values of the appropriate expression involving I, selected from table 1, against Qstd. The choice of expression from table 1 depends on the flow rate measurement device used (see Section 7.4.1) and also on whether the calibration curve is to incorporate geographic average barometric pressure (Pa) and seasonal average temperature (Ta) for the site to approximate actual pressure and temperature. Where Pa and Ta can be determined for a site for a seasonal period such that the actual barometric pressure and temperature at the site do not vary by more than ±60 mm Hg (8 kPa) from Pa or ±15°C from Ta, respectively, then using Pa and Ta avoids the need for subsequent pressure and temperature calculation when the sampler is used. The geographic average barometric pressure (Pa) may be estimated from an altitude-pressure table or by making an (approximate) elevation correction of −26 mm Hg (−3.46 kPa) for each 305 m (1,000 ft) above sea level (760 mm Hg or 101 kPa). The seasonal average temperature (Ta) may be estimated from weather station or other records. Be sure to use consistent units (mm Hg or kPa) for barometric pressure.
9.3.10 Draw the sampler calibration curve or calculate the linear least squares slope (m), intercept (b), and correlation coefficient of the calibration curve: [Expression from table 1]= mQstd + b. See Figures 3 and 5. Calibration curves should be readable to 0.02 std m 3/min.
9.3.11 For a sampler equipped with a flow controller, the flow controlling mechanism should be re-enabled and set to a flow near the lower flow limit to allow maximum control range. The sample flow rate should be verified at this time with a clean filter installed. Then add two or more filters to the sampler to see if the flow controller maintains a constant flow; this is particularly important at high altitudes where the range of the flow controller may be reduced.
9.4 Alternate calibration of flow-controlled samplers. A flow-controlled sampler may be calibrated solely at its controlled flow rate, provided that previous operating history of the sampler demonstrates that the flow rate is stable and reliable. In this case, the flow indicator may remain uncalibrated but should be used to indicate any relative change between initial and final flows, and the sampler should be recalibrated more often to minimize potential loss of samples because of controller malfunction.
9.4.1 Set the flow controller for a flow near the lower limit of the flow range to allow maximum control range.
9.4.2 Install a clean filter in the sampler and carry out steps 9.3.2, 9.3.3, 9.3.4, 9.3.6, and 9.3.7.
9.4.3 Following calibration, add one or two additional clean filters to the sampler, reconnect the transfer standard, and operate the sampler to verify that the controller maintains the same calibrated flow rate; this is particularly important at high altitudes where the flow control range may be reduced.
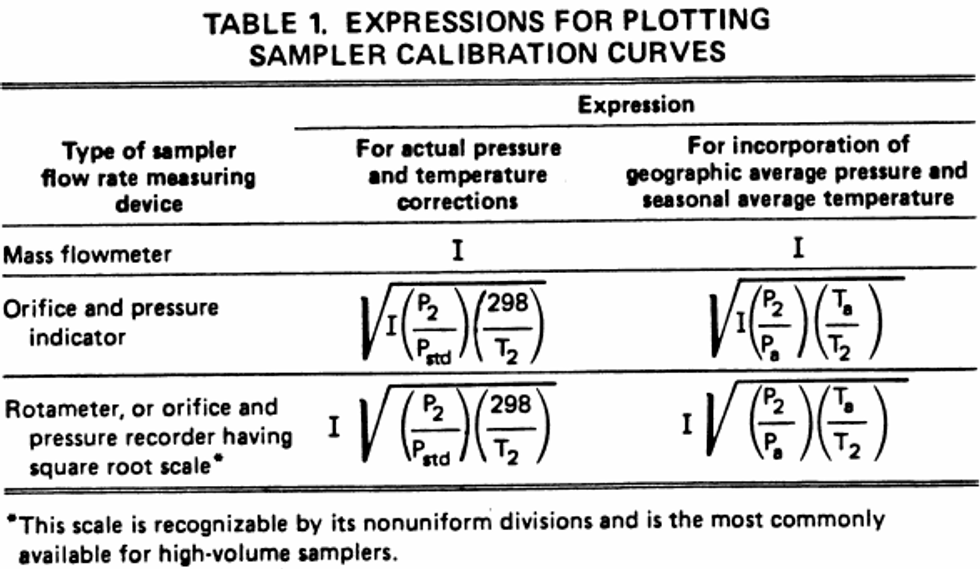

10.0 Calculations of TSP Concentration.
10.1 Determine the average sampler flow rate during the sampling period according to either 10.1.1 or 10.1.2 below.
10.1.1 For a sampler without a continuous flow recorder, determine the appropriate expression to be used from table 2 corresponding to the one from table 1 used in step 9.3.9. Using this appropriate expression, determine Qstd for the initial flow rate from the sampler calibration curve, either graphically or from the transposed regression equation:
Qstd =
1/m ([Appropriate expression from table 2]−b)
Similarly, determine Qstd from the final flow reading, and calculate the average flow Qstd as one-half the sum of the initial and final flow rates.
10.1.2 For a sampler with a continuous flow recorder, determine the average flow rate device reading, I, for the period. Determine the appropriate expression from table 2 corresponding to the one from table 1 used in step 9.3.9. Then using this expression and the average flow rate reading, determine Qstd from the sampler calibration curve, either graphically or from the transposed regression equation:
Qstd =
1/m ([Appropriate expression from table 2]−b)
If the trace shows substantial flow change during the sampling period, greater accuracy may be achieved by dividing the sampling period into intervals and calculating an average reading before determining Qstd.
10.2 Calculate the total air volume sampled as:
V − Qstd × t
where:
V = total air volume sampled, in standard volume units, std m 3/;
Qstd = average standard flow rate, std m 3/min;
t = sampling time, min.
10.3 Calculate and report the particulate matter concentration as:
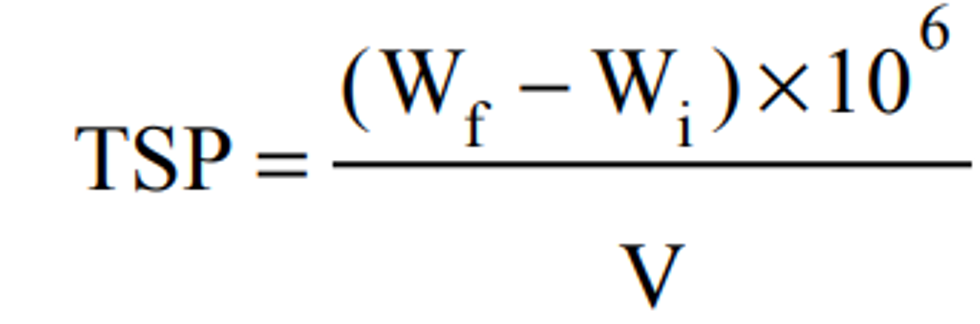
where:
TSP = mass concentration of total suspended particulate matter, µg/std m 3;
Wi = initial weight of clean filter, g;
Wf = final weight of exposed filter, g;
V = air volume sampled, converted to standard conditions, std m 3,
10 6 = conversion of g to µg.
10.4 If desired, the actual particulate matter concentration (see Section 2.2) can be calculated as follows:
(TSP)a = TSP (P3/Pstd)(298/T3)
where:
(TSP)a = actual concentration at field conditions, µg/m 3;
TSP = concentration at standard conditions, µg/std m 3;
P3 = average barometric pressure during sampling period, mm Hg;
Pstd = 760 mn Hg (or 101 kPa);
T3 = average ambient temperature during sampling period, K.
11.0 References.
1. Quality Assurance Handbook for Air Pollution Measurement Systems, Volume I, Principles. EPA-600/9-76-005, U.S. Environmental Protection Agency, Research Triangle Park, NC 27711, 1976.
2. Quality Assurance Handbook for Air Pollution Measurement Systems, Volume II, Ambient Air Specific Methods. EPA-600/4-77-027a, U.S. Environmental Protection Agency, Research Triangle Park, NC 27711, 1977.
3. Wedding, J. B., A. R. McFarland, and J. E. Cernak. Large Particle Collection Characteristics of Ambient Aerosol Samplers. Environ. Sci. Technol. 11:387-390, 1977.
4. McKee, H. C., et al. Collaborative Testing of Methods to Measure Air Pollutants, I. The High-Volume Method for Suspended Particulate Matter. J. Air Poll. Cont. Assoc., 22 (342), 1972.
5. Clement, R. E., and F. W. Karasek. Sample Composition Changes in Sampling and Analysis of Organic Compounds in Aerosols. The Intern. J. Environ. Anal. Chem., 7:109, 1979.
6. Lee, R. E., Jr., and J. Wagman. A Sampling Anomaly in the Determination of Atmospheric Sulfuric Concentration. Am. Ind. Hygiene Assoc. J., 27:266, 1966.
7. Appel, B. R., et al. Interference Effects in Sampling Particulate Nitrate in Ambient Air. Atmospheric Environment, 13:319, 1979.
8. Tierney, G. P., and W. D. Conner. Hygroscopic Effects on Weight Determinations of Particulates Collected on Glass-Fiber Filters. Am. Ind. Hygiene Assoc. J., 28:363, 1967.
9. Chahal, H. S., and D. J. Romano. High-Volume Sampling Effect of Windborne Particulate Matter Deposited During Idle Periods. J. Air Poll. Cont. Assoc., Vol. 26 (885), 1976.
10. Patterson, R. K. Aerosol Contamination from High-Volume Sampler Exhaust. J. Air Poll. Cont. Assoc., Vol. 30 (169), 1980.
11. EPA Test Procedures for Determining pH and Integrity of High-Volume Air Filters. QAD/M-80.01. Available from the Methods Standardization Branch, Quality Assurance Division, Environmental Monitoring Systems Laboratory (MD-77), U.S. Environmental Protection Agency, Research Triangle Park, NC 27711, 1980.
12. Smith, F., P. S. Wohlschlegel, R. S. C. Rogers, and D. J. Mulligan. Investigation of Flow Rate Calibration Procedures Associated with the High-Volume Method for Determination of Suspended Particulates. EPA-600/4-78-047, U.S. Environmental Protection Agency, Research Triangle Park, NC, June 1978XXXX.
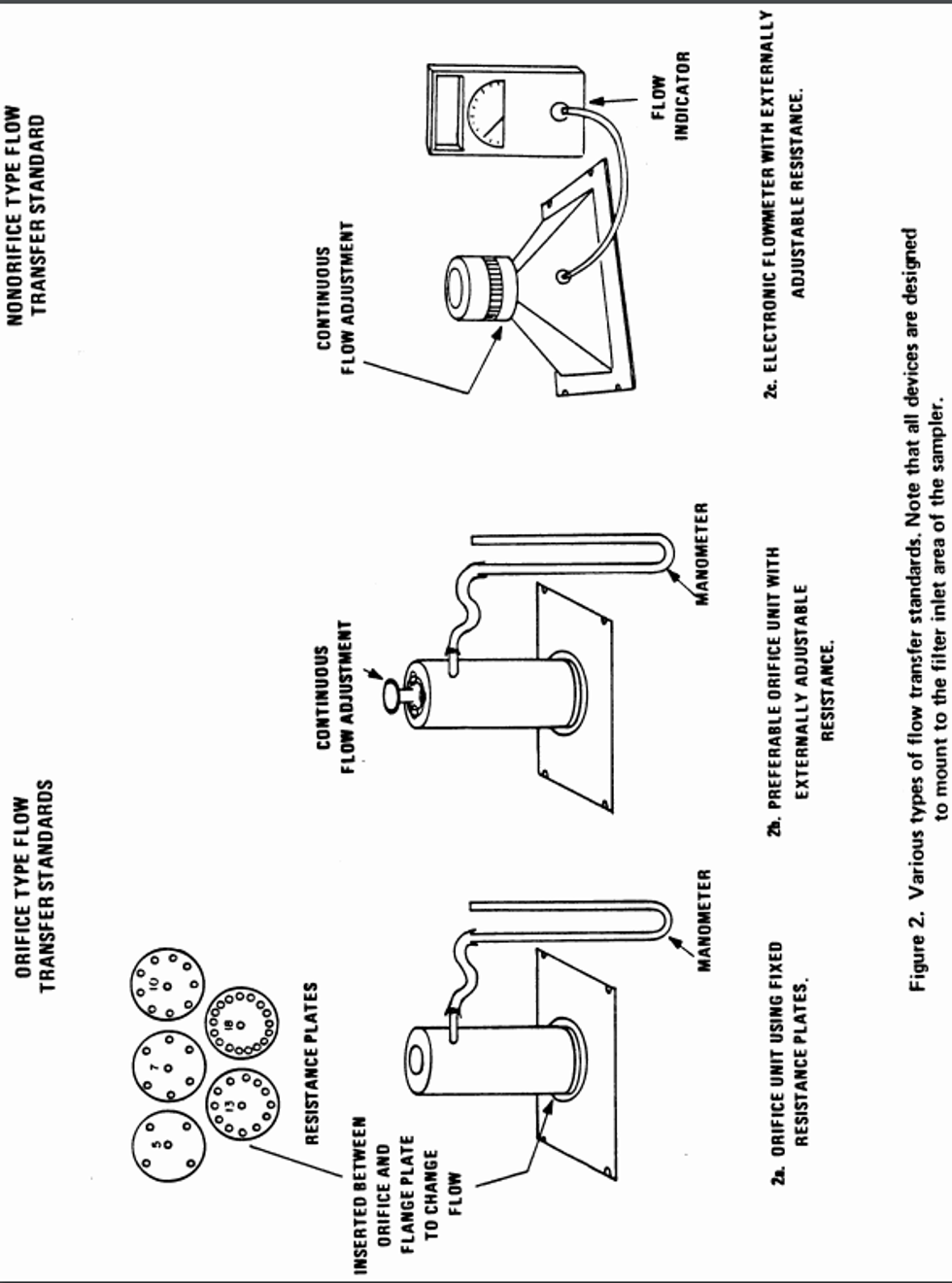

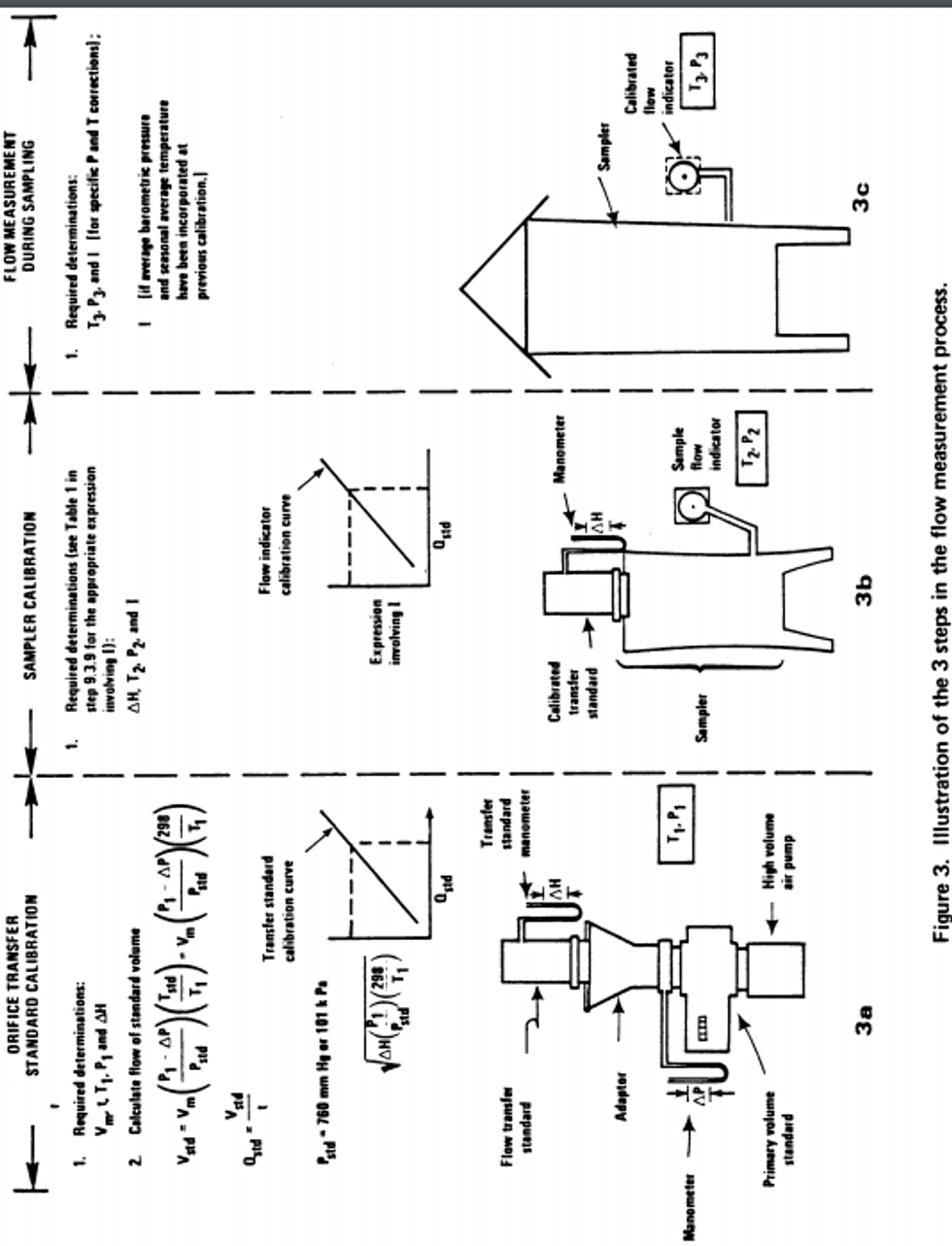


[47 FR 54912, Dec. 6, 1982; 48 FR 17355, Apr. 22, 1983]
Appendix C to Part 50 - Measurement Principle and Calibration Procedure for the Measurement of Carbon Monoxide in the Atmosphere (Non-Dispersive Infrared Photometry)
1.0 Applicability
1.1 This non-dispersive infrared photometry (NDIR) Federal Reference Method (FRM) provides measurements of the concentration of carbon monoxide (CO) in ambient air for determining compliance with the primary and secondary National Ambient Air Quality Standards (NAAQS) for CO as specified in §50.8 of this chapter. The method is applicable to continuous sampling and measurement of ambient CO concentrations suitable for determining 1-hour or longer average measurements. The method may also provide measurements of shorter averaging times, subject to specific analyzer performance limitations. Additional CO monitoring quality assurance procedures and guidance are provided in part 58, appendix A, of this chapter and in reference 1 of this appendix C.
2.0 Measurement Principle
2.1 Measurements of CO in ambient air are based on automated measurement of the absorption of infrared radiation by CO in an ambient air sample drawn into an analyzer employing non-wavelength-dispersive, infrared photometry (NDIR method). Infrared energy from a source in the photometer is passed through a cell containing the air sample to be analyzed, and the quantitative absorption of energy by CO in the sample cell is measured by a suitable detector. The photometer is sensitized specifically to CO by employing CO gas in a filter cell in the optical path, which, when compared to a differential optical path without a CO filter cell, limits the measured absorption to one or more of the characteristic wavelengths at which CO strongly absorbs. However, to meet measurement performance requirements, various optical filters, reference cells, rotating gas filter cells, dual-beam configurations, moisture traps, or other means may also be used to further enhance sensitivity and stability of the photometer and to minimize potential measurement interference from water vapor, carbon dioxide (CO2), or other species. Also, various schemes may be used to provide a suitable zero reference for the photometer, and optional automatic compensation may be provided for the actual pressure and temperature of the air sample in the measurement cell. The measured infrared absorption, converted to a digital reading or an electrical output signal, indicates the measured CO concentration.
2.2 The measurement system is calibrated by referencing the analyzer's CO measurements to CO concentration standards traceable to a National Institute of Standards and Technology (NIST) primary standard for CO, as described in the associated calibration procedure specified in section 4 of this reference method.
2.3 An analyzer implementing this measurement principle will be considered a reference method only if it has been designated as a reference method in accordance with §53.11 of this chapter.
2.4 Sampling considerations. The use of a particle filter in the sample inlet line of a CO FRM analyzer is optional and left to the discretion of the user unless such a filter is specified or recommended by the analyzer manufacturer in the analyzer's associated operation or instruction manual.
3.0 Interferences
3.1 The NDIR measurement principle is potentially susceptible to interference from water vapor and CO2, which have some infrared absorption at wavelengths in common with CO and normally exist in the atmosphere. Various instrumental techniques can be used to effectively minimize these interferences.
4.0 Calibration Procedures
4.1 Principle. Either of two methods may be selected for dynamic multipoint calibration of FRM CO analyzers, using test gases of accurately known CO concentrations obtained from one or more compressed gas cylinders certified as CO transfer standards:
4.1.1 Dilution method: A single certified standard cylinder of CO is quantitatively diluted as necessary with zero air to obtain the various calibration concentration standards needed.
4.1.2 Multiple-cylinder method: Multiple, individually certified standard cylinders of CO are used for each of the various calibration concentration standards needed.
4.1.3 Additional information on calibration may be found in Section 12 of reference 1.
4.2 Apparatus. The major components and typical configurations of the calibration systems for the two calibration methods are shown in Figures 1 and 2. Either system may be made up using common laboratory components, or it may be a commercially manufactured system. In either case, the principal components are as follows:
4.2.1 CO standard gas flow control and measurement devices (or a combined device) capable of regulating and maintaining the standard gas flow rate constant to within ±2 percent and measuring the gas flow rate accurate to within ±2 percent, properly calibrated to a NIST-traceable standard.
4.2.2 For the dilution method (Figure 1), dilution air flow control and measurement devices (or a combined device) capable of regulating and maintaining the air flow rate constant to within ±2 percent and measuring the air flow rate accurate to within ±2 percent, properly calibrated to a NIST-traceable standard.
4.2.3 Standard gas pressure regulator(s) for the standard CO cylinder(s), suitable for use with a high-pressure CO gas cylinder and having a non-reactive diaphragm and internal parts and a suitable delivery pressure.
4.2.4 Mixing chamber for the dilution method of an inert material and of proper design to provide thorough mixing of CO standard gas and diluent air streams.
4.2.5 Output sampling manifold, constructed of an inert material and of sufficient diameter to ensure an insignificant pressure drop at the analyzer connection. The system must have a vent designed to ensure nearly atmospheric pressure at the analyzer connection port and to prevent ambient air from entering the manifold.
4.3 Reagents
4.3.1 CO gas concentration transfer standard(s) of CO in air, containing an appropriate concentration of CO suitable for the selected operating range of the analyzer under calibration and traceable to a NIST standard reference material (SRM). If the CO analyzer has significant sensitivity to CO2, the CO standard(s) should also contain 350 to 400 ppm CO2 to replicate the typical CO2 concentration in ambient air. However, if the zero air dilution ratio used for the dilution method is not less than 100:1 and the zero air contains ambient levels of CO2, then the CO standard may be contained in nitrogen and need not contain CO2.
4.3.2 For the dilution method, clean zero air, free of contaminants that could cause a detectable response on or a change in sensitivity of the CO analyzer. The zero air should contain <0.1 ppm CO.
4.4 Procedure Using the Dilution Method
4.4.1 Assemble or obtain a suitable dynamic dilution calibration system such as the one shown schematically in Figure 1. Generally, all calibration gases including zero air must be introduced into the sample inlet of the analyzer. However, if the analyzer has special, approved zero and span inlets and automatic valves to specifically allow introduction of calibration standards at near atmospheric pressure, such inlets may be used for calibration in lieu of the sample inlet. For specific operating instructions, refer to the manufacturer's manual.
4.4.2 Ensure that there are no leaks in the calibration system and that all flowmeters are properly and accurately calibrated, under the conditions of use, if appropriate, against a reliable volume or flow rate standard such as a soap-bubble meter or wet-test meter traceable to a NIST standard. All volumetric flow rates should be corrected to the same temperature and pressure such as 298.15 K (25°C) and 760 mm Hg (101 kPa), using a correction formula such as the following:
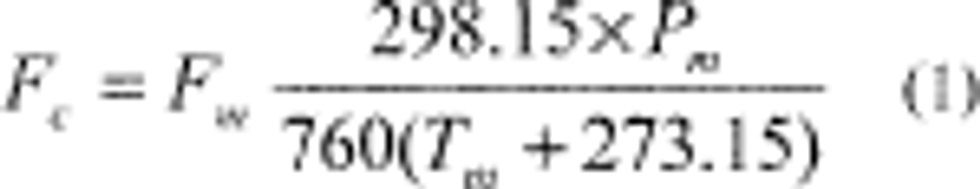
Where:
Fc = corrected flow rate (L/min at 25°C and 760 mm Hg),
Fm = measured flow rate (at temperature Tm and pressure Pm),
Pm = measured pressure in mm Hg (absolute), and
Tm = measured temperature in degrees Celsius.
4.4.3 Select the operating range of the CO analyzer to be calibrated. Connect the measurement signal output of the analyzer to an appropriate readout instrument to allow the analyzer's measurement output to be continuously monitored during the calibration. Where possible, this readout instrument should be the same one used to record routine monitoring data, or, at least, an instrument that is as closely representative of that system as feasible.
4.4.4 Connect the inlet of the CO analyzer to the output-sampling manifold of the calibration system.
4.4.5 Adjust the calibration system to deliver zero air to the output manifold. The total air flow must exceed the total demand of the analyzer(s) connected to the output manifold to ensure that no ambient air is pulled into the manifold vent. Allow the analyzer to sample zero air until a stable response is obtained. After the response has stabilized, adjust the analyzer zero reading.
4.4.6 Adjust the zero air flow rate and the CO gas flow rate from the standard CO cylinder to provide a diluted CO concentration of approximately 80 percent of the measurement upper range limit (URL) of the operating range of the analyzer. The total air flow rate must exceed the total demand of the analyzer(s) connected to the output manifold to ensure that no ambient air is pulled into the manifold vent. The exact CO concentration is calculated from:
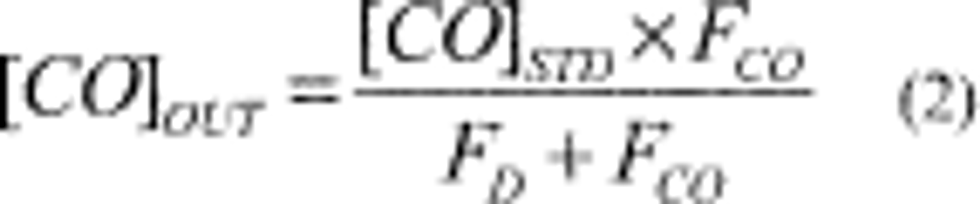
Where:
[CO]OUT = diluted CO concentration at the output manifold (ppm),
[CO]STD = concentration of the undiluted CO standard (ppm),
FCO = flow rate of the CO standard (L/min), and
FD = flow rate of the dilution air (L/min).
Sample this CO concentration until a stable response is obtained. Adjust the analyzer span control to obtain the desired analyzer response reading equivalent to the calculated standard concentration. If substantial adjustment of the analyzer span control is required, it may be necessary to recheck the zero and span adjustments by repeating steps 4.4.5 and 4.4.6. Record the CO concentration and the analyzer's final response.
4.4.7 Generate several additional concentrations (at least three evenly spaced points across the remaining scale are suggested to verify linearity) by decreasing FCO or increasing FD. Be sure the total flow exceeds the analyzer's total flow demand. For each concentration generated, calculate the exact CO concentration using equation (2). Record the concentration and the analyzer's stable response for each concentration. Plot the analyzer responses (vertical or y-axis) versus the corresponding CO concentrations (horizontal or x-axis). Calculate the linear regression slope and intercept of the calibration curve and verify that no point deviates from this line by more than 2 percent of the highest concentration tested.
4.5 Procedure Using the Multiple-Cylinder Method. Use the procedure for the dilution method with the following changes:
4.5.1 Use a multi-cylinder, dynamic calibration system such as the typical one shown in Figure 2.
4.5.2 The flowmeter need not be accurately calibrated, provided the flow in the output manifold can be verified to exceed the analyzer's flow demand.
4.5.3 The various CO calibration concentrations required in Steps 4.4.5, 4.4.6, and 4.4.7 are obtained without dilution by selecting zero air or the appropriate certified standard cylinder.
4.6 Frequency of Calibration. The frequency of calibration, as well as the number of points necessary to establish the calibration curve and the frequency of other performance checking, will vary by analyzer. However, the minimum frequency, acceptance criteria, and subsequent actions are specified in reference 1, appendix D, “Measurement Quality Objectives and Validation Template for CO” (page 5 of 30). The user's quality control program should provide guidelines for initial establishment of these variables and for subsequent alteration as operational experience is accumulated. Manufacturers of CO analyzers should include in their instruction/operation manuals information and guidance as to these variables and on other matters of operation, calibration, routine maintenance, and quality control.
5.0 Reference
1. QA Handbook for Air Pollution Measurement Systems - Volume II. Ambient Air Quality Monitoring Program. U.S. EPA. EPA-454/B-08-003 (2008).
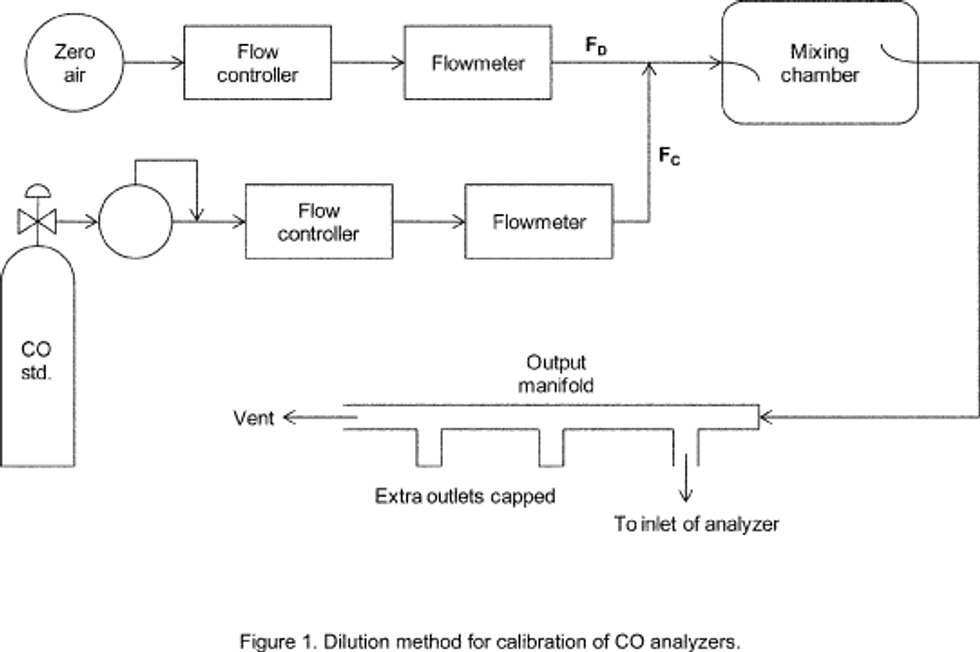
[76 FR 54323, Aug. 31, 2011]
Appendix D to Part 50 - Reference Measurement Principle and Calibration Procedure for the Measurement of Ozone in the Atmosphere (Chemiluminescence Method)
1.0 Applicability.
1.1 This chemiluminescence method provides reference measurements of the concentration of ozone (O3) in ambient air for determining compliance with the national primary and secondary ambient air quality standards for O3 as specified in 40 CFR part 50. This automated method is applicable to the measurement of ambient O3 concentrations using continuous (real-time) sampling and analysis. Additional quality assurance procedures and guidance are provided in 40 CFR part 58, appendix A, and in Reference 14.
2.0 Measurement Principle.
2.1 This reference method is based on continuous automated measurement of the intensity of the characteristic chemiluminescence released by the gas phase reaction of O3 in sampled air with either ethylene (C2H4) or nitric oxide (NO) gas. An ambient air sample stream and a specific flowing concentration of either C2H4 (ET-CL method) or NO (NO-CL method) are mixed in a measurement cell, where the resulting chemiluminescence is quantitatively measured by a sensitive photo-detector. References 8-11 describe the chemiluminescence measurement principle.
2.2 The measurement system is calibrated by referencing the instrumental chemiluminescence measurements to certified O 3 standard concentrations generated in a dynamic flow system and assayed by ultraviolet (UV) photometry to be traceable to a National Institute of Standards and Technology (NIST) standard reference photometer for O 3 (see section 4, Calibration Procedure, below) with a specified ozone absorption cross-section value. The absorption cross-section value stated in section 4.1 and section 4.5.3.10 of this appendix (304.39 atm −1 cm −1 ± 0.94 atm −1 cm −1 ) will be implemented January 1, 2025, with an additional year to complete implementation (January 1, 2026). Until January 1, 2025, the previous ozone absorption cross-section value, 308 ± 4 atm −1 cm −1 , will be used. After January 1, 2025, both cross-section values, 304.39 ± 0.94 atm −1 cm −1 and 308 ± 4 atm −1 cm −1 , may be used. After January 1, 2026, only the cross-section value of 304.39 ± 0.94 atm −1 cm −1 may be used.
2.3 An analyzer implementing this measurement principle is shown schematically in Figure 1. Designs implementing this measurement principle must include: an appropriately designed mixing and measurement cell; a suitable quantitative photometric measurement system with adequate sensitivity and wavelength specificity for O3; a pump, flow control, and sample conditioning system for sampling the ambient air and moving it into and through the measurement cell; a sample air dryer as necessary to meet the water vapor interference limit requirement specified in subpart B of §53.11 of this chapter; a means to supply, meter, and mix a constant, flowing stream of either C2H4 or NO gas of fixed concentration with the sample air flow in the measurement cell; suitable electronic control and measurement processing capability; and other associated apparatus as may be necessary. The analyzer must be designed and constructed to provide accurate, repeatable, and continuous measurements of O3 concentrations in ambient air, with measurement performance that meets the requirements specified in subpart B of §53.11 of this chapter.
2.4 An analyzer implementing this measurement principle and calibration procedure will be considered a federal reference method (FRM) only if it has been designated as a reference method in accordance with §53.11 of this chapter.
2.5 Sampling considerations. The use of a particle filter on the sample inlet line of a chemiluminescence O3 FRM analyzer is required to prevent buildup of particulate matter in the measurement cell and inlet components. This filter must be changed weekly (or at least often as specified in the manufacturer's operation/instruction manual), and the sample inlet system used with the analyzer must be kept clean, to avoid loss of O3 in the O3 sample air prior to the concentration measurement.
3.0 Interferences.
3.1 Except as described in 3.2 below, the chemiluminescence measurement system is inherently free of significant interferences from other pollutant substances that may be present in ambient air.
3.2 A small sensitivity to variations in the humidity of the sample air is minimized by a sample air dryer. Potential loss of O3 in the inlet air filter and in the air sample handling components of the analyzer and associated exterior air sampling components due to buildup of airborne particulate matter is minimized by filter replacement and cleaning of the other inlet components.
4.0 Calibration Procedure.
4.1 Principle. The calibration procedure is based on the photometric assay of O 3 concentrations in a dynamic flow system. The concentration of O 3 in an absorption cell is determined from a measurement of the amount of 254 nm light absorbed by the sample. This determination requires knowledge of (1) the absorption coefficient (α) of O 3 at 254 nm, (2) the optical path length (l) through the sample, (3) the transmittance of the sample at a nominal wavelength of 254 nm, and (4) the temperature (T) and pressure (P) of the sample. The transmittance is defined as the ratio I/I 0, where I is the intensity of light which passes through the cell and is sensed by the detector when the cell contains an O 3 sample, and I 0 is the intensity of light which passes through the cell and is sensed by the detector when the cell contains zero air. It is assumed that all conditions of the system, except for the contents of the absorption cell, are identical during measurement of I and I 0. The quantities defined above are related by the Beer-Lambert absorption law,
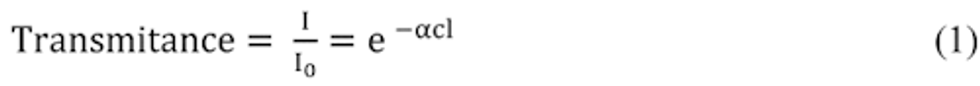
Where:
α = absorption coefficient of O 3 at 254 nm = 304.39 atm −1 cm −1 , with an uncertainty of 0.94 atm −1 cm −1 at 0°C and 1 atm. 1, 2, 3, 4, 5, 6, 7, 15
c = O 3 concentration in atmospheres, and
l = optical path length in cm.
A stable O 3 generator is used to produce O 3 concentrations over the required calibration concentration range. Each O 3 concentration is determined from the measurement of the transmittance (I/I 0) of the sample at 254 nm with a photometer of path length l and calculated from the equation,
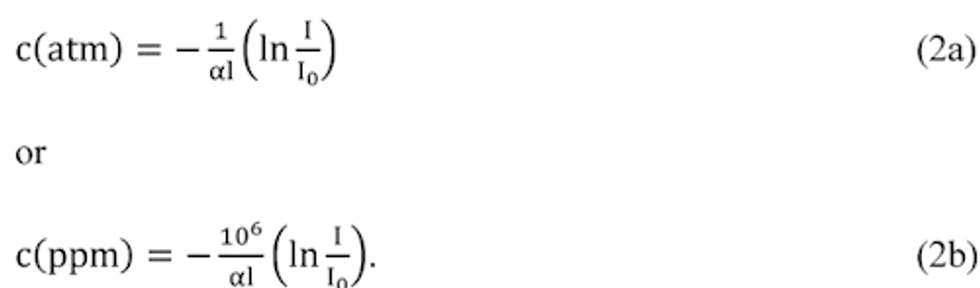
The calculated O 3 concentrations must be corrected for O 3 losses, which may occur in the photometer, and for the temperature and pressure of the sample.
4.2 Applicability. This procedure is applicable to the calibration of ambient air O3 analyzers, either directly or by means of a transfer standard certified by this procedure. Transfer standards must meet the requirements and specifications set forth in Reference 12.
4.3 Apparatus. A complete UV calibration system consists of an O3 generator, an output port or manifold, a photometer, an appropriate source of zero air, and other components as necessary. The configuration must provide a stable O3 concentration at the system output and allow the photometer to accurately assay the output concentration to the precision specified for the photometer (4.3.1). Figure 2 shows a commonly used configuration and serves to illustrate the calibration procedure, which follows. Other configurations may require appropriate variations in the procedural steps. All connections between components in the calibration system downstream of the O3 generator must be of glass, Teflon, or other relatively inert materials. Additional information regarding the assembly of a UV photometric calibration apparatus is given in Reference 13. For certification of transfer standards which provide their own source of O3, the transfer standard may replace the O3 generator and possibly other components shown in Figure 2; see Reference 12 for guidance.
4.3.1 UV photometer. The photometer consists of a low-pressure mercury discharge lamp, (optional) collimation optics, an absorption cell, a detector, and signal-processing electronics, as illustrated in Figure 2. It must be capable of measuring the transmittance, I/I0, at a wavelength of 254 nm with sufficient precision such that the standard deviation of the concentration measurements does not exceed the greater of 0.005 ppm or 3% of the concentration. Because the low-pressure mercury lamp radiates at several wavelengths, the photometer must incorporate suitable means to assure that no O3 is generated in the cell by the lamp, and that at least 99.5% of the radiation sensed by the detector is 254 nm radiation. (This can be readily achieved by prudent selection of optical filter and detector response characteristics.) The length of the light path through the absorption cell must be known with an accuracy of at least 99.5%. In addition, the cell and associated plumbing must be designed to minimize loss of O3 from contact with cell walls and gas handling components. See Reference 13 for additional information.
4.3.2 Air flow controllers. Air flow controllers are devices capable of regulating air flows as necessary to meet the output stability and photometer precision requirements.
4.3.3 Ozone generator. The ozone generator used must be capable of generating stable levels of O3 over the required concentration range.
4.3.4 Output manifold. The output manifold must be constructed of glass, Teflon, or other relatively inert material, and should be of sufficient diameter to insure a negligible pressure drop at the photometer connection and other output ports. The system must have a vent designed to insure atmospheric pressure in the manifold and to prevent ambient air from entering the manifold.
4.3.5 Two-way valve. A manual or automatic two-way valve, or other means is used to switch the photometer flow between zero air and the O3 concentration.
4.3.6 Temperature indicator. A device to indicate temperature must be used that is accurate to ±1°C.
4.3.7 Barometer or pressure indicator. A device to indicate barometric pressure must be used that is accurate to ±2 torr.
4.4 Reagents.
4.4.1 Zero air. The zero air must be free of contaminants which would cause a detectable response from the O3 analyzer, and it must be free of NO, C2H4, and other species which react with O3. A procedure for generating suitable zero air is given in Reference 13. As shown in Figure 2, the zero air supplied to the photometer cell for the I0 reference measurement must be derived from the same source as the zero air used for generation of the O3 concentration to be assayed (I measurement). When using the photometer to certify a transfer standard having its own source of O3, see Reference 12 for guidance on meeting this requirement.
4.5 Procedure.
4.5.1 General operation. The calibration photometer must be dedicated exclusively to use as a calibration standard. It must always be used with clean, filtered calibration gases, and never used for ambient air sampling. A number of advantages are realized by locating the calibration photometer in a clean laboratory where it can be stationary, protected from the physical shock of transportation, operated by a responsible analyst, and used as a common standard for all field calibrations via transfer standards.
4.5.2 Preparation. Proper operation of the photometer is of critical importance to the accuracy of this procedure. Upon initial operation of the photometer, the following steps must be carried out with all quantitative results or indications recorded in a chronological record, either in tabular form or plotted on a graphical chart. As the performance and stability record of the photometer is established, the frequency of these steps may be reduced to be consistent with the documented stability of the photometer and the guidance provided in Reference 12.
4.5.2.1 Instruction manual. Carry out all set up and adjustment procedures or checks as described in the operation or instruction manual associated with the photometer.
4.5.2.2 System check. Check the photometer system for integrity, leaks, cleanliness, proper flow rates, etc. Service or replace filters and zero air scrubbers or other consumable materials, as necessary.
4.5.2.3 Linearity. Verify that the photometer manufacturer has adequately established that the linearity error of the photometer is less than 3%, or test the linearity by dilution as follows: Generate and assay an O3 concentration near the upper range limit of the system or appropriate calibration scale for the instrument, then accurately dilute that concentration with zero air and re-assay it. Repeat at several different dilution ratios. Compare the assay of the original concentration with the assay of the diluted concentration divided by the dilution ratio, as follows

Where:
E = linearity error, percent
A1 = assay of the original concentration
A2 = assay of the diluted concentration
R = dilution ratio = flow of original concentration divided by the total flow
The linearity error must be less than 5%. Since the accuracy of the measured flow-rates will affect the linearity error as measured this way, the test is not necessarily conclusive. Additional information on verifying linearity is contained in Reference 13.
4.5.2.4 Inter-comparison. The photometer must be inter-compared annually, either directly or via transfer standards, with a NIST standard reference photometer (SRP) or calibration photometers used by other agencies or laboratories.
4.5.2.5 Ozone losses. Some portion of the O3 may be lost upon contact with the photometer cell walls and gas handling components. The magnitude of this loss must be determined and used to correct the calculated O3 concentration. This loss must not exceed 5%. Some guidelines for quantitatively determining this loss are discussed in Reference 13.
4.5.3 Assay of O3concentrations. The operator must carry out the following steps to properly assay O3 concentrations.
4.5.3.1 Allow the photometer system to warm up and stabilize.
4.5.3.2 Verify that the flow rate through the photometer absorption cell, F, allows the cell to be flushed in a reasonably short period of time (2 liter/min is a typical flow). The precision of the measurements is inversely related to the time required for flushing, since the photometer drift error increases with time.
4.5.3.3 Ensure that the flow rate into the output manifold is at least 1 liter/min greater than the total flow rate required by the photometer and any other flow demand connected to the manifold.
4.5.3.4 Ensure that the flow rate of zero air, Fz, is at least 1 liter/min greater than the flow rate required by the photometer.
4.5.3.5 With zero air flowing in the output manifold, actuate the two-way valve to allow the photometer to sample first the manifold zero air, then Fz. The two photometer readings must be equal (I = I0).
Note:
In some commercially available photometers, the operation of the two-way valve and various other operations in section 4.5.3 may be carried out automatically by the photometer.
4.5.3.6 Adjust the O3 generator to produce an O3 concentration as needed.
4.5.3.7 Actuate the two-way valve to allow the photometer to sample zero air until the absorption cell is thoroughly flushed and record the stable measured value of Io.
4.5.3.8 Actuate the two-way valve to allow the photometer to sample the O3 concentration until the absorption cell is thoroughly flushed and record the stable measured value of I.
4.5.3.9 Record the temperature and pressure of the sample in the photometer absorption cell. (See Reference 13 for guidance.)
4.5.3.10. Calculate the O 3 concentration from equation 4. An average of several determinations will provide better precision.
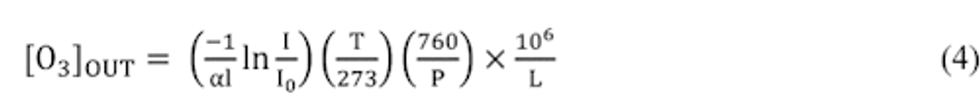
Where:
[O 3 ] OUT = O 3 concentration, ppm
α = absorption coefficient of O 3 at 254 nm = 304.39 atm −1 cm −1 at 0 °C and 1 atm
l = optical path length, cm
T = sample temperature, K
P = sample pressure, torr
L = correction factor for O 3 losses from 4.5.2.5 = (1−fraction of O 3 lost).
Note:
Some commercial photometers may automatically evaluate all or part of equation 4. It is the operator's responsibility to verify that all of the information required for equation 4 is obtained, either automatically by the photometer or manually. For “automatic” photometers which evaluate the first term of equation 4 based on a linear approximation, a manual correction may be required, particularly at higher O3 levels. See the photometer instruction manual and Reference 13 for guidance.
4.5.3.11 Obtain additional O3 concentration standards as necessary by repeating steps 4.5.3.6 to 4.5.3.10 or by Option 1.
4.5.4 Certification of transfer standards. A transfer standard is certified by relating the output of the transfer standard to one or more O3 calibration standards as determined according to section 4.5.3. The exact procedure varies depending on the nature and design of the transfer standard. Consult Reference 12 for guidance.
4.5.5 Calibration of ozone analyzers. Ozone analyzers must be calibrated as follows, using O3 standards obtained directly according to section 4.5.3 or by means of a certified transfer standard.
4.5.5.1 Allow sufficient time for the O3 analyzer and the photometer or transfer standard to warm-up and stabilize.
4.5.5.2 Allow the O3 analyzer to sample zero air until a stable response is obtained and then adjust the O3 analyzer's zero control. Offsetting the analyzer's zero adjustment to +5% of scale is recommended to facilitate observing negative zero drift (if any). Record the stable zero air response as “Z”.
4.5.5.3 Generate an O3 concentration standard of approximately 80% of the desired upper range limit (URL) of the O3 analyzer. Allow the O3 analyzer to sample this O3 concentration standard until a stable response is obtained.
4.5.5.4 Adjust the O3 analyzer's span control to obtain the desired response equivalent to the calculated standard concentration. Record the O3 concentration and the corresponding analyzer response. If substantial adjustment of the span control is necessary, recheck the zero and span adjustments by repeating steps 4.5.5.2 to 4.5.5.4.
4.5.5.5 Generate additional O3 concentration standards (a minimum of 5 are recommended) over the calibration scale of the O3 analyzer by adjusting the O3 source or by Option 1. For each O3 concentration standard, record the O3 concentration and the corresponding analyzer response.
4.5.5.6 Plot the O3 analyzer responses (vertical or Y-axis) versus the corresponding O3 standard concentrations (horizontal or X-axis). Compute the linear regression slope and intercept and plot the regression line to verify that no point deviates from this line by more than 2 percent of the maximum concentration tested.
4.5.5.7 Option 1: The various O3 concentrations required in steps 4.5.3.11 and 4.5.5.5 may be obtained by dilution of the O3 concentration generated in steps 4.5.3.6 and 4.5.5.3. With this option, accurate flow measurements are required. The dynamic calibration system may be modified as shown in Figure 3 to allow for dilution air to be metered in downstream of the O3 generator. A mixing chamber between the O3 generator and the output manifold is also required. The flow rate through the O3 generator (Fo) and the dilution air flow rate (FD) are measured with a flow or volume standard that is traceable to a NIST flow or volume calibration standard. Each O3 concentration generated by dilution is calculated from:
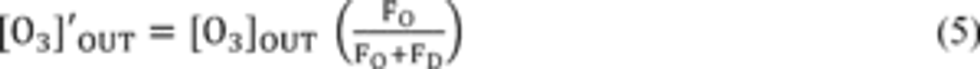
Where:
[O3]′OUT = diluted O3 concentration, ppm
FO = flow rate through the O3 generator, liter/min
FD = diluent air flow rate, liter/min
Note:
Additional information on calibration and pollutant standards is provided in Section 12 of Reference 14.
5.0 Frequency of Calibration.
5.1 The frequency of calibration, as well as the number of points necessary to establish the calibration curve, and the frequency of other performance checking will vary by analyzer; however, the minimum frequency, acceptance criteria, and subsequent actions are specified in Appendix D of Reference 14: Measurement Quality Objectives and Validation Templates. The user's quality control program shall provide guidelines for initial establishment of these variables and for subsequent alteration as operational experience is accumulated. Manufacturers of analyzers should include in their instruction/operation manuals information and guidance as to these variables and on other matters of operation, calibration, routine maintenance, and quality control.
6.0 References.
1. E.C.Y. Inn and Y. Tanaka, “Absorption coefficient of Ozone in the Ultraviolet and Visible Regions”, J. Opt. Soc. Am., 43, 870 (1953).
2. A. G. Hearn, “Absorption of Ozone in the Ultraviolet and Visible Regions of the Spectrum”, Proc. Phys. Soc. (London), 78, 932 (1961).
3. W. B. DeMore and O. Raper, “Hartley Band Extinction Coefficients of Ozone in the Gas Phase and in Liquid Nitrogen, Carbon Monoxide, and Argon”, J. Phys. Chem., 68, 412 (1964).
4. M. Griggs, “Absorption Coefficients of Ozone in the Ultraviolet and Visible Regions”, J. Chem. Phys., 49, 857 (1968).
5. K. H. Becker, U. Schurath, and H. Seitz, “Ozone Olefin Reactions in the Gas Phase. 1. Rate Constants and Activation Energies”, Int'l Jour. of Chem. Kinetics, VI, 725 (1974).
6. M. A. A. Clyne and J. A. Coxom, “Kinetic Studies of Oxy-halogen Radical Systems”, Proc. Roy. Soc., A303, 207 (1968).
7. J. W. Simons, R. J. Paur, H. A. Webster, and E. J. Bair, “Ozone Ultraviolet Photolysis. VI. The Ultraviolet Spectrum”, J. Chem. Phys., 59, 1203 (1973).
8. Ollison, W.M.; Crow, W.; Spicer, C.W. “Field testing of new-technology ambient air ozone monitors.” J. Air Waste Manage. Assoc., 63 (7), 855-863 (2013).
9. Parrish, D.D.; Fehsenfeld, F.C. “Methods for gas-phase measurements of ozone, ozone precursors and aerosol precursors.” Atmos. Environ., 34 (12-14), 1921-1957(2000).
10. Ridley, B.A.; Grahek, F.E.; Walega, J.G. “A small, high-sensitivity, medium-response ozone detector suitable for measurements from light aircraft.” J. Atmos. Oceanic Technol., 9 (2), 142-148(1992).
11. Boylan, P., Helmig, D., and Park, J.H. “Characterization and mitigation of water vapor effects in the measurement of ozone by chemiluminescence with nitric oxide.” Atmos. Meas. Tech. 7, 1231-1244 (2014).
12. Transfer Standards for Calibration of Ambient Air Monitoring Analyzers for Ozone, EPA publication number EPA-454/B-13-004, October 2013. EPA, Office of Air Quality Planning and Standards, Research Triangle Park, NC 27711. [Available at www.epa.gov/ttnamti1/files/ambient/qaqc/OzoneTransferStandardGuidance.pdf.]
13. Technical Assistance Document for the Calibration of Ambient Ozone Monitors, EPA publication number EPA–454/B–22–003, January 2023.
14. QA Handbook for Air Pollution Measurement Systems—Volume II. Ambient Air Quality Monitoring Program. EPA–454/B–17–001, January 2017.
15. Hodges, J.T., Viallon, J., Brewer, P.J., Drouin, B.J., Gorshelev, V., Janssen, C., Lee, S., Possolo, A., Smith, M.A.H., Walden, and Wielgosz, R.I., Recommendation of a consensus value of the ozone absorption cross-section at 253.65 nm based on a literature review, Metrologia, 56 (2019) 034001. [Available at https://doi.org/10.1088/1681-7575/ab0bdd.]
7.0 Figures.
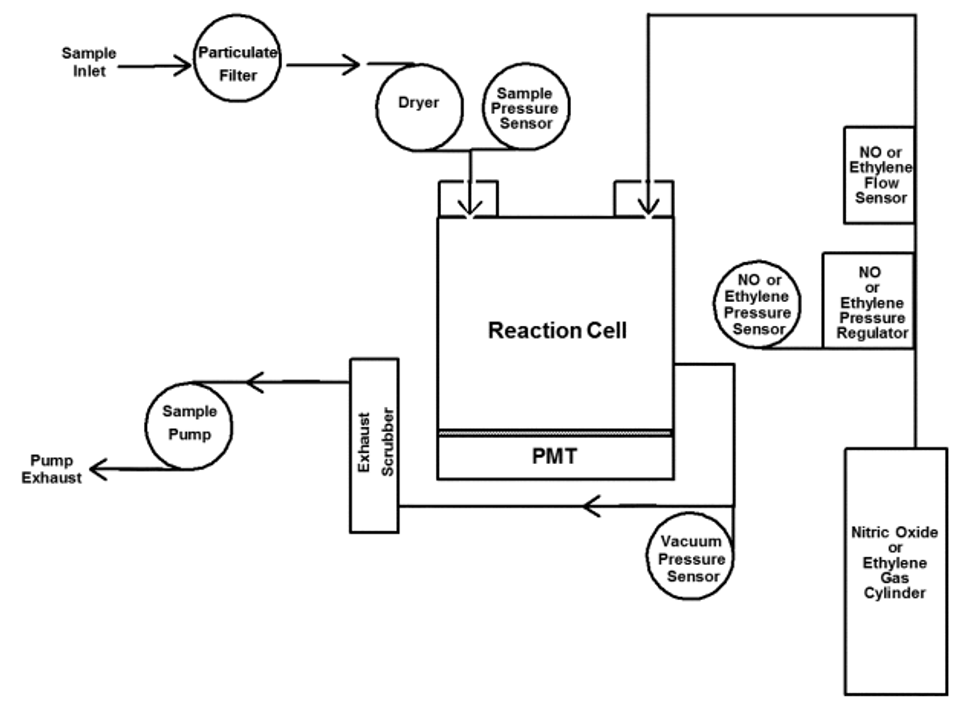
Figure 1. Gas-phase chemiluminescence analyzer schematic diagram, where PMT means photomultiplier tube.
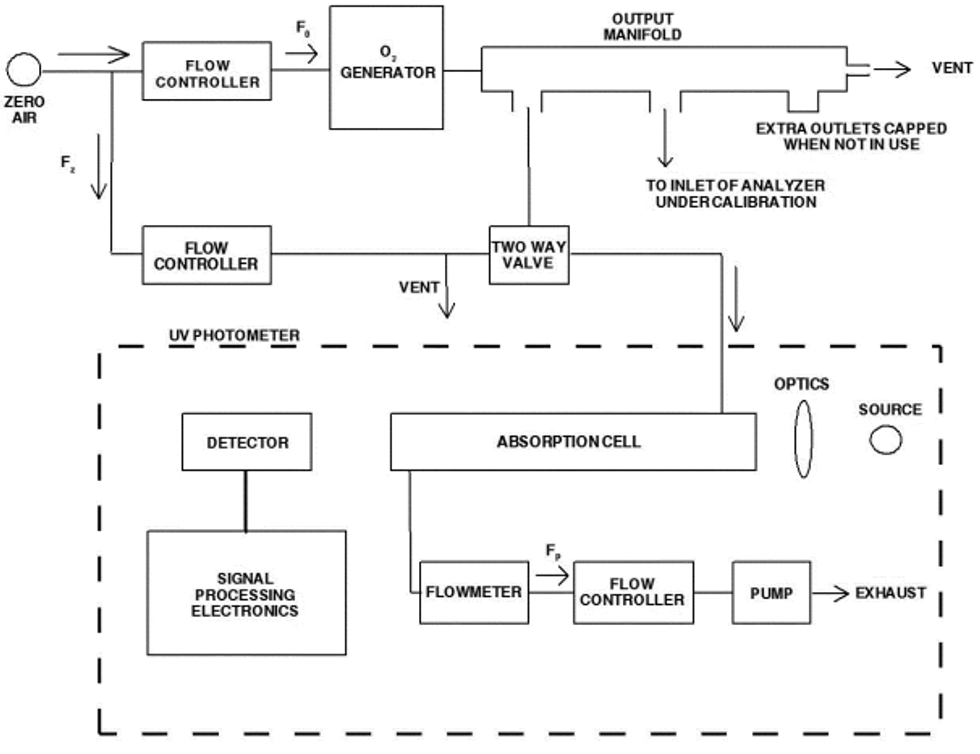
Figure 2. Schematic diagram of a typical UV photometric calibration system.
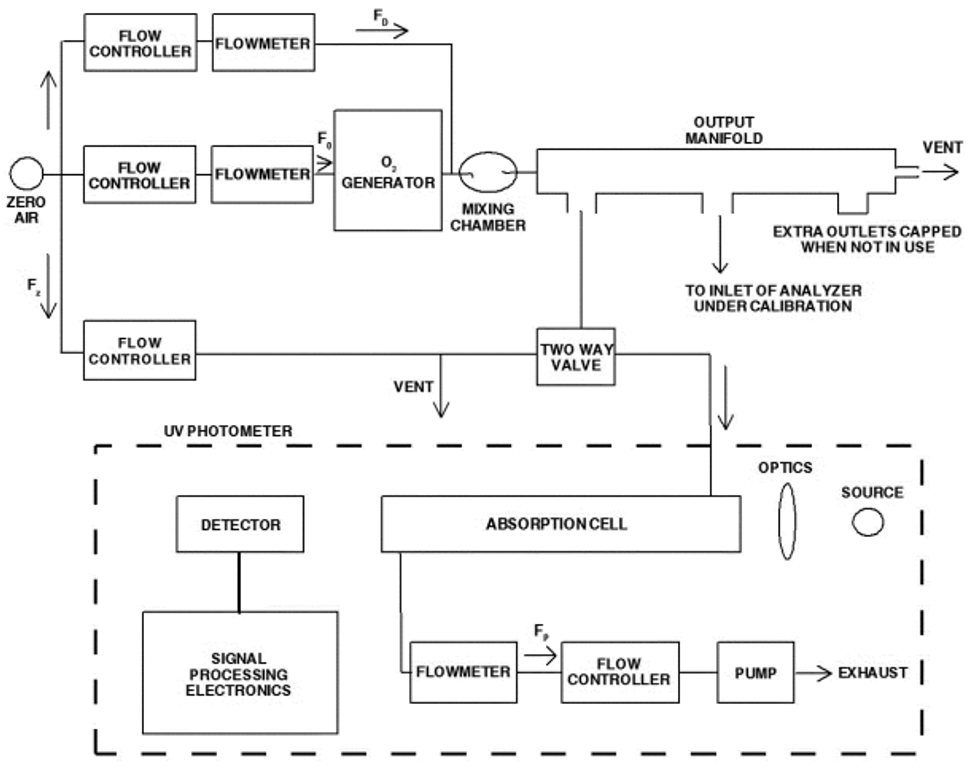
Figure 3. Schematic diagram of a typical UV photometric calibration system (Option 1).
[80 FR 65453, Oct. 26, 2015; 88 FR 70598, Oct. 12, 2023]
Appendix E to Part 50 [Reserved]
Appendix F to Part 50 - Measurement Principle and Calibration Procedure for the Measurement of Nitrogen Dioxide in the Atmosphere (Gas Phase Chemiluminescence)
Principle and Applicability
1. Atmospheric concentrations of nitrogen dioxide (NO2) are measured indirectly by photometrically measuring the light intensity, at wavelengths greater than 600 nanometers, resulting from the chemiluminescent reaction of nitric oxide (NO) with ozone (O3). (1,2,3) NO2 is first quantitatively reduced to NO(4,5,6) by means of a converter. NO, which commonly exists in ambient air together with NO2, passes through the converter unchanged causing a resultant total NOX concentration equal to NO + NO2. A sample of the input air is also measured without having passed through the converted. This latter NO measurement is subtracted from the former measurement (NO + NO2) to yield the final NO2 measurement. The NO and NO + NO2 measurements may be made concurrently with dual systems, or cyclically with the same system provided the cycle time does not exceed 1 minute.
2. Sampling considerations.
2.1 Chemiluminescence NO/NOX/NO2 analyzers will respond to other nitrogen containing compounds, such as peroxyacetyl nitrate (PAN), which might be reduced to NO in the thermal converter. (7) Atmospheric concentrations of these potential interferences are generally low relative to NO2 and valid NO2 measurements may be obtained. In certain geographical areas, where the concentration of these potential interferences is known or suspected to be high relative to NO2, the use of an equivalent method for the measurement of NO2 is recommended.
2.2 The use of integrating flasks on the sample inlet line of chemiluminescence NO/NOX/NO2 analyzers is optional and left to couraged. The sample residence time between the sampling point and the analyzer should be kept to a minimum to avoid erroneous NO2 measurements resulting from the reaction of ambient levels of NO and O3 in the sampling system.
2.3 The use of particulate filters on the sample inlet line of chemiluminescence NO/NOX/NO2 analyzers is optional and left to the discretion of the user or the manufacturer.
Use of the filter should depend on the analyzer's susceptibility to interference, malfunction, or damage due to particulates. Users are cautioned that particulate matter concentrated on a filter may cause erroneous NO2 measurements and therefore filters should be changed frequently.
3. An analyzer based on this principle will be considered a reference method only if it has been designated as a reference method in accordance with §53.11 of this chapter.
Calibration
1. Alternative A - Gas phase titration (GPT) of an NO standard with O3.
Major equipment required: Stable O3 generator. Chemiluminescence NO/NOX/NO2 analyzer with strip chart recorder(s). NO concentration standard.
1.1 Principle. This calibration technique is based upon the rapid gas phase reaction between NO and O3 to produce stoichiometric quantities of NO2 in accordance with the following equation: (8)

The quantitative nature of this reaction is such that when the NO concentration is known, the concentration of NO2 can be determined. Ozone is added to excess NO in a dynamic calibration system, and the NO channel of the chemiluminescence NO/NOX/NO2 analyzer is used as an indicator of changes in NO concentration. Upon the addition of O3, the decrease in NO concentration observed on the calibrated NO channel is equivalent to the concentration of NO2 produced. The amount of NO2 generated may be varied by adding variable amounts of O3 from a stable uncalibrated O3 generator. (9)
1.2 Apparatus. Figure 1, a schematic of a typical GPT apparatus, shows the suggested configuration of the components listed below. All connections between components in the calibration system downstream from the O3 generator should be of glass, Teflon ®, or other non-reactive material.
1.2.1 Air flow controllers. Devices capable of maintaining constant air flows within ±2% of the required flowrate.
1.2.2 NO flow controller. A device capable of maintaining constant NO flows within ±2% of the required flowrate. Component parts in contact with the NO should be of a non-reactive material.
1.2.3 Air flowmeters. Calibrated flowmeters capable of measuring and monitoring air flowrates with an accuracy of ±2% of the measured flowrate.
1.2.4 NO flowmeter. A calibrated flowmeter capable of measuring and monitoring NO flowrates with an accuracy of ±2% of the measured flowrate. (Rotameters have been reported to operate unreliably when measuring low NO flows and are not recommended.)
1.2.5 Pressure regulator for standard NO cylinder. This regulator must have a nonreactive diaphragm and internal parts and a suitable delivery pressure.
1.2.6 Ozone generator. The generator must be capable of generating sufficient and stable levels of O3 for reaction with NO to generate NO2 concentrations in the range required. Ozone generators of the electric discharge type may produce NO and NO2 and are not recommended.
1.2.7 Valve. A valve may be used as shown in Figure 1 to divert the NO flow when zero air is required at the manifold. The valve should be constructed of glass, Teflon ®, or other nonreactive material.
1.2.8 Reaction chamber. A chamber, constructed of glass, Teflon ®, or other nonreactive material, for the quantitative reaction of O3 with excess NO. The chamber should be of sufficient volume (VRC) such that the residence time (tR) meets the requirements specified in 1.4. For practical reasons, tR should be less than 2 minutes.
1.2.9 Mixing chamber. A chamber constructed of glass, Teflon ®, or other nonreactive material and designed to provide thorough mixing of reaction products and diluent air. The residence time is not critical when the dynamic parameter specification given in 1.4 is met.
1.2.10 Output manifold. The output manifold should be constructed of glass, Teflon ®, or other non-reactive material and should be of sufficient diameter to insure an insignificant pressure drop at the analyzer connection. The system must have a vent designed to insure atmospheric pressure at the manifold and to prevent ambient air from entering the manifold.
1.3 Reagents.
1.3.1 NO concentration standard. Gas cylinder standard containing 50 to 100 ppm NO in N2 with less than 1 ppm NO2. This standard must be traceable to a National Bureau of Standards (NBS) NO in N2 Standard Reference Material (SRM 1683 or SRM 1684), an NBS NO2 Standard Reference Material (SRM 1629), or an NBS/EPA-approved commercially available Certified Reference Material (CRM). CRM's are described in Reference 14, and a list of CRM sources is available from the address shown for Reference 14. A recommended protocol for certifying NO gas cylinders against either an NO SRM or CRM is given in section 2.0.7 of Reference 15. Reference 13 gives procedures for certifying an NO gas cylinder against an NBS NO2 SRM and for determining the amount of NO2 impurity in an NO cylinder.
1.3.2 Zero air. Air, free of contaminants which will cause a detectable response on the NO/NOX/NO2 analyzer or which might react with either NO, O3, or NO2 in the gas phase titration. A procedure for generating zero air is given in reference 13.
1.4 Dynamic parameter specification.
1.4.1 The O3 generator air flowrate (F0) and NO flowrate (FNO) (see Figure 1) must be adjusted such that the following relationship holds:
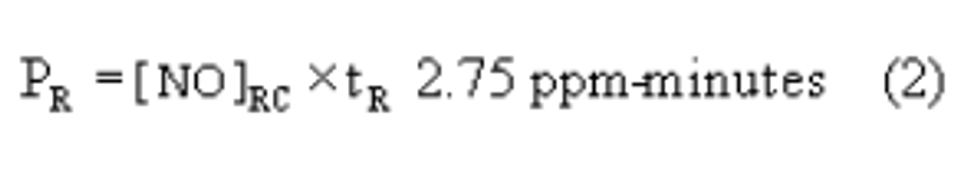
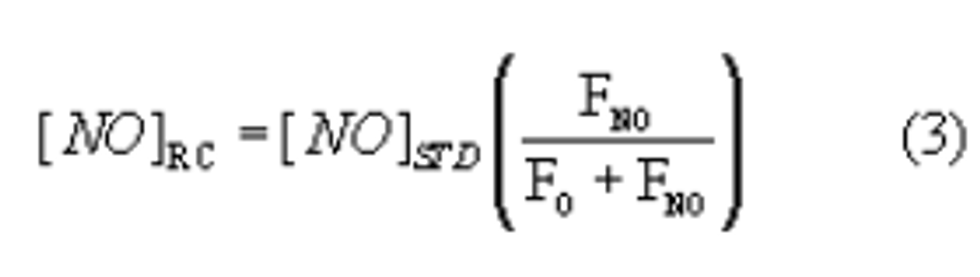
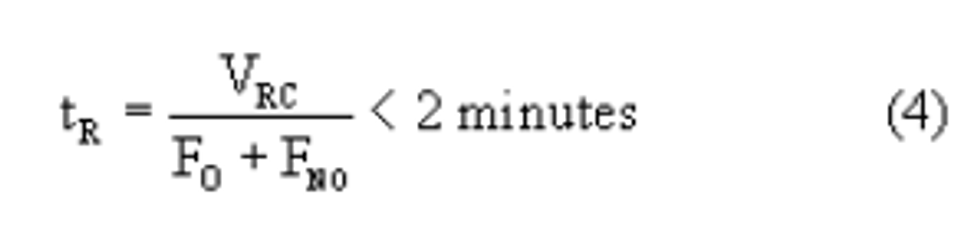
where:
PR = dynamic parameter specification, determined empirically, to insure complete reaction of the available O3, ppm-minute
[NO]RC = NO concentration in the reaction chamber, ppm
R = residence time of the reactant gases in the reaction chamber, minute
[NO]STD = concentration of the undiluted NO standard, ppm
FNO = NO flowrate, scm 3/min
FO = O3 generator air flowrate, scm 3/min
VRC = volume of the reaction chamber, scm 3
1.4.2 The flow conditions to be used in the GPT system are determined by the following procedure:
(a) Determine FT, the total flow required at the output manifold (FT = analyzer demand plus 10 to 50% excess).
(b) Establish [NO]OUT as the highest NO concentration (ppm) which will be required at the output manifold. [NO]OUT should be approximately equivalent to 90% of the upper range limit (URL) of the NO2 concentration range to be covered.
(c) Determine FNO as
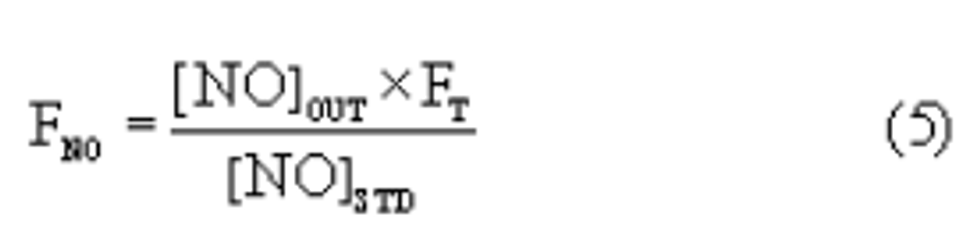
(d) Select a convenient or available reaction chamber volume. Initially, a trial VRC may be selected to be in the range of approximately 200 to 500 scm 3.
(e) Compute FO as
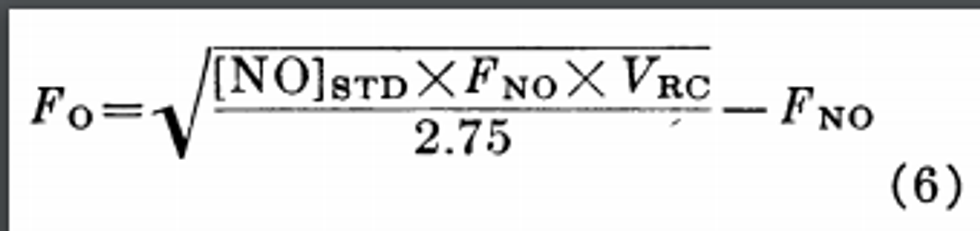
(f) Compute tR as

Verify that tR <2 minutes. If not, select a reaction chamber with a smaller VRC.
(g) Compute the diluent air flowrate as

where:
FD = diluent air flowrate, scm 3/min
(h) If FO turns out to be impractical for the desired system, select a reaction chamber having a different VRC and recompute FO and FD.
Note:
A dynamic parameter lower than 2.75 ppm-minutes may be used if it can be determined empirically that quantitative reaction of O3 with NO occurs. A procedure for making this determination as well as a more detailed discussion of the above requirements and other related considerations is given in reference 13.
1.5 Procedure.
1.5.1 Assemble a dynamic calibration system such as the one shown in Figure 1.
1.5.2 Insure that all flowmeters are calibrated under the conditions of use against a reliable standard such as a soap-bubble meter or wet-test meter. All volumetric flowrates should be corrected to 25°C and 760 mm Hg. A discussion on the calibration of flowmeters is given in reference 13.
1.5.3 Precautions must be taken to remove O2 and other contaminants from the NO pressure regulator and delivery system prior to the start of calibration to avoid any conversion of the standard NO to NO2. Failure to do so can cause significant errors in calibration. This problem may be minimized by (1) carefully evacuating the regulator, when possible, after the regulator has been connected to the cylinder and before opening the cylinder valve; (2) thoroughly flushing the regulator and delivery system with NO after opening the cylinder valve; (3) not removing the regulator from the cylinder between calibrations unless absolutely necessary. Further discussion of these procedures is given in reference 13.
1.5.4 Select the operating range of the NO/NOX/NO2 analyzer to be calibrated. In order to obtain maximum precision and accuracy for NO2 calibration, all three channels of the analyzer should be set to the same range. If operation of the NO and NOX channels on higher ranges is desired, subsequent recalibration of the NO and NOX channels on the higher ranges is recommended.
Note:
Some analyzer designs may require identical ranges for NO, NOX, and NO2 during operation of the analyzer.
1.5.5 Connect the recorder output cable(s) of the NO/NOX/NO2 analyzer to the input terminals of the strip chart recorder(s). All adjustments to the analyzer should be performed based on the appropriate strip chart readings. References to analyzer responses in the procedures given below refer to recorder responses.
1.5.6 Determine the GPT flow conditions required to meet the dynamic parameter specification as indicated in 1.4.
1.5.7 Adjust the diluent air and O3 generator air flows to obtain the flows determined in section 1.4.2. The total air flow must exceed the total demand of the analyzer(s) connected to the output manifold to insure that no ambient air is pulled into the manifold vent. Allow the analyzer to sample zero air until stable NO, NOX, and NO2 responses are obtained. After the responses have stabilized, adjust the analyzer zero control(s).
Note:
Some analyzers may have separate zero controls for NO, NOX, and NO2. Other analyzers may have separate zero controls only for NO and NOX, while still others may have only one zero control common to all three channels.
Offsetting the analyzer zero adjustments to + 5 percent of scale is recommended to facilitate observing negative zero drift. Record the stable zero air responses as ZNO, Znox, and Zno2.
1.5.8 Preparation of NO and NOXcalibration curves.
1.5.8.1 Adjustment of NO span control. Adjust the NO flow from the standard NO cylinder to generate an NO concentration of approximately 80 percent of the upper range limit (URL) of the NO range. This exact NO concentration is calculated from:
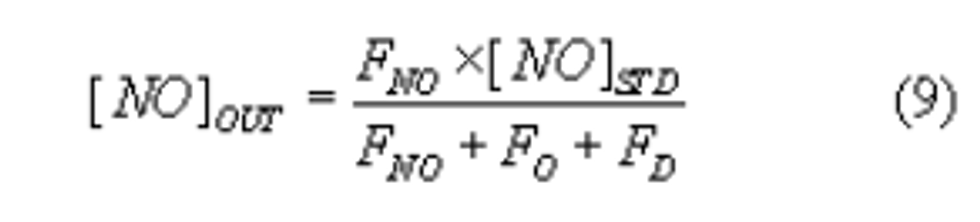
where:
[NO]OUT = diluted NO concentration at the output manifold, ppm
Sample this NO concentration until the NO and NOX responses have stabilized. Adjust the NO span control to obtain a recorder response as indicated below:
recorder response (percent scale) =
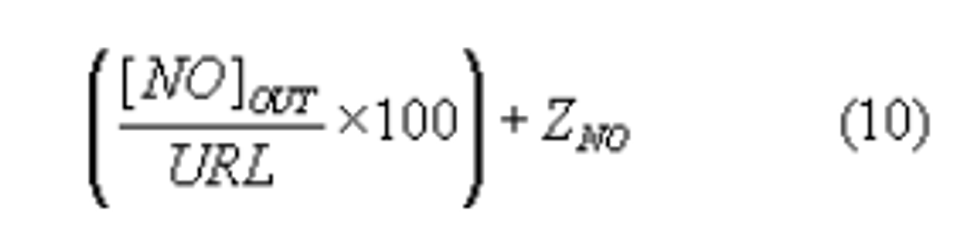
where:
URL = nominal upper range limit of the NO channel, ppm
Note:
Some analyzers may have separate span controls for NO, NOX, and NO2. Other analyzers may have separate span controls only for NO and NOX, while still others may have only one span control common to all three channels. When only one span control is available, the span adjustment is made on the NO channel of the analyzer.
If substantial adjustment of the NO span control is necessary, it may be necessary to recheck the zero and span adjustments by repeating steps 1.5.7 and 1.5.8.1. Record the NO concentration and the analyzer's NO response.
1.5.8.2 Adjustment of NOXspan control. When adjusting the analyzer's NOX span control, the presence of any NO2 impurity in the standard NO cylinder must be taken into account. Procedures for determining the amount of NO2 impurity in the standard NO cylinder are given in reference 13. The exact NOX concentration is calculated from:

where:
[NOX]OUT = diluted NOX concentration at the output manifold, ppm
[NO2]IMP = concentration of NO2 impurity in the standard NO cylinder, ppm
Adjust the NOX span control to obtain a recorder response as indicated below:
recorder response (% scale) =
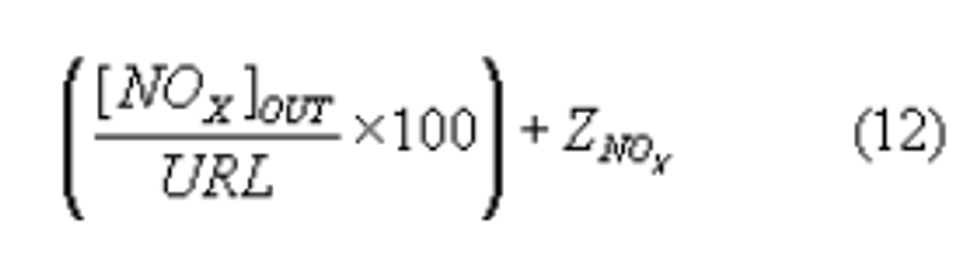
Note:
If the analyzer has only one span control, the span adjustment is made on the NO channel and no further adjustment is made here for NOX.
If substantial adjustment of the NOX span control is necessary, it may be necessary to recheck the zero and span adjustments by repeating steps 1.5.7 and 1.5.8.2. Record the NOX concentration and the analyzer's NOX response.
1.5.8.3 Generate several additional concentrations (at least five evenly spaced points across the remaining scale are suggested to verify linearity) by decreasing FNO or increasing FD. For each concentration generated, calculate the exact NO and NOX concentrations using equations (9) and (11) respectively. Record the analyzer's NO and NOX responses for each concentration. Plot the analyzer responses versus the respective calculated NO and NOX concentrations and draw or calculate the NO and NOX calibration curves. For subsequent calibrations where linearity can be assumed, these curves may be checked with a two-point calibration consisting of a zero air point and NO and NOX concentrations of approximately 80% of the URL.
1.5.9 Preparation of NO2calibration curve.
1.5.9.1 Assuming the NO2 zero has been properly adjusted while sampling zero air in step 1.5.7, adjust FO and FD as determined in section 1.4.2. Adjust FNO to generate an NO concentration near 90% of the URL of the NO range. Sample this NO concentration until the NO and NOX responses have stabilized. Using the NO calibration curve obtained in section 1.5.8, measure and record the NO concentration as [NO]orig. Using the NOX calibration curve obtained in section 1.5.8, measure and record the NOX concentration as [NOX]orig.
1.5.9.2 Adjust the O3 generator to generate sufficient O3 to produce a decrease in the NO concentration equivalent to approximately 80% of the URL of the NO2 range. The decrease must not exceed 90% of the NO concentration determined in step 1.5.9.1. After the analyzer responses have stabilized, record the resultant NO and NOX concentrations as [NO]rem and [NOX]rem.
1.5.9.3 Calculate the resulting NO2 concentration from:
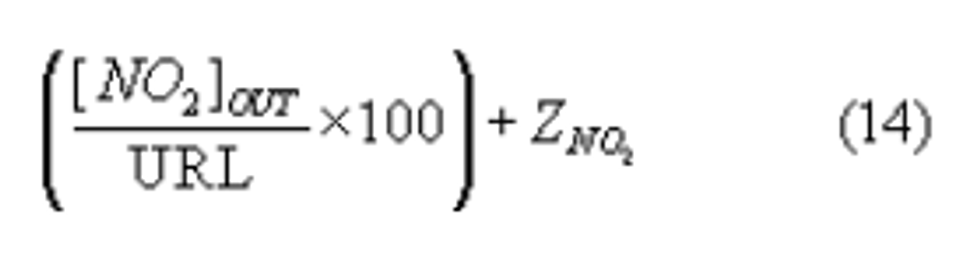
where:
[NO2]OUT = diluted NO2 concentration at the output manifold, ppm
[NO]orig = original NO concentration, prior to addition of O3, ppm
[NO]rem = NO concentration remaining after addition of O3, ppm
Adjust the NO2 span control to obtain a recorder response as indicated below:
recorder response (% scale) =

Note:
If the analyzer has only one or two span controls, the span adjustments are made on the NO channel or NO and NOX channels and no further adjustment is made here for NO2.
If substantial adjustment of the NO2 span control is necessary, it may be necessary to recheck the zero and span adjustments by repeating steps 1.5.7 and 1.5.9.3. Record the NO2 concentration and the corresponding analyzer NO2 and NOX responses.
1.5.9.4 Maintaining the same FNO, FO, and FD as in section 1.5.9.1, adjust the ozone generator to obtain several other concentrations of NO2 over the NO2 range (at least five evenly spaced points across the remaining scale are suggested). Calculate each NO2 concentration using equation (13) and record the corresponding analyzer NO2 and NOX responses. Plot the analyzer's NO2 responses versus the corresponding calculated NO2 concentrations and draw or calculate the NO2 calibration curve.
1.5.10 Determination of converter efficiency.
1.5.10.1 For each NO2 concentration generated during the preparation of the NO2 calibration curve (see section 1.5.9) calculate the concentration of NO2 converted from:
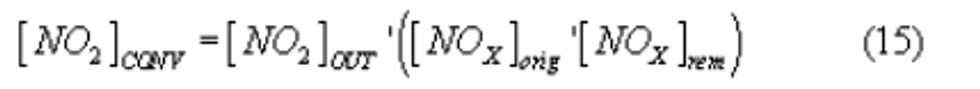
where:
[NO2]CONV = concentration of NO2 converted, ppm
[NOX]orig = original NOX concentration prior to addition of O3, ppm
[NOX]rem = NOX concentration remaining after addition of O3, ppm
Note:
Supplemental information on calibration and other procedures in this method are given in reference 13.
Plot [NO2]CONV (y-axis) versus [NO2]OUT (x-axis) and draw or calculate the converter efficiency curve. The slope of the curve times 100 is the average converter efficiency, EC The average converter efficiency must be greater than 96%; if it is less than 96%, replace or service the converter.
2. Alternative B - NO2 permeation device.
Major equipment required:
Stable O3 generator.
Chemiluminescence NO/NOX/NO2 analyzer with strip chart recorder(s).
NO concentration standard.
NO2 concentration standard.
2.1 Principle. Atmospheres containing accurately known concentrations of nitrogen dioxide are generated by means of a permeation device. (10) The permeation device emits NO2 at a known constant rate provided the temperature of the device is held constant (±0.1°C) and the device has been accurately calibrated at the temperature of use. The NO2 emitted from the device is diluted with zero air to produce NO2 concentrations suitable for calibration of the NO2 channel of the NO/NOX/NO2 analyzer. An NO concentration standard is used for calibration of the NO and NOX channels of the analyzer.
2.2 Apparatus. A typical system suitable for generating the required NO and NO2 concentrations is shown in Figure 2. All connections between components downstream from the permeation device should be of glass, Teflon ®, or other non-reactive material.
2.2.1 Air flow controllers. Devices capable of maintaining constant air flows within ±2% of the required flowrate.
2.2.2 NO flow controller. A device capable of maintaining constant NO flows within ±2% of the required flowrate. Component parts in contact with the NO must be of a non-reactive material.
2.2.3 Air flowmeters. Calibrated flowmeters capable of measuring and monitoring air flowrates with an accuracy of ±2% of the measured flowrate.
2.2.4 NO flowmeter. A calibrated flowmeter capable of measuring and monitoring NO flowrates with an accuracy of ±2% of the measured flowrate. (Rotameters have been reported to operate unreliably when measuring low NO flows and are not recommended.)
2.2.5 Pressure regulator for standard NO cylinder. This regulator must have a non-reactive diaphragm and internal parts and a suitable delivery pressure.
2.2.6 Drier. Scrubber to remove moisture from the permeation device air system. The use of the drier is optional with NO2 permeation devices not sensitive to moisture. (Refer to the supplier's instructions for use of the permeation device.)
2.2.7 Constant temperature chamber. Chamber capable of housing the NO2 permeation device and maintaining its temperature to within ±0.1°C.
2.2.8 Temperature measuring device. Device capable of measuring and monitoring the temperature of the NO2 permeation device with an accuracy of ±0.05°C.
2.2.9 Valves. A valve may be used as shown in Figure 2 to divert the NO2 from the permeation device when zero air or NO is required at the manifold. A second valve may be used to divert the NO flow when zero air or NO2 is required at the manifold.
The valves should be constructed of glass, Teflon ®, or other nonreactive material.
2.2.10 Mixing chamber. A chamber constructed of glass, Teflon ®, or other nonreactive material and designed to provide thorough mixing of pollutant gas streams and diluent air.
2.2.11 Output manifold. The output manifold should be constructed of glass, Teflon ®, or other non-reactive material and should be of sufficient diameter to insure an insignificant pressure drop at the analyzer connection. The system must have a vent designed to insure atmospheric pressure at the manifold and to prevent ambient air from entering the manifold.
2.3 Reagents.
2.3.1 Calibration standards. Calibration standards are required for both NO and NO2. The reference standard for the calibration may be either an NO or NO2 standard, and must be traceable to a National Bureau of Standards (NBS) NO in N2 Standard Reference Material (SRM 1683 or SRM 1684), and NBS NO2 Standard Reference Material (SRM 1629), or an NBS/EPA-approved commercially available Certified Reference Material (CRM). CRM's are described in Reference 14, and a list of CRM sources is available from the address shown for Reference 14. Reference 15 gives recommended procedures for certifying an NO gas cylinder against an NO SRM or CRM and for certifying an NO2 permeation device against an NO2 SRM. Reference 13 contains procedures for certifying an NO gas cylinder against an NO2 SRM and for certifying an NO2 permeation device against an NO SRM or CRM. A procedure for determining the amount of NO2 impurity in an NO cylinder is also contained in Reference 13. The NO or NO2 standard selected as the reference standard must be used to certify the other standard to ensure consistency between the two standards.
2.3.1.1 NO2Concentration standard. A permeation device suitable for generating NO2 concentrations at the required flow-rates over the required concentration range. If the permeation device is used as the reference standard, it must be traceable to an SRM or CRM as specified in 2.3.1. If an NO cylinder is used as the reference standard, the NO2 permeation device must be certified against the NO standard according to the procedure given in Reference 13. The use of the permeation device should be in strict accordance with the instructions supplied with the device. Additional information regarding the use of permeation devices is given by Scaringelli et al. (11) and Rook et al. (12).
2.3.1.2 NO Concentration standard. Gas cylinder containing 50 to 100 ppm NO in N2 with less than 1 ppm NO2. If this cylinder is used as the reference standard, the cylinder must be traceable to an SRM or CRM as specified in 2.3.1. If an NO2 permeation device is used as the reference standard, the NO cylinder must be certified against the NO2 standard according to the procedure given in Reference 13. The cylinder should be recertified on a regular basis as determined by the local quality control program.
2.3.3 Zero air. Air, free of contaminants which might react with NO or NO2 or cause a detectable response on the NO/NOX/NO2 analyzer. When using permeation devices that are sensitive to moisture, the zero air passing across the permeation device must be dry to avoid surface reactions on the device. (Refer to the supplier's instructions for use of the permeation device.) A procedure for generating zero air is given in reference 13.
2.4 Procedure.
2.4.1 Assemble the calibration apparatus such as the typical one shown in Figure 2.
2.4.2 Insure that all flowmeters are calibrated under the conditions of use against a reliable standard such as a soap bubble meter or wet-test meter. All volumetric flowrates should be corrected to 25°C and 760 mm Hg. A discussion on the calibration of flowmeters is given in reference 13.
2.4.3 Install the permeation device in the constant temperature chamber. Provide a small fixed air flow (200-400 scm 3/min) across the device. The permeation device should always have a continuous air flow across it to prevent large buildup of NO2 in the system and a consequent restabilization period. Record the flowrate as FP. Allow the device to stabilize at the calibration temperature for at least 24 hours. The temperature must be adjusted and controlled to within ±0.1°C or less of the calibration temperature as monitored with the temperature measuring device.
2.4.4 Precautions must be taken to remove O2 and other contaminants from the NO pressure regulator and delivery system prior to the start of calibration to avoid any conversion of the standard NO to NO2. Failure to do so can cause significant errors in calibration. This problem may be minimized by
(1) Carefully evacuating the regulator, when possible, after the regulator has been connected to the cylinder and before opening the cylinder valve;
(2) Thoroughly flushing the regulator and delivery system with NO after opening the cylinder valve;
(3) Not removing the regulator from the cylinder between calibrations unless absolutely necessary. Further discussion of these procedures is given in reference 13.
2.4.5 Select the operating range of the NO/NOX NO2 analyzer to be calibrated. In order to obtain maximum precision and accuracy for NO2 calibration, all three channels of the analyzer should be set to the same range. If operation of the NO and NOX channels on higher ranges is desired, subsequent recalibration of the NO and NOX channels on the higher ranges is recommended.
Note:
Some analyzer designs may require identical ranges for NO, NOX, and NO2 during operation of the analyzer.
2.4.6 Connect the recorder output cable(s) of the NO/NOX/NO2 analyzer to the input terminals of the strip chart recorder(s). All adjustments to the analyzer should be performed based on the appropriate strip chart readings. References to analyzer responses in the procedures given below refer to recorder responses.
2.4.7 Switch the valve to vent the flow from the permeation device and adjust the diluent air flowrate, FD, to provide zero air at the output manifold. The total air flow must exceed the total demand of the analyzer(s) connected to the output manifold to insure that no ambient air is pulled into the manifold vent. Allow the analyzer to sample zero air until stable NO, NOX, and NO2 responses are obtained. After the responses have stabilized, adjust the analyzer zero control(s).
Note:
Some analyzers may have separate zero controls for NO, NOX, and NO2. Other analyzers may have separate zero controls only for NO and NOX, while still others may have only one zero common control to all three channels.
Offsetting the analyzer zero adjustments to + 5% of scale is recommended to facilitate observing negative zero drift. Record the stable zero air responses as ZNO, ZNOX, and ZNO2.
2.4.8 Preparation of NO and NOXcalibration curves.
2.4.8.1 Adjustment of NO span control. Adjust the NO flow from the standard NO cylinder to generate an NO concentration of approximately 80% of the upper range limit (URL) of the NO range. The exact NO concentration is calculated from:
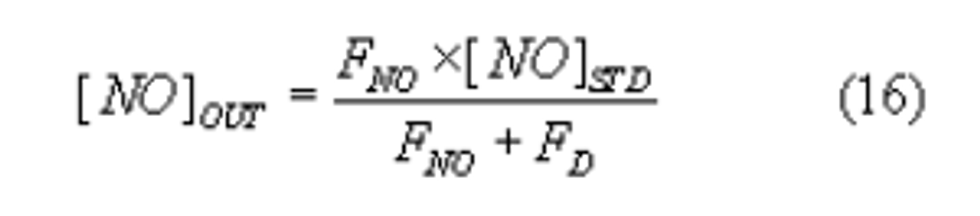
where:
[NO]OUT = diluted NO concentration at the output manifold, ppm
FNO = NO flowrate, scm 3/min
[NO]STD = concentration of the undiluted NO standard, ppm
FD = diluent air flowrate, scm 3/min
Sample this NO concentration until the NO and NOX responses have stabilized. Adjust the NO span control to obtain a recorder response as indicated below:
recorder response (% scale) =
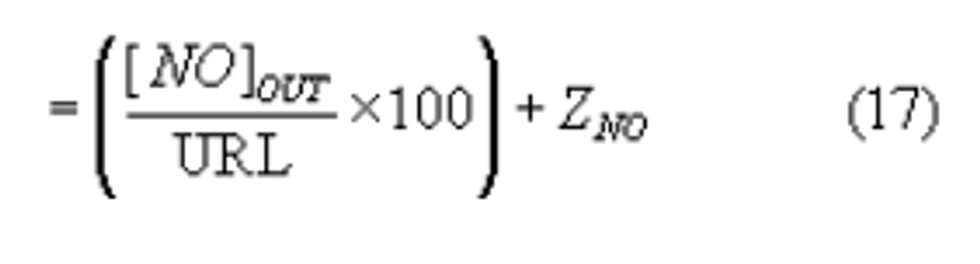
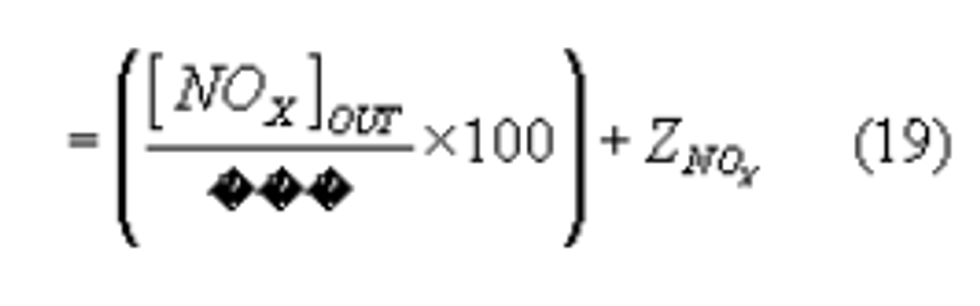
where:
URL = nominal upper range limit of the NO channel, ppm
Note:
Some analyzers may have separate span controls for NO, NOX, and NO2. Other analyzers may have separate span controls only for NO and NOX, while still others may have only one span control common to all three channels. When only one span control is available, the span adjustment is made on the NO channel of the analyzer.
If substantial adjustment of the NO span control is necessary, it may be necessary to recheck the zero and span adjustments by repeating steps 2.4.7 and 2.4.8.1. Record the NO concentration and the analyzer's NO response.
2.4.8.2 Adjustment of NOXspan control. When adjusting the analyzer's NOX span control, the presence of any NO2 impurity in the standard NO cylinder must be taken into account. Procedures for determining the amount of NO2 impurity in the standard NO cylinder are given in reference 13. The exact NOX concentration is calculated from:
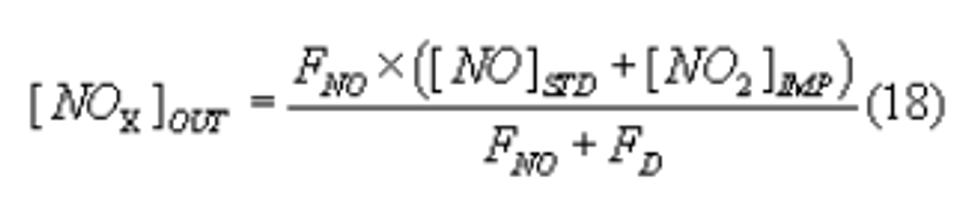
where:
[NOX]OUT = diluted NOX cencentration at the output manifold, ppm
[NO2]IMP = concentration of NO2 impurity in the standard NO cylinder, ppm
Adjust the NOX span control to obtain a convenient recorder response as indicated below:
recorder response (% scale)
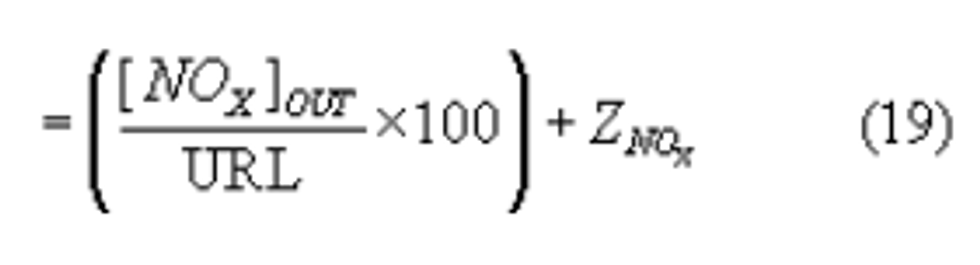
Note:
If the analyzer has only one span control, the span adjustment is made on the NO channel and no further adjustment is made here for NOX.
If substantial adjustment of the NOX span control is necessary, it may be necessary to recheck the zero and span adjustments by repeating steps 2.4.7 and 2.4.8.2. Record the NOX concentration and the analyzer's NOX response.
2.4.8.3 Generate several additional concentrations (at least five evenly spaced points across the remaining scale are suggested to verify linearity) by decreasing FNO or increasing FD. For each concentration generated, calculate the exact NO and NOX concentrations using equations (16) and (18) respectively. Record the analyzer's NO and NOX responses for each concentration. Plot the analyzer responses versus the respective calculated NO and NOX concentrations and draw or calculate the NO and NOX calibration curves. For subsequent calibrations where linearity can be assumed, these curves may be checked with a two-point calibration consisting of a zero point and NO and NOX concentrations of approximately 80 percent of the URL.
2.4.9 Preparation of NO2calibration curve.
2.4.9.1 Remove the NO flow. Assuming the NO2 zero has been properly adjusted while sampling zero air in step 2.4.7, switch the valve to provide NO2 at the output manifold.
2.4.9.2 Adjust FD to generate an NO2 concentration of approximately 80 percent of the URL of the NO2 range. The total air flow must exceed the demand of the analyzer(s) under calibration. The actual concentration of NO2 is calculated from:
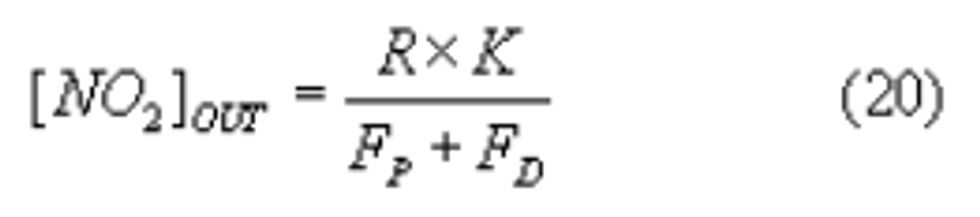
where:
[NO2]OUT = diluted NO2 concentration at the output manifold, ppm
R = permeation rate, µg/min
K = 0.532 µl NO2/µg NO2 (at 25°C and 760 mm Hg)
Fp = air flowrate across permeation device, scm 3/min
FD = diluent air flowrate, scm 3/min
Sample this NO2 concentration until the NOX and NO2 responses have stabilized. Adjust the NO2 span control to obtain a recorder response as indicated below:
recorder response (% scale)
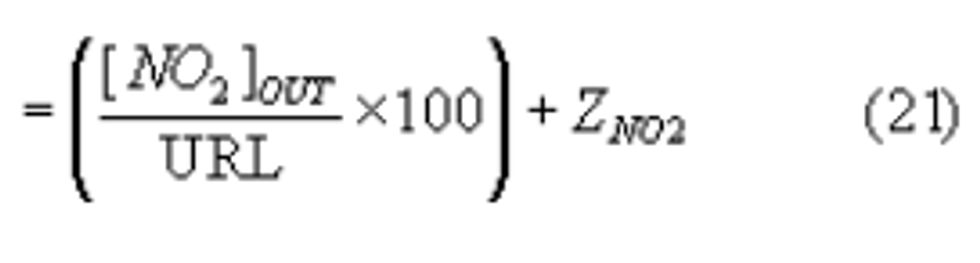
Note:
If the analyzer has only one or two span controls, the span adjustments are made on the NO channel or NO and NOX channels and no further adjustment is made here for NO2.
If substantial adjustment of the NO2 span control is necessary it may be necessary to recheck the zero and span adjustments by repeating steps 2.4.7 and 2.4.9.2. Record the NO2 concentration and the analyzer's NO2 response. Using the NOX calibration curve obtained in step 2.4.8, measure and record the NOX concentration as [NOX]M.
2.4.9.3 Adjust FD to obtain several other concentrations of NO2 over the NO2 range (at least five evenly spaced points across the remaining scale are suggested). Calculate each NO2 concentration using equation (20) and record the corresponding analyzer NO2 and NOX responses. Plot the analyzer's NO2 responses versus the corresponding calculated NO2 concentrations and draw or calculate the NO2 calibration curve.
2.4.10 Determination of converter efficiency.
2.4.10.1 Plot [NOX]M (y-axis) versus [NO2]OUT (x-axis) and draw or calculate the converter efficiency curve. The slope of the curve times 100 is the average converter efficiency, EC. The average converter efficiency must be greater than 96 percent; if it is less than 96 percent, replace or service the converter.
Note:
Supplemental information on calibration and other procedures in this method are given in reference 13.
3. Frequency of calibration. The frequency of calibration, as well as the number of points necessary to establish the calibration curve and the frequency of other performance checks, will vary from one analyzer to another. The user's quality control program should provide guidelines for initial establishment of these variables and for subsequent alteration as operational experience is accumulated. Manufacturers of analyzers should include in their instruction/operation manuals information and guidance as to these variables and on other matters of operation, calibration, and quality control.
References
1. A. Fontijn, A. J. Sabadell, and R. J. Ronco, “Homogeneous Chemiluminescent Measurement of Nitric Oxide with Ozone,” Anal. Chem., 42, 575 (1970).
2. D. H. Stedman, E. E. Daby, F. Stuhl, and H. Niki, “Analysis of Ozone and Nitric Oxide by a Chemiluminiscent Method in Laboratory and Atmospheric Studies of Photochemical Smog,” J. Air Poll. Control Assoc., 22, 260 (1972).
3. B. E. Martin, J. A. Hodgeson, and R. K. Stevens, “Detection of Nitric Oxide Chemiluminescence at Atmospheric Pressure,” Presented at 164th National ACS Meeting, New York City, August 1972.
4. J. A. Hodgeson, K. A. Rehme, B. E. Martin, and R. K. Stevens, “Measurements for Atmospheric Oxides of Nitrogen and Ammonia by Chemiluminescence,” Presented at 1972 APCA Meeting, Miami, FL, June 1972.
5. R. K. Stevens and J. A. Hodgeson, “Applications of Chemiluminescence Reactions to the Measurement of Air Pollutants,” Anal. Chem., 45, 443A (1973).
6. L. P. Breitenbach and M. Shelef, “Development of a Method for the Analysis of NO2 and NH3 by NO-Measuring Instruments,” J. Air Poll. Control Assoc., 23, 128 (1973).
7. A. M. Winer, J. W. Peters, J. P. Smith, and J. N. Pitts, Jr., “Response of Commercial Chemiluminescent NO-NO2 Analyzers to Other Nitrogen-Containing Compounds,” Environ. Sci. Technol., 8, 1118 (1974).
8. K. A. Rehme, B. E. Martin, and J. A. Hodgeson, Tentative Method for the Calibration of Nitric Oxide, Nitrogen Dioxide, and Ozone Analyzers by Gas Phase Titration,” EPA-R2-73-246, March 1974.
9. J. A. Hodgeson, R. K. Stevens, and B. E. Martin, “A Stable Ozone Source Applicable as a Secondary Standard for Calibration of Atmospheric Monitors,” ISA Transactions, 11, 161 (1972).
10. A. E. O'Keeffe and G. C. Ortman, “Primary Standards for Trace Gas Analysis,” Anal. Chem., 38, 760 (1966).
11. F. P. Scaringelli, A. E. O'Keeffe, E. Rosenberg, and J. P. Bell, “Preparation of Known Concentrations of Gases and Vapors with Permeation Devices Calibrated Gravimetrically,” Anal. Chem., 42, 871 (1970).
12. H. L. Rook, E. E. Hughes, R. S. Fuerst, and J. H. Margeson, “Operation Characteristics of NO2 Permeation Devices,” Presented at 167th National ACS Meeting, Los Angeles, CA, April 1974.
13. E. C. Ellis, “Technical Assistance Document for the Chemiluminescence Measurement of Nitrogen Dioxide,” EPA-E600/4-75-003 (Available in draft form from the United States Environmental Protection Agency, Department E (MD-76), Environmental Monitoring and Support Laboratory, Research Triangle Park, NC 27711).
14. A Procedure for Establishing Traceability of Gas Mixtures to Certain National Bureau of Standards Standard Reference Materials. EPA-600/7-81-010, Joint publication by NBS and EPA. Available from the U.S. Environmental Protection Agency, Environmental Monitoring Systems Laboratory (MD-77), Research Triangle Park, NC 27711, May 1981.
15. Quality Assurance Handbook for Air Pollution Measurement Systems, Volume II, Ambient Air Specific Methods. The U.S. Environmental Protection Agency, Environmental Monitoring Systems Laboratory, Research Triangle Park, NC 27711. Publication No. EAP-600/4-77-027a.
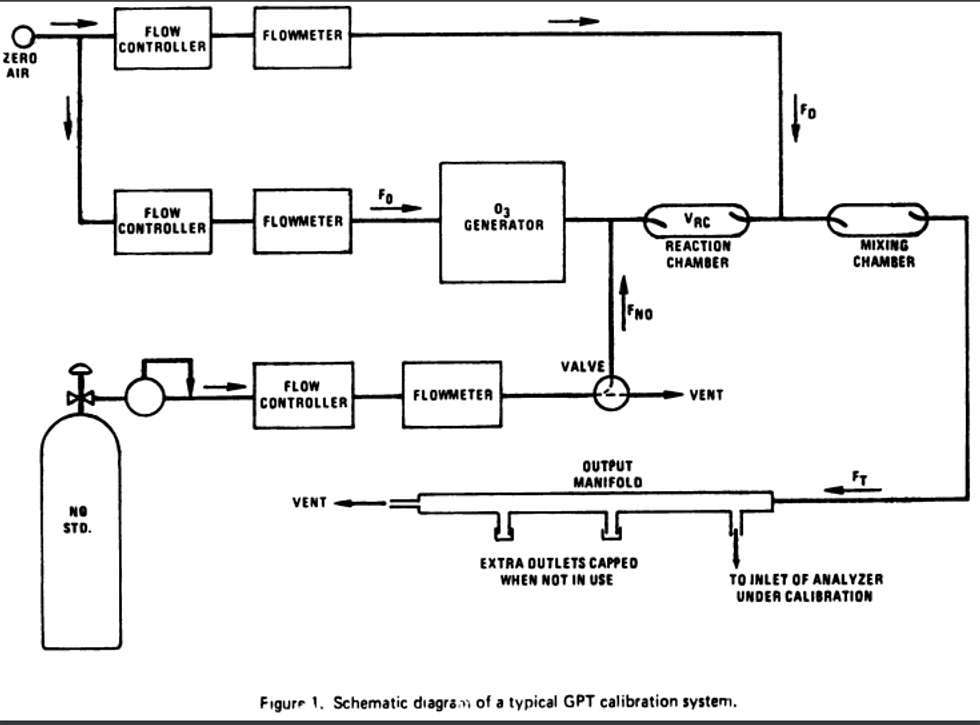
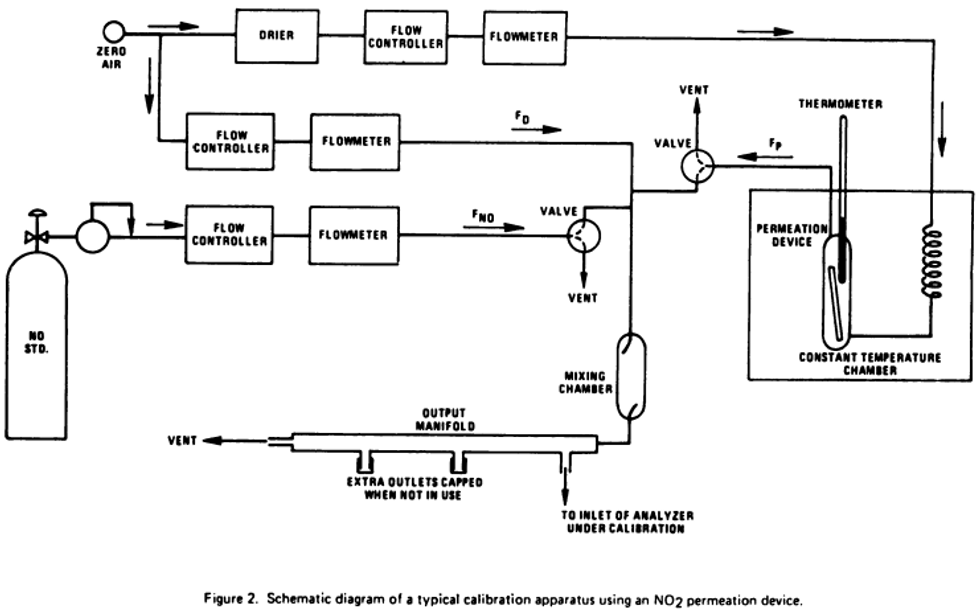
[41 FR 52688, Dec. 1, 1976, as amended at 48 FR 2529, Jan. 20, 1983]
Appendix G to Part 50 - Reference Method for the Determination of Lead in Total Suspended Particulate Matter
1.0 Scope and Applicability
Based on review of the air quality criteria and national ambient air quality standard (NAAQS) for lead (Pb) completed in 2008, the EPA made revisions to the primary and secondary NAAQS for Pb to protect public health and welfare. The EPA revised the level from 1.5 µg/m 3 to 0.15 µg/m 3 while retaining the current indicator of Pb in total suspended particulate matter (Pb-TSP).
Pb-TSP is collected for 24 hours on a TSP filter as described in Appendix B of part 50, the Reference Method for the Determination of Suspended Particulate Matter in the Atmosphere (High-Volume Method). This method is for the analysis of Pb from TSP filters by Inductively Coupled Plasma Mass Spectrometry (ICP-MS) using a heated ultrasonic bath with nitric acid (HNO3) and hydrochloric acid (HCl) or a heated block (hot block) digester with HNO3 for filter extraction.
This method is based on the EPA's Office of Solid Waste (SW-846) Method 6020A - Inductively Coupled Plasma Mass Spectrometry (U.S. EPA, 2007). Wording in certain sections of this method is paraphrased or taken directly from Method 6020A.
1.1 ICP-MS is applicable for the sub-µg/mL (ppb) determination of Pb in a wide variety of matrices. Results reported for monitoring or compliance purposes are calculated in µg/m 3 at local conditions (LC). This procedure describes a method for the acid extraction of Pb in particulate matter collected on glass fiber, quartz, or PTFE filters and measurement of the extracted Pb using ICP-MS.
1.2 Due to variations in the isotopic abundance of Pb, the value for total Pb must be based on the sum of the signal intensities for isotopic masses, 206, 207, and 208. Most instrument software packages are able to sum the primary isotope signal intensities automatically.
1.3 ICP-MS requires the use of an internal standard. 115In (Indium), 165Ho (Holmium), and 209Bi (Bismuth) are recommended internal standards for the determination of Pb.
1.4 Use of this method is restricted to use by, or under supervision of, properly trained and experienced laboratory personnel. Requirements include training and experience in inorganic sample preparation, including acid extraction, and also knowledge in the recognition and in the correction of spectral, chemical and physical interference in ICP-MS.
2.0 Summary of Method
2.1 This method describes the acid extraction of Pb in particulate matter collected on glass fiber, quartz, or PTFE ambient air filters with subsequent measurement of Pb by ICP-MS. Estimates of the Method Detection Limit (MDL) or sensitivity of the method are provided in Tables 1, 3 and 5 and determined using Pb-spiked filters or filter strips analyzed in accordance with the guidance provided in 40 CFR 136, Appendix B - Determination and procedures for the Determination of the Method Detection Limit - Revision 1.1. The analytical range of the method is 0.00024 µg/m 3 to 0.60 µg/m 3, and based on the low and high calibration curve standards and a nominal filter sample volume of 2000 m 3.
2.2 This method includes two extraction methods. In the first method, a solution of HNO3 and HCl is added to the filters or filter strips in plastic digestion tubes and the tubes are placed in a heated ultrasonic bath for one hour to facilitate the extraction of Pb. Following ultrasonication, the samples are brought to a final volume of 40 mL (50 mL for PTFE filters), vortex mixed or shaken vigorously, and centrifuged prior to aliquots being taken for ICP-MS analysis. In the second method, a solution of dilute HNO3 is added to the filter strips in plastic digestion tubes and the tubes placed into the hot block digester. The filter strip is completely covered by the solution. The tubes are covered with polypropylene watch glasses and refluxed. After reflux, the samples are diluted to a final volume of 50 mL with reagent water and mixed before analysis.
2.3 Calibration standards and check standards are prepared to matrix match the acid composition of the samples. ICP-MS analysis is then performed. With this method, the samples are first aspirated and the aerosol thus created is transported by a flow of argon gas into the plasma torch. The ions produced (e.g., Pb+ 1) in the plasma are extracted via a differentially-pumped vacuum interface and are separated on the basis of their mass-to-charge ratio. The ions are quantified by a channel electron multiplier or a Faraday detector and the signal collected is processed by the instrument's software. Interferences must be assessed and corrected for, if present.
3.0 Definitions
Pb - Elemental or ionic lead
HNO3 - Nitric acid
HCl - Hydrochloric acid
ICP-MS - Inductively Coupled Plasma Mass Spectrometer
MDL - Method detection limit
RSD - Relative standard deviation
RPD - Relative percent difference
CB - Calibration Blank
CAL - Calibration Standard
ICB - Initial calibration blank
CCB - Continuing calibration blank
ICV - Initial calibration verification
CCV - Continuing calibration verification
LLCV - Lower Level Calibration Verification, serves as the lower level ICV and lower level CCV
RB - Reagent blank
RBS - Reagent blank spike
MSDS - Material Safety Data Sheet
NIST - National Institute of Standards and Technology
D.I. water - Deionized water
SRM - NIST Standard Reference Material
CRM - Certified Reference Material
EPA - Environmental Protection Agency
v/v - Volume to volume ratio
4.0 Interferences
4.1 Reagents, glassware, plasticware, and other sample processing hardware may yield artifacts and/or interferences to sample analysis. If reagent blanks, filter blanks, or quality control blanks yield results above the detection limit, the source of contamination must be identified. All containers and reagents used in the processing of the samples must be checked for contamination prior to sample extraction and analysis. Reagents shall be diluted to match the final concentration of the extracts and analyzed for Pb. Labware shall be rinsed with dilute acid solution and the solution analyzed. Once a reagent or labware article (such as extraction tubes) from a manufacturer has been successfully screened, additional screening is not required unless contamination is suspected.
4.2 Isobaric elemental interferences in ICP-MS are caused by isotopes of different elements forming atomic ions with the same nominal mass-to-charge ratio (m/z) as the species of interest. There are no species found in ambient air that will result in isobaric interference with the three Pb isotopes (206, 207, and 208) being measured. Polyatomic interferences occur when two or more elements combine to form an ion with the same mass-to-charge ratio as the isotope being measured. Pb is not subject to interference from common polyatomic ions and no correction is required.
4.3 The distribution of Pb isotopes is not constant. The analysis of total Pb should be based on the summation of signal intensities for the isotopic masses 206, 207, and 208. In most cases, the instrument software can perform the summation automatically.
4.4 Physical interferences are associated with the sample nebulization and transport processes as well as with ion-transmission efficiencies. Dissolved solids can deposit on the nebulizer tip of a pneumatic nebulizer and on the interface skimmers of the ICP-MS. Nebulization and transport processes can be affected if a matrix component causes a change in surface tension or viscosity. Changes in matrix composition can cause significant signal suppression or enhancement. These interferences are compensated for by use of internal standards. Sample dilution will reduce the effects of high levels of dissolved salts, but calibration standards must be prepared in the extraction medium and diluted accordingly.
4.5 Memory interferences are related to sample transport and result when there is carryover from one sample to the next. Sample carryover can result from sample deposition on the sample and skimmer cones and from incomplete rinsing of the sample solution from the plasma torch and the spray chamber between samples. These memory effects are dependent upon both the analyte being measured and sample matrix and can be minimized through the use of suitable rinse times.
5.0 Health and Safety Cautions
5.1 The toxicity or carcinogenicity of reagents used in this method has not been fully established. Each chemical should be regarded as a potential health hazard and exposure to these compounds should be as low as reasonably achievable. Each laboratory is responsible for maintaining a current file of OSHA regulations regarding the safe handling of the chemicals specified in this method. A reference file of material safety data sheets (MSDSs) should be available to all personnel involved in the chemical analysis. Specifically, concentrated HNO3 presents various hazards and is moderately toxic and extremely irritating to skin and mucus membranes. Use this reagent in a fume hood whenever possible and if eye or skin contact occurs, flush with large volumes of water. Always wear safety glasses or a shield for eye protection, protective clothing, and observe proper mixing when working with these reagents.
5.2 Concentrated HNO3 and HCl are moderately toxic and extremely irritating to the skin. Use these reagents in a fume hood, and if eye and skin contact occurs, flush with large volumes of water. Always wear safety glasses or a shield for eye protection when working with these reagents. The component of this procedure requiring the greatest care is HNO3. HNO3 is a strong, corrosive, oxidizing agent that requires protection of the eyes, skin, and clothing. Items to be worn during use of this reagent include:
1. Safety goggles (or safety glasses with side shields),
2. Acid resistant rubber gloves, and
3. A protective garment such as a laboratory apron. HNO3 spilled on clothing will destroy the fabric; contact with the skin underneath will result in a burn.
It is also essential that an eye wash fountain or eye wash bottle be available during performance of this method. An eye wash bottle has a spout that covers the eye. If acid or any other corrosive gets into the eye, the water in this bottle is squirted onto the eye to wash out the harmful material. Eye washing should be performed with large amounts of water immediately after exposure. Medical help should be sought immediately after washing. If either acid, but especially HNO3, is spilled onto the skin, wash immediately with large amounts of water. Medical attention is not required unless the burn appears to be significant. Even after washing and drying, HNO3 may leave the skin slightly brown in color; this will heal and fade with time.
5.3 Pb salts and Pb solutions are toxic. Great care must be taken to ensure that samples and standards are handled properly; wash hands thoroughly after handling.
5.4 Care must be taken when using the ultrasonic bath and hot block digester as they are capable of causing mild burns. Users should refer to the safety guidance provided by the manufacturer of their specific equipment.
5.5 Analytical plasma sources emit radio frequency radiation in addition to intense ultra violet (UV) radiation. Suitable precautions should be taken to protect personnel from such hazards. The inductively coupled plasma should only be viewed with proper eye protection from UV emissions.
6.0 Equipment
6.1 Thermo Scientific X-Series ICP-MS or equivalent. The system must be capable of providing resolution better or equal to 1.0 atomic mass unit (amu) at 10 percent peak height. The system must have a mass range from at least 7 to 240 amu that allows for the application of the internal standard technique. For the measurement of Pb, an instrument with a collision or reaction cell is not required.
6.2 Ultrasonic Extraction Equipment
6.2.1 Heated ultrasonic bath capable of maintaining a temperature of 80°C; VWR Model 750HT, 240W, or equivalent. Ultrasonic bath must meet the following performance criteria:
1. Cut a strip of aluminum foil almost the width of the tank and double the depth.
2. Turn the ultrasonic bath on and lower the foil into the bath vertically until almost touching the bottom of the tank and hold for 10 seconds.
3. Remove the foil from the tank and observe the distribution of perforations and small pin prick holes. The indentations should be fine and evenly distributed. The even distribution of indentations indicates the ultrasonic bath is acceptable for use.
6.2.2 Laboratory centrifuge, Beckman GS-6, or equivalent.
6.2.3 Vortex mixer, VWR Signature Digital Vortex Mixer, VWR Catalog No. 14005-824, or equivalent.
6.3 Hot block extraction equipment
6.3.1 Hot block digester, SCP Science DigiPrep Model MS, No. 010-500-205 block digester capable of maintaining a temperature of 95°C, or equivalent.
6.4 Materials and Supplies
Argon gas supply, 99.99 percent purity or better. National Welders Microbulk, or equivalent.
Plastic digestion tubes with threaded caps for extraction and storage, SCP Science DigiTUBE® Item No. 010-500-063, or equivalent.
Disposable polypropylene ribbed watch glasses (for heated block extraction), SCP Science Item No. 010-500-081, or equivalent.
Pipette, Rainin EDP2, 100 µL, ±1 percent accuracy, ≤1 percent RSD (precision), with disposable tips, or equivalent.
Pipette, Rainin EDP2, 1000 µL, ±1 percent accuracy, ≤1 percent RSD (precision), with disposable tips, or equivalent.
Pipette, Rainin EDP2, 1-10 mL, ±1 percent accuracy, ≤1 percent RSD (precision), with disposable tips, or equivalent.
Pipette, Thermo Lab Systems, 5 mL, ±1 percent accuracy, ≤1 percent RSD (precision), with disposable tips, or equivalent.
Plastic tweezer, VWR Catalog No. 89026-420, or equivalent.
Laboratory marker.
Ceramic knife, Kyocera LK-25, and non-metal ruler or other suitable cutting tools for making straight cuts for accurately measured strips.
Blank labels or labeling tape, VWR Catalog No. 36425-045, or equivalent.
Graduated cylinder, 1 L, VWR 89000-260, or equivalent.
Volumetric flask, Class A, 1 L, VWR Catalog No. 89025-778, or equivalent.
Millipore Element deionized water system, or equivalent, capable of generating water with a resistivity of ≥17.9 MΩ-cm).
Disposable syringes, 10-mL, with 0.45 micron filters (must be Pb-free).
Plastic or PTFE wash bottles.
Glassware, Class A - volumetric flasks, pipettes, and graduated cylinders.
Glass fiber, quartz, or PTFE filters from the same filter manufacturer and lot used for sample collection for use in the determination of the MDL and for laboratory blanks.
7.0 Reagents and Standards
7.1 Reagent - or trace metals-grade chemicals must be used in all tests. Unless otherwise indicated, it is intended that all reagents conform to the specifications of the Committee on Analytical Reagents of the American Chemical Society, where such specifications are available.
7.2 Concentrated nitric acid, 67-70 percent, SCP Science Catalog No. 250-037-177, or equivalent.
7.3 Concentrated hydrochloric acid (for the ultrasonic extraction method), 33-36 percent, SCP Science Catalog No. 250-037-175, or equivalent.
7.4 Deionized water - All references to deionized water in the method refer to deionized water with a resistivity ≥17.9 MΩ-cm.
7.5 Standard stock solutions may be commercially purchased for each element or as a multi-element mix. Internal standards may be purchased as a mixed multi-element solution. The manufacturer's expiration date and storage conditions must be adhered to.
7.5.1 Lead standard, 1000 µg/mL, NIST traceable, commercially available with certificate of analysis. High Purity Standards Catalog No. 100028-1, or equivalent.
7.5.2 Indium (In) standard, 1000 µg/mL, NIST traceable, commercially available with certificate of analysis. High Purity Standards Catalog No. 100024-1, or equivalent.
7.5.3 Bismuth (Bi) standard, 1000 µg/mL, NIST traceable, commercially available with certificate of analysis. High Purity Standards Catalog No. 100006-1, or equivalent.
7.5.4 Holmium (Ho) standard, 1000 µg/mL, NIST traceable, commercially available with certificate of analysis. High Purity Standards Catalog No. 100023-1, or equivalent.
7.5.5 Second source lead standard, 1000 µg/mL, NIST traceable, commercially available with certificate of analysis. Must be from a different vendor or lot than the standard described in 7.5.1. Inorganic Ventures Catalog No. CGPB-1, or equivalent.
7.5.6 Standard Reference Materials, NIST SRM 2583, 2586, 2587 or 1648, or equivalent. 5
5 Certificates of Analysis for these SRMs can be found at: http://www.nist.gov/srm/index.cfm.
Note: The In, Bi, and Ho internal standards may also be purchased as 10 µg/mL standards. Calibration standards are prepared by diluting stock standards to the appropriate levels in the same acid concentrations as in the final sample volume. The typical range for calibration standards is 0.001 to 2.00 µg/mL. At a minimum, the curve must contain a blank and five Pb containing calibration standards. The calibration standards are stored at ambient laboratory temperature. Calibration standards must be prepared weekly and verified against a freshly prepared ICV using a NIST-traceable source different from the calibration standards.
7.6 Internal standards may be added to the test solution or by on-line addition. The nominal concentration for an internal standard is 0.010 µg/mL (10 ppb). Bismuth (Bi) or holmium (Ho) are the preferred internal standards for Pb, but indium (In) may be used in the event the sample contains Bi and high recoveries are observed.
7.7 Three laboratory blank solutions are required for analysis: (1) The calibration blank is used in the construction of the calibration curve and as a periodic check of system cleanliness (ICB and CCB); (2) the reagent blank (RB) is carried through the extraction process to assess possible contamination; and (3) the rinse blank is run between samples to clean the sample introduction system. If RBs or laboratory blanks yield results above the detection limit, the source of contamination must be identified. Screening of labware and reagents is addressed in Section 4.1.
7.7.1 The calibration blank is prepared in the same acid matrix as the calibration standards and samples and contains all internal standards used in the analysis.
7.7.2 The RB contains all reagents used in the extraction and is carried through the extraction procedure at the same time as the samples.
7.7.3 The rinse blank is a solution of 1 to 2 percent HNO3 (v/v) in reagent grade water. A sufficient volume should be prepared to flush the system between all standards and samples analyzed.
7.7.4 The EPA currently provides glass fiber, quartz, and PTFE filters to air monitoring agencies as requested annually. As part of the procurement process, these filters are tested for acceptance by the EPA. The current acceptance criteria for glass fiber and quartz filters is 15 µg per filter or 0.0075 µg/m 3 using a nominal sample volume of 2000 m 3 and 4.8 ng/cm 2 or 0.0024 µg/m 3 for PTFE filters using a nominal sample volume of 24 m 3. Acceptance test results for filters obtained by the EPA are typically well below the criterion specified and also below the recently revised Pb method performance detection limit of 0.0075 µg/m 3; therefore, blank subtraction should not be performed.
7.7.5 If filters are not provided by the EPA for sample collection and analysis, filter lot blanks should be analyzed for Pb content. For large filter lots (>500 filters), randomly select 20 to 30 filters from the lot and analyze the filter or filter strips for Pb. For smaller filter lots, a lesser number of filters can be analyzed. Glass, quartz and PTFE filters must not have levels of Pb above the criteria specified in section 7.7.4 and, therefore, blank correction should not be performed. If acceptance testing shows levels of Pb above the criteria in Section 7.7.4, corrective action must be taken to reduce the levels before proceeding.
7.8 The Initial Calibration Verification (ICV), Lower Level Calibration Verification (LLCV), and Continuing Calibration Verification (CCV) solutions are prepared from a different Pb source than the calibration curve standards and at a concentration that is either at or below the midpoint on the calibration curve, but within the calibration range. Both are prepared in the same acid matrix as the calibration standards. Note that the same solution may be used for both the ICV and CCV. The ICV/CCV and LLCV solutions must be prepared fresh daily.
7.9 Tuning Solution. Prepare a tuning solution according to the instrument manufacturer's recommendations. This solution will be used to verify the mass calibration and resolution of the instrument.
8.0 Quality Control (QC)
8.1 Standard QC practices shall be employed to assess the validity of the data generated, including: MDL, RB, duplicate samples, spiked samples, serial dilutions, ICV, CCV, LLCV, ICB, CCB, and SRMs/CRMs.
8.2 MDLs must be calculated in accordance with 40 CFR part 136, Appendix B. RBs with low-level standard spikes are used to estimate the MDL. The low-level standard spike is added to at least 7 individual filter strips and then carried through the entire extraction procedure. This will result in at least 7 individual samples to be used for the MDL. The recommended range for spiking the strips is 1 to 5 times the estimated MDL.
8.3 For each batch of samples, one RB and one reagent blank spike (RBS) that is spiked at the same level as the sample spike (see Section 8.6) must be prepared and carried throughout the entire process. The results of the RB must be below 0.001 µg/mL. The recovery for the RBS must be within ±20 percent of the expected value. If the RB yields a result above 0.001 µg/mL, the source of contamination must be identified and the extraction and analysis repeated. Reagents and labware must be suspected as sources of contamination. Screening of reagents and labware is addressed in Section 4.1.
8.4 Any samples that exceed the highest calibration standard must be diluted and rerun so that the concentration falls within the curve. The minimum dilution will be 1 to 5 with matrix matched acid solution.
8.5 The internal standard response must be monitored during the analysis. If the internal standard response falls below 70 percent or rises above 120 percent of expected due to possible matrix effects, the sample must be diluted and reanalyzed. The minimum dilution will be 1 to 5 with matrix matched acid solution. If the first dilution does not correct the problem, additional dilutions must be run until the internal standard falls within the specified range.
8.6 For every batch of samples prepared, there must be one duplicate and one spike sample prepared. The spike added is to be at a level that falls within the calibration curve, normally the midpoint of the curve. The initial plus duplicate sample must yield a relative percent difference ≤20 percent. The spike must be within ±20 percent of the expected value.
8.7 For each batch of samples, one extract must be diluted five-fold and analyzed. The corrected dilution result must be within ±10 percent of the undiluted result. The sample chosen for the serial dilution shall have a concentration at or above 10X the lowest standard in the curve to ensure the diluted value falls within the curve. If the serial dilution fails, chemical or physical interference should be suspected.
8.8 ICB, ICV, LLCV, CCB and CCV samples are to be run as shown in the following table.
| Sample | Frequency | Performance specification |
|---|---|---|
| ICB | Prior to first sample | Less than 0.001 µg/mL. |
| ICV | Prior to first sample | Within 90 to 110 percent of the expected value. |
| LLCV | Daily, before first sample and after last sample | ±10 percent of the expected value. |
| CCB | After every 10 extracted samples | Less than 0.001 µg/mL. |
| CCV | After every 10 extracted samples | Within 90-110 percent of the expected value. |
If any of these QC samples fails to meet specifications, the source of the unacceptable performance must be determined, the problem corrected, and any samples not bracketed by passing QC samples must be reanalyzed.
8.9 For each batch of samples, one certified reference material (CRM) must be combined with a blank filter strip and carried through the entire extraction procedure. The result must be within ±10 percent of the expected value.
8.10 For each run, a LLCV must be analyzed. The LLCV must be prepared at a concentration not more than three times the lowest calibration standard and at a concentration not used in the calibration curve. The LLCV is used to assess performance at the low end of the curve. If the LLCV fails (±10 percent of the expected value) the run must be terminated, the problem corrected, the instrument recalibrated, and the analysis repeated.
8.11 Pipettes used for volumetric transfer must have the calibration checked at least once every 6 months and pass ±1 percent accuracy and ≤1 percent RSD (precision) based on five replicate readings. The pipettes must be checked weekly for accuracy with a single replicate. Any pipette that does not meet ±1 percent accuracy on the weekly check must be removed from service, repaired, and pass a full calibration check before use.
8.12 Samples with physical deformities are not quantitatively analyzable. The analyst should visually check filters prior to proceeding with preparation for holes, tears, or non-uniform deposit which would prevent representative sampling. Document any deformities and qualify the data with flags appropriately. Care must be taken to protect filters from contamination. Filters must be kept covered prior to sample preparation.
9.0 ICP MS Calibration
Follow the instrument manufacturer's instructions for the routine maintenance, cleaning, and ignition procedures for the specific ICP-MS instrument being used.
9.1 Ignite the plasma and wait for at least one half hour for the instrument to warm up before beginning any pre-analysis steps.
9.2 For the Thermo X-Series with Xt cones, aspirate a 10 ng/mL tuning solution containing In, Bi, and Ce (Cerium). Monitor the intensities of In, Bi, Ce, and CeO (Cerium oxide) and adjust the instrument settings to achieve the highest In and Bi counts while minimizing the CeO/Ce oxide ratio. For other instruments, follow the manufacturer's recommended practice. Tune to meet the instrument manufacturer's specifications. After tuning, place the sample aspiration probe into a 2 percent HNO3 rinse solution for at least 5 minutes to flush the system.
9.3 Aspirate a 5 ng/mL solution containing Co, In, and Bi to perform a daily instrument stability check. Run 10 replicates of the solution. The percent RSD for the replicates must be less than 3 percent at all masses. If the percent RSD is greater than 3 percent, the sample introduction system, pump tubing, and tune should be examined, and the analysis repeated. Place the sample aspiration probe into a 2 percent HNO3 rinse solution for at least 5 minutes to flush the system.
9.4 Load the calibration standards in the autosampler and analyze using the same method parameters that will be used to analyze samples. The curve must include one blank and at least 5 Pb-containing calibration standards. The correlation coefficient must be at least 0.998 for the curve to be accepted. The lowest standard must recover ±15 percent of the expected value and the remaining standards must recover ±10 percent of the expected value to be accepted.
9.5 Immediately after the calibration curve is completed, analyze an ICV and an ICB. The ICV must be prepared from a different source of Pb than the calibration standards. The ICV must recover 90-110 percent of the expected value for the run to continue. The ICB must be less than 0.001 µg/mL. If either the ICV or the ICB fails, the run must be terminated, the problem identified and corrected, and the analysis re-started.
9.6 A LLCV, CCV and a CCB must be run after the ICV and ICB. A CCV and CCB must be run at a frequency of not less than every 10 extracted samples. A typical analytical run sequence would be: Calibration blank, Calibration standards, ICV, ICB, LLCV, CCV, CCB, Extracts 1-10, CCV, CCB, Extracts 11-20, CCV, CCB, Extracts 21-30, CCV, CCB, LLCV, CCV, CCB. Extracts are any field sample or QC samples that have been carried through the extraction process. The CCV solution is prepared from a different source than the calibration standards and may be the same as the ICV solution. The LLCV must be within ±10 percent of expected value. The CCV value must be within ±10 percent of expected for the run to continue. The CCB must be less than 0.001 µg/mL. If either the CCV, LLCV, or CCB fails, the run must be terminated, the problem identified and corrected, and the analysis re-started from the last passing CCV/LLCV/CCB set.
9.7 A LLCV, CCV, and CCB set must be run at the end of the analysis. The LLCV must be within ±30 percent of expected value. If either the CCV, LLCV, or CCB fails, the run must be terminated, the problem identified and corrected, and the analysis re-started from the last passing CCV/LLCV/CCB set.
10.0 Heated Ultrasonic Filter Strip Extraction
All plasticware (e.g., Nalgene) and glassware used in the extraction procedures is soaked in 1 percent HNO3 (v/v) for at least 24 hours and rinsed with reagent water prior to use. All mechanical pipettes used must be calibrated to ±1 percent accuracy and ≤1 percent RSD at a minimum of once every 6 months.
10.1 Sample Preparation - Heated Ultrasonic Bath
10.1.1 Extraction solution (1.03M HNO3 + 2.23M HCl). Prepare by adding 500 mL of deionized water to a 1000 mL flask, adding 64.4 mL of concentrated HNO3 and 182 mL of concentrated HCl, shaking to mix, allowing solution to cool, diluting to volume with reagent water, and inverting several times to mix. Extraction solution must be prepared at least weekly.
10.1.2 Use a ceramic knife and non-metal ruler, or other cutting device that will not contaminate the filter with Pb. Cut a 3/4 inch × 8 inch strip from the glass fiber or quartz filter by cutting a strip from the edge of the filter where it has been folded along the 10 inch side at least 1 inch from the right or left side to avoid the un-sampled area covered by the filter holder. The filters must be carefully handled to avoid dislodging deposits.
10.1.3 Using plastic tweezers, roll the filter strip up in a coil and place the rolled strip in the bottom of a labeled 50 mL extraction tube. In a fume hood, add 15.00 ±0.15 mL of the extraction solution (see Section 10.1.1) using a calibrated mechanical pipette. Ensure that the extraction solution completely covers the filter strip.
10.1.4 Loosely cap the 50 mL extraction tube and place it upright in a plastic rack. When all samples have been prepared, place the racks in an uncovered heated ultrasonic water bath that has been preheated to 80 ±5°C and ensure that the water level in the ultrasonic is above the level of the extraction solution in the tubes but well below the level of the extraction tube caps to avoid contamination. Start the ultrasonic bath and allow the unit to run for 1 hour ±5 minutes at 80 ±5°C.
10.1.5 Remove the rack(s) from the ultrasonic bath and allow the racks to cool.
10.1.6 Add 25.00 ±0.25 mL of D.I. water with a calibrated mechanical pipette to bring the sample to a final volume of 40.0 ±0.4 mL. Tightly cap the tubes, and vortex mix or shake vigorously. Place the extraction tubes in an appropriate holder and centrifuge for 20 minutes at 2500 revolutions per minute (RPM).
CAUTION - Make sure that the centrifuge holder has a flat bottom to support the flat bottomed extraction tubes.
10.1.7 Pour an aliquot of the solution into an autosampler vial for ICP-MS analysis to avoid the potential for contamination. Do not pipette an aliquot of solution into the autosampler vial.
10.1.8 Decant the extract to a clean tube, cap tightly, and store the sample extract at ambient laboratory temperature. Extracts may be stored for up to 6 months from the date of extraction.
10.2 47 mm PTFE Filter Extraction - Heated Ultrasonic Bath
10.2.1 Extraction solution (1.03M HNO3 + 2.23M HCl). Prepare by adding 500 mL of D.I. water to a 1000mL flask, adding 64.4 mL of concentrated HNO3 and 182 mL of concentrated HCl, shaking to mix, allowing solution to cool, diluting to volume with reagent water, and inverting several times to mix. Extraction solution must be prepared at least weekly.
10.2.2 Using plastic tweezers, bend the PTFE filter into a U-shape and insert the filter into a labeled 50 mL extraction tube with the particle loaded side facing the center of the tube. Gently push the filter to the bottom of the extraction tube. In a fume hood, add 25.00 ±0.15 mL of the extraction solution (see Section 10.2.1) using a calibrated mechanical pipette. Ensure that the extraction solution completely covers the filter.
10.2.3 Loosely cap the 50 mL extraction tube and place it upright in a plastic rack. When all samples have been prepared, place the racks in an uncovered heated ultrasonic water bath that has been preheated to 80 ±5°C and ensure that the water level in the ultrasonic is above the level of the extraction solution in the tubes, but well below the level of the extraction tube caps to avoid contamination. Start the ultrasonic bath and allow the unit to run for 1 hour ±5 minutes at 80 ±5°C.
10.2.4 Remove the rack(s) from the ultrasonic bath and allow the racks to cool.
10.2.5 Add 25.00 ±0.25 mL of D.I. water with a calibrated mechanical pipette to bring the sample to a final volume of 50.0 ±0.4 mL. Tightly cap the tubes, and vortex mix or shake vigorously. Allow samples to stand for one hour to allow complete diffusion of the extracted Pb. The sample is now ready for analysis.
Note: Although PTFE filters have only been extracted using the ultrasonic extraction procedure in the development of this FRM, PTFE filters are inert and have very low Pb content. No issues are expected with the extraction of PTFE filters using the heated block digestion method. However, prior to using PTFE filters in the heated block extraction method, extraction method performance test using CRMs must be done to confirm performance (see Section 8.9).
11.0 Hot Block Filter Strip Extraction
All plasticware (e.g., Nalgene) and glassware used in the extraction procedures is soaked in 1 percent HNO3 for at least 24 hours and rinsed with reagent water prior to use. All mechanical pipettes used must be calibrated to ±1 percent accuracy and ≤1 percent RSD at a minimum of once every 6 months.
11.1 Sample Preparation - Hot Block Digestion
11.1.1 Extraction solution (1:19, v/v HNO3). Prepare by adding 500 mL of D.I. water to a 1000 mL flask, adding 50 mL of concentrated HNO3, shaking to mix, allowing solution to cool, diluting to volume with reagent water, and inverting several times to mix. The extraction solution must be prepared at least weekly.
11.1.2 Use a ceramic knife and non-metal ruler, or other cutting device that will not contaminate the filter with Pb. Cut a 1-inch × 8-inch strip from the glass fiber or quartz filter. Cut a strip from the edge of the filter where it has been folded along the 10-inch side at least 1 inch from the right or left side to avoid the un-sampled area covered by the filter holder. The filters must be carefully handled to avoid dislodging particle deposits.
11.1.3 Using plastic tweezers, roll the filter strip up in a coil and place the rolled strip in the bottom of a labeled 50 mL extraction tube. In a fume hood, add 20.0 ±0.15 mL of the extraction solution (see Section 11.1.1) using a calibrated mechanical pipette. Ensure that the extraction solution completely covers the filter strip.
11.1.4 Place the extraction tube in the heated block digester and cover with a disposable polyethylene ribbed watch glass. Heat at 95 ±5°C for 1 hour and ensure that the sample does not evaporate to dryness. For proper heating, adjust the temperature control of the hot block such that an uncovered vessel containing 50 mL of water placed in the center of the hot block can be maintained at a temperature approximately, but no higher than 85C. Once the vessel is covered with a ribbed watch glass, the temperature of the water will increase to approximately 95°C.
11.1.5 Remove the rack(s) from the heated block digester and allow the samples to cool.
11.1.6 Bring the samples to a final volume of 50 mL with D.I. water. Tightly cap the tubes, and vortex mix or shake vigorously for at least 5 seconds. Set aside (with the filter strip in the tube) for at least 30 minutes to allow the HNO3 trapped in the filter to diffuse into the extraction solution.
11.1.7 Shake thoroughly (with the filter strip in the digestion tube) and let settle for at least one hour. The sample is now ready for analysis.
12.0 Measurement Procedure
12.1 Follow the instrument manufacturer's startup procedures for the ICP-MS.
12.2 Set instrument parameters to the appropriate operating conditions as presented in the instrument manufacturer's operating manual and allow the instrument to warm up for at least 30 minutes.
12.3 Calibrate the instrument per Section 9.0 of this method.
12.4 Verify the instrument is suitable for analysis as defined in Sections 9.2 and 9.3.
12.5 As directed in Section 8.0 of this method, analyze an ICV and ICB immediately after the calibration curve followed by a LLCV, then CCV and CCB. The acceptance requirements for these parameters are presented in Section 8.8.
12.6 Analyze a CCV and a CCB after every 10 extracted samples.
12.7 Analyze a LLCV, CCV and CCB at the end of the analysis.
12.8 A typical sample run will include field samples, field sample duplicates, spiked field sample extracts, serially diluted samples, the set of QC samples listed in Section 8.8 above, and one or more CRMs or SRMs.
12.9 Any samples that exceed the highest standard in the calibration curve must be diluted and reanalyzed so that the diluted concentration falls within the calibration curve.
13.0 Results
13.1 The filter results must be initially reported in µg/mL as analyzed. Any additional dilutions must be accounted for. The internal standard recoveries must be included in the result calculation; this is done by the ICP-MS software for most commercially-available instruments. Final results should be reported in µg Pb/m 3 to three significant figures as follows:
C = ((µg Pb/mL * Vf * A)* D))/Vs
Where:
C = Concentration, µg Pb/m 3
µg Pb/mL = Lead concentration in solution
Vf = Total extraction solution volume
A = Area correction; 3/4″ × 8″ strip = 5.25 in 2 analyzed, A = 12.0 or 1″ × 8″ strip = 7 in 2 analyzed, A = 9.0
D = dilution factor (if required)
Vs = Actual volume of air sampled
The calculation assumes the use of a standard 8-inch × 10-inch TSP filter which has a sampled area of 9-inch × 7-inch (63.0 in 2) due to the 1/2-inch filter holder border around the outer edge. The 3/4-inch × 8-inch strip has a sampled area of 3/4-inch × 7-inch (5.25 in 2). The 1-inch × 8-inch strip has a sampled area of 1-inch × 7-inch (7.0 in 2). If filter lot blanks are provided for analysis, refer to Section 7.7.5 of this method for guidance on testing.
14.0 Method Performance
Information in this section is an example of typical performance results achieved by this method. Actual performance must be demonstrated by each individual laboratory and instrument.
14.1 Performance data have been collected to estimate MDLs for this method. MDLs were determined in accordance with 40 CFR 136, Appendix B. MDLs were estimated for glass fiber, quartz, and PTFE filters using seven reagent/filter blank solutions spiked with low level Pb at three times the estimated MDL of 0.001 µg/mL. Tables 1, 3, and 5 shows the MDLs estimated using both the ultrasonic and hot block extraction methods for glass fiber and quartz filters and the ultrasonic method for PTFE filters. The MDLs are well below the EPA requirement of five percent of the current Pb NAAQS or 0.0075 µg/m 3. These MDLs are provided to demonstrate the adequacy of the method's performance for Pb in TSP. Each laboratory using this method should determine MDLs in their laboratory and verify them annually. It is recommended that laboratories also perform the optional iterative procedure in 40 CFR 136, Appendix B to verify the reasonableness of the estimated MDL and subsequent MDL determinations.
14.2 Extraction method recovery tests with glass fiber and quartz filter strips, and PTFE filters spiked with NIST SRMs were performed using the ultrasonic/HNO3 and HCl filter extraction methods and measurement of the dissolved Pb with ICP-MS. Tables 2, 4, and 6 show recoveries obtained with these SRM. The recoveries for all SRMs were ≥90 percent at the 95 percent confidence level.
| Ultrasonic
extraction method | Hotblock
extraction method | |
|---|---|---|
| µg/m 3 | µg/m 3 | |
| n = 1 | 0.0000702 | 0.000533 |
| n = 2 | 0.0000715 | 0.000482 |
| n = 3 | 0.0000611 | 0.000509 |
| n = 4 | 0.0000587 | 0.000427 |
| n = 5 | 0.0000608 | 0.000449 |
| n = 6 | 0.0000607 | 0.000539 |
| n = 7 | 0.0000616 | 0.000481 |
| Average | 0.0000635 | 0.000489 |
| Standard Deviation | 0.0000051 | 0.000042 |
| MDL** | 0.0000161 | 0.000131 |
| * Assumes 2000 m
3 of air sampled. ** MDL is 3.143 times the standard deviation of the results for seven sample replicates analyzed. | ||
| Extraction method | Recovery, ICP-MS, (percent) | |||
|---|---|---|---|---|
| NIST 1547 plant | NIST 2709 soil | NIST 2583 dust | NIST 2582 paint | |
| Ultrasonic Bath | 100 ±4 | 98 ±1 | 103 ±8 | 101 ±0 |
| Block Digestion | 92 ±7 | 98 ±3 | 103 ±4 | 94 ±4 |
| Ultrasonic
extraction method | Hotblock
extraction method | |
|---|---|---|
| µg/m 3* | µg/m 3* | |
| n = 1 | 0.000533 | 0.000274 |
| n = 2 | 0.000552 | 0.000271 |
| n = 3 | 0.000534 | 0.000281 |
| n = 4 | 0.000684 | 0.000269 |
| n = 5 | 0.000532 | 0.000278 |
| n = 6 | 0.000532 | 0.000272 |
| n = 7 | 0.000552 | 0.000261 |
| Average | 0.000560 | 0.000272 |
| Standard Deviation | 0.000055 | 0.000007 |
| MDL** | 0.000174 | 0.000021 |
| * Assumes 2000 m
3 of air sampled. ** MDL is 3.143 times the standard deviation of the results for seven sample replicates analyzed. | ||
| Extraction method | Recovery, ICP-MS, (percent) | |||
|---|---|---|---|---|
| NIST 1547 plant | NIST 2709 soil | NIST 2583 dust | NIST 2582 paint | |
| Ultrasonic Bath | 101 ±6 | 95 ±1 | 91 ±5 | 93 ±1 |
| Block Digestion | 106 ±3 | 104 ±3 | 92 ±6 | 95 ±2 |
| Ultrasonic
extraction method | |
|---|---|
| µg/m 3* | |
| n = 1 | 0.001775 |
| n = 2 | 0.001812 |
| n = 3 | 0.001773 |
| n = 4 | 0.001792 |
| n = 5 | 0.001712 |
| n = 6 | 0.001767 |
| n = 7 | 0.001778 |
| Average | 0.001773 |
| Standard Deviation | 0.000031 |
| MDL** | 0.000097 |
| * Assumes 24 m
3 of air sampled. ** MDL is 3.143 times the standard deviation of the results for seven sample replicates analyzed. | |
| Extraction method | Recovery, ICP-MS, (percent) | |||
|---|---|---|---|---|
| NIST 1547 plant | NIST 2709 soil | NIST 2583 dust | NIST 2582 paint | |
| Ultrasonic Bath | 104 ±5 | 93 ±1 | 108 ±11 | 96 ±3 |
15.0 Pollution Prevention
15.1 Pollution prevention encompasses any technique that reduces or eliminates the quantity and/or toxicity of waste at the point of generation. Numerous opportunities for pollution prevention exist in laboratory operations. Whenever feasible, laboratory personnel should use pollution prevention techniques to address their waste generation. The sources of pollution generated with this procedure are waste acid extracts and Pb-containing solutions.
15.2 For information about pollution prevention that may be applicable to laboratories and research institutions, consult Less is Better: Laboratory Chemical Management for Waste Reduction, available from the American Chemical Society's Department of Government Relations and Science Policy, 1155 16th St. NW., Washington, DC 20036, www.acs.org.
16.0 Waste Management
16.1 Laboratory waste management practices must be conducted consistent with all applicable rules and regulations. Laboratories are urged to protect air, water, and land by minimizing all releases from hood and bench operations, complying with the letter and spirit of any sewer and discharge permits and regulations, and by complying with all solid and hazardous waste regulation. For further information on waste management, consult The Waste Management Manual for Laboratory Personnel available from the American Chemical Society listed in Section 15.2 of this method.
16.2 Waste HNO3, HCl, and solutions containing these reagents and/or Pb must be placed in labeled bottles and delivered to a commercial firm that specializes in removal of hazardous waste.
17.0 References
FACDQ (2007). Report of the Federal Advisory Committee on Detection and Quantitation Approaches and Uses in Clean Water Act Programs, submitted to the U.S. EPA December 2007. Available: http://water.epa.gov/scitech/methods/cwa/det/upload/final-report-200712.pdf.
Rice J (2013). Results from the Development of a New Federal Reference Method (FRM) for Lead in Total Suspended Particulate (TSP) Matter. Docket # EPA-HQ-OAR-2012-0210.
U.S. EPA (2007). Method 6020A - Inductively Coupled Plasma Mass Spectrometry. U.S. Environmental Protection Agency. Revision 1, February 2007. Available: http://www.epa.gov/osw/hazard/testmethods/sw846/pdfs/6020a.pdf.
U.S. EPA (2011). A Laboratory Study of Procedures Evaluated by the Federal Advisory Committee on Detection and Quantitation Approaches and Uses in Clean Water Act Programs. December 2011. Available: http://water.epa.gov/scitech/methods/cwa/det/upload/fac_report_2009.pdf.
[78 FR 40004, July 3, 2013]
Appendix H to Part 50 - Interpretation of the 1-Hour Primary and Secondary National Ambient Air Quality Standards for Ozone
1. General
This appendix explains how to determine when the expected number of days per calendar year with maximum hourly average concentrations above 0.12 ppm (235 µg/m 3) is equal to or less than 1. An expanded discussion of these procedures and associated examples are contained in the “Guideline for Interpretation of Ozone Air Quality Standards.” For purposes of clarity in the following discussion, it is convenient to use the term “exceedance” to describe a daily maximum hourly average ozone measurement that is greater than the level of the standard. Therefore, the phrase “expected number of days with maximum hourly average ozone concentrations above the level of the standard” may be simply stated as the “expected number of exceedances.”
The basic principle in making this determination is relatively straightforward. Most of the complications that arise in determining the expected number of annual exceedances relate to accounting for incomplete sampling. In general, the average number of exceedances per calendar year must be less than or equal to 1. In its simplest form, the number of exceedances at a monitoring site would be recorded for each calendar year and then averaged over the past 3 calendar years to determine if this average is less than or equal to 1.
2. Interpretation of Expected Exceedances
The ozone standard states that the expected number of exceedances per year must be less than or equal to 1. The statistical term “expected number” is basically an arithmetic average. The following example explains what it would mean for an area to be in compliance with this type of standard. Suppose a monitoring station records a valid daily maximum hourly average ozone value for every day of the year during the past 3 years. At the end of each year, the number of days with maximum hourly concentrations above 0.12 ppm is determined and this number is averaged with the results of previous years. As long as this average remains “less than or equal to 1,” the area is in compliance.
3. Estimating the Number of Exceedances for a Year
In general, a valid daily maximum hourly average value may not be available for each day of the year, and it will be necessary to account for these missing values when estimating the number of exceedances for a particular calendar year. The purpose of these computations is to determine if the expected number of exceedances per year is less than or equal to 1. Thus, if a site has two or more observed exceedances each year, the standard is not met and it is not necessary to use the procedures of this section to account for incomplete sampling.
The term “missing value” is used here in the general sense to describe all days that do not have an associated ozone measurement. In some cases, a measurement might actually have been missed but in other cases no measurement may have been scheduled for that day. A daily maximum ozone value is defined to be the highest hourly ozone value recorded for the day. This daily maximum value is considered to be valid if 75 percent of the hours from 9:01 a.m. to 9:00 p.m. (LST) were measured or if the highest hour is greater than the level of the standard.
In some areas, the seasonal pattern of ozone is so pronounced that entire months need not be sampled because it is extremely unlikely that the standard would be exceeded. Any such waiver of the ozone monitoring requirement would be handled under provisions of 40 CFR, part 58. Some allowance should also be made for days for which valid daily maximum hourly values were not obtained but which would quite likely have been below the standard. Such an allowance introduces a complication in that it becomes necessary to define under what conditions a missing value may be assumed to have been less than the level of the standard. The following criterion may be used for ozone:
A missing daily maximum ozone value may be assumed to be less than the level of the standard if the valid daily maxima on both the preceding day and the following day do not exceed 75 percent of the level of the standard.
Let z denote the number of missing daily maximum values that may be assumed to be less than the standard. Then the following formula shall be used to estimate the expected number of exceedances for the year:

(*Indicates multiplication.)
where:
e = the estimated number of exceedances for the year,
N = the number of required monitoring days in the year,
n = the number of valid daily maxima,
v = the number of daily values above the level of the standard, and
z = the number of days assumed to be less than the standard level.
This estimated number of exceedances shall be rounded to one decimal place (fractional parts equal to 0.05 round up).
It should be noted that N will be the total number of days in the year unless the appropriate Regional Administrator has granted a waiver under the provisions of 40 CFR part 58.
The above equation may be interpreted intuitively in the following manner. The estimated number of exceedances is equal to the observed number of exceedances (v) plus an increment that accounts for incomplete sampling. There were (N-n) missing values for the year but a certain number of these, namely z, were assumed to be less than the standard. Therefore, (N-n-z) missing values are considered to include possible exceedances. The fraction of measured values that are above the level of the standard is v/n. It is assumed that this same fraction applies to the (N-n-z) missing values and that (v/n)*(N-n-z) of these values would also have exceeded the level of the standard.
[44 FR 8220, Feb. 8, 1979, as amended at 62 FR 38895, July 18, 1997]
Appendix I to Part 50 - Interpretation of the 8-Hour Primary and Secondary National Ambient Air Quality Standards for Ozone
1. General.
This appendix explains the data handling conventions and computations necessary for determining whether the national 8-hour primary and secondary ambient air quality standards for ozone specified in §50.10 are met at an ambient ozone air quality monitoring site. Ozone is measured in the ambient air by a reference method based on appendix D of this part. Data reporting, data handling, and computation procedures to be used in making comparisons between reported ozone concentrations and the level of the ozone standard are specified in the following sections. Whether to exclude, retain, or make adjustments to the data affected by stratospheric ozone intrusion or other natural events is subject to the approval of the appropriate Regional Administrator.
2. Primary and Secondary Ambient Air Quality Standards for Ozone.
2.1 Data Reporting and Handling Conventions.
2.1.1 Computing 8-hour averages. Hourly average concentrations shall be reported in parts per million (ppm) to the third decimal place, with additional digits to the right being truncated. Running 8-hour averages shall be computed from the hourly ozone concentration data for each hour of the year and the result shall be stored in the first, or start, hour of the 8-hour period. An 8-hour average shall be considered valid if at least 75% of the hourly averages for the 8-hour period are available. In the event that only 6 (or 7) hourly averages are available, the 8-hour average shall be computed on the basis of the hours available using 6 (or 7) as the divisor. (8-hour periods with three or more missing hours shall not be ignored if, after substituting one-half the minimum detectable limit for the missing hourly concentrations, the 8-hour average concentration is greater than the level of the standard.) The computed 8-hour average ozone concentrations shall be reported to three decimal places (the insignificant digits to the right of the third decimal place are truncated, consistent with the data handling procedures for the reported data.)
2.1.2 Daily maximum 8-hour average concentrations. (a) There are 24 possible running 8-hour average ozone concentrations for each calendar day during the ozone monitoring season. (Ozone monitoring seasons vary by geographic location as designated in part 58, appendix D to this chapter.) The daily maximum 8-hour concentration for a given calendar day is the highest of the 24 possible 8-hour average concentrations computed for that day. This process is repeated, yielding a daily maximum 8-hour average ozone concentration for each calendar day with ambient ozone monitoring data. Because the 8-hour averages are recorded in the start hour, the daily maximum 8-hour concentrations from two consecutive days may have some hourly concentrations in common. Generally, overlapping daily maximum 8-hour averages are not likely, except in those non-urban monitoring locations with less pronounced diurnal variation in hourly concentrations.
(b) An ozone monitoring day shall be counted as a valid day if valid 8-hour averages are available for at least 75% of possible hours in the day (i.e., at least 18 of the 24 averages). In the event that less than 75% of the 8-hour averages are available, a day shall also be counted as a valid day if the daily maximum 8-hour average concentration for that day is greater than the level of the ambient standard.
2.2 Primary and Secondary Standard-related Summary Statistic. The standard-related summary statistic is the annual fourth-highest daily maximum 8-hour ozone concentration, expressed in parts per million, averaged over three years. The 3-year average shall be computed using the three most recent, consecutive calendar years of monitoring data meeting the data completeness requirements described in this appendix. The computed 3-year average of the annual fourth-highest daily maximum 8-hour average ozone concentrations shall be expressed to three decimal places (the remaining digits to the right are truncated.)
2.3 Comparisons with the Primary and Secondary Ozone Standards. (a) The primary and secondary ozone ambient air quality standards are met at an ambient air quality monitoring site when the 3-year average of the annual fourth-highest daily maximum 8-hour average ozone concentration is less than or equal to 0.08 ppm. The number of significant figures in the level of the standard dictates the rounding convention for comparing the computed 3-year average annual fourth-highest daily maximum 8-hour average ozone concentration with the level of the standard. The third decimal place of the computed value is rounded, with values equal to or greater than 5 rounding up. Thus, a computed 3-year average ozone concentration of 0.085 ppm is the smallest value that is greater than 0.08 ppm.
(b) This comparison shall be based on three consecutive, complete calendar years of air quality monitoring data. This requirement is met for the three year period at a monitoring site if daily maximum 8-hour average concentrations are available for at least 90%, on average, of the days during the designated ozone monitoring season, with a minimum data completeness in any one year of at least 75% of the designated sampling days. When computing whether the minimum data completeness requirements have been met, meteorological or ambient data may be sufficient to demonstrate that meteorological conditions on missing days were not conducive to concentrations above the level of the standard. Missing days assumed less than the level of the standard are counted for the purpose of meeting the data completeness requirement, subject to the approval of the appropriate Regional Administrator.
(c) Years with concentrations greater than the level of the standard shall not be ignored on the ground that they have less than complete data. Thus, in computing the 3-year average fourth maximum concentration, calendar years with less than 75% data completeness shall be included in the computation if the average annual fourth maximum 8-hour concentration is greater than the level of the standard.
(d) Comparisons with the primary and secondary ozone standards are demonstrated by examples 1 and 2 in paragraphs (d)(1) and (d) (2) respectively as follows:
(1) As shown in example 1, the primary and secondary standards are met at this monitoring site because the 3-year average of the annual fourth-highest daily maximum 8-hour average ozone concentrations (i.e., 0.084 ppm) is less than or equal to 0.08 ppm. The data completeness requirement is also met because the average percent of days with valid ambient monitoring data is greater than 90%, and no single year has less than 75% data completeness.
| Year | Percent Valid Days | 1st Highest Daily Max 8-hour Conc. (ppm) | 2nd Highest Daily Max 8-hour Conc. (ppm) | 3rd Highest Daily Max 8-hour Conc. (ppm) | 4th Highest Daily Max 8-hour Conc. (ppm) | 5th Highest Daily Max 8-hour Conc. (ppm) |
|---|---|---|---|---|---|---|
| 1993 | 100% | 0.092 | 0.091 | 0.090 | 0.088 | 0.085 |
| 1994 | 96% | 0.090 | 0.089 | 0.086 | 0.084 | 0.080 |
| 1995 | 98% | 0.087 | 0.085 | 0.083 | 0.080 | 0.075 |
| Average | 98% |
(2) As shown in example 2, the primary and secondary standards are not met at this monitoring site because the 3-year average of the fourth-highest daily maximum 8-hour average ozone concentrations (i.e., 0.093 ppm) is greater than 0.08 ppm. Note that the ozone concentration data for 1994 is used in these computations, even though the data capture is less than 75%, because the average fourth-highest daily maximum 8-hour average concentration is greater than 0.08 ppm.
| Year | Percent Valid Days | 1st Highest Daily Max 8-hour Conc. (ppm) | 2nd Highest Daily Max 8-hour Conc. (ppm) | 3rd Highest Daily Max 8-hour Conc. (ppm) | 4th Highest Daily Max 8-hour Conc. (ppm) | 5th Highest Daily Max 8-hour Conc. (ppm) |
|---|---|---|---|---|---|---|
| 1993 | 96% | 0.105 | 0.103 | 0.103 | 0.102 | 0.102 |
| 1994 | 74% | 0.090 | 0.085 | 0.082 | 0.080 | 0.078 |
| 1995 | 98% | 0.103 | 0.101 | 0.101 | 0.097 | 0.095 |
| Average | 89% |
3. Design Values for Primary and Secondary Ambient Air Quality Standards for Ozone. The air quality design value at a monitoring site is defined as that concentration that when reduced to the level of the standard ensures that the site meets the standard. For a concentration-based standard, the air quality design value is simply the standard-related test statistic. Thus, for the primary and secondary ozone standards, the 3-year average annual fourth-highest daily maximum 8-hour average ozone concentration is also the air quality design value for the site.
[62 FR 38895, July 18, 1997]
Appendix J to Part 50 - Reference Method for the Determination of Particulate Matter as PM10 in the Atmosphere
1.0 Applicability.
1.1 This method provides for the measurement of the mass concentration of particulate matter with an aerodynamic diameter less than or equal to a nominal 10 micrometers (PM1O) in ambient air over a 24-hour period for purposes of determining attainment and maintenance of the primary and secondary national ambient air quality standards for particulate matter specified in §50.6 of this chapter. The measurement process is nondestructive, and the PM10 sample can be subjected to subsequent physical or chemical analyses. Quality assurance procedures and guidance are provided in part 58, appendices A and B, of this chapter and in References 1 and 2.
2.0 Principle.
2.1 An air sampler draws ambient air at a constant flow rate into a specially shaped inlet where the suspended particulate matter is inertially separated into one or more size fractions within the PM10 size range. Each size fraction in the PM1O size range is then collected on a separate filter over the specified sampling period. The particle size discrimination characteristics (sampling effectiveness and 50 percent cutpoint) of the sampler inlet are prescribed as performance specifications in §53.11 of this chapter.
2.2 Each filter is weighed (after moisture equilibration) before and after use to determine the net weight (mass) gain due to collected PM10. The total volume of air sampled, corrected to EPA reference conditions (25 C, 101.3 kPa), is determined from the measured flow rate and the sampling time. The mass concentration of PM10 in the ambient air is computed as the total mass of collected particles in the PM10 size range divided by the volume of air sampled, and is expressed in micrograms per standard cubic meter (µg/std m 3). For PM10 samples collected at temperatures and pressures significantly different from EPA reference conditions, these corrected concentrations sometimes differ substantially from actual concentrations (in micrograms per actual cubic meter), particularly at high elevations. Although not required, the actual PM10 concentration can be calculated from the corrected concentration, using the average ambient temperature and barometric pressure during the sampling period.
2.3 A method based on this principle will be considered a reference method only if (a) the associated sampler meets the requirements specified in this appendix and the requirements in §53.11 of this chapter, and (b) the method has been designated as a reference method in accordance with §53.11 of this chapter.
3.0 Range.
3.1 The lower limit of the mass concentration range is determined by the repeatability of filter tare weights, assuming the nominal air sample volume for the sampler. For samplers having an automatic filter-changing mechanism, there may be no upper limit. For samplers that do not have an automatic filter-changing mechanism, the upper limit is determined by the filter mass loading beyond which the sampler no longer maintains the operating flow rate within specified limits due to increased pressure drop across the loaded filter. This upper limit cannot be specified precisely because it is a complex function of the ambient particle size distribution and type, humidity, filter type, and perhaps other factors. Nevertheless, all samplers should be capable of measuring 24-hour PM10 mass concentrations of at least 300 µg/std m 3 while maintaining the operating flow rate within the specified limits.
4.0 Precision.
4.1 The precision of PM10 samplers must be 5 µg/m 3 for PM10 concentrations below 80 µg/m 3 and 7 percent for PM10 concentrations above 80 µg/m 3, as required by §53.11 of this chapter, which prescribes a test procedure that determines the variation in the PM10 concentration measurements of identical samplers under typical sampling conditions. Continual assessment of precision via collocated samplers is required by §53.11 of this chapter for PM10 samplers used in certain monitoring networks.
5.0 Accuracy.
5.1 Because the size of the particles making up ambient particulate matter varies over a wide range and the concentration of particles varies with particle size, it is difficult to define the absolute accuracy of PM10 samplers. Part 53 of this chapter provides a specification for the sampling effectiveness of PM10 samplers. This specification requires that the expected mass concentration calculated for a candidate PM10 sampler, when sampling a specified particle size distribution, be within ±10 percent of that calculated for an ideal sampler whose sampling effectiveness is explicitly specified. Also, the particle size for 50 percent sampling effectiveness is required to be 10 ±0.5 micrometers. Other specifications related to accuracy apply to flow measurement and calibration, filter media, analytical (weighing) procedures, and artifact. The flow rate accuracy of PM10 samplers used in certain monitoring networks is required by §53.11 of this chapter to be assessed periodically via flow rate audits.
6.0 Potential Sources of Error.
6.1 Volatile Particles. Volatile particles collected on filters are often lost during shipment and/or storage of the filters prior to the post-sampling weighing 3. Although shipment or storage of loaded filters is sometimes unavoidable, filters should be reweighed as soon as practical to minimize these losses.
6.2 Artifacts. Positive errors in PM10 concentration measurements may result from retention of gaseous species on filters. 4 5 Such errors include the retention of sulfur dioxide and nitric acid. Retention of sulfur dioxide on filters, followed by oxidation to sulfate, is referred to as artifact sulfate formation, a phenomenon which increases with increasing filter alkalinity. 6 Little or no artifact sulfate formation should occur using filters that meet the alkalinity specification in section 7.2.4. Artifact nitrate formation, resulting primarily from retention of nitric acid, occurs to varying degrees on many filter types, including glass fiber, cellulose ester, and many quartz fiber filters. 5 7 8 9 10 Loss of true atmospheric particulate nitrate during or following sampling may also occur due to dissociation or chemical reaction. This phenomenon has been observed on Teflon ® filters 8 and inferred for quartz fiber filters. 11 12 The magnitude of nitrate artifact errors in PM10 mass concentration measurements will vary with location and ambient temperature; however, for most sampling locations, these errors are expected to be small.
6.3 Humidity. The effects of ambient humidity on the sample are unavoidable. The filter equilibration procedure in section 9.0 is designed to minimize the effects of moisture on the filter medium.
6.4 Filter Handling. Careful handling of filters between presampling and postsampling weighings is necessary to avoid errors due to damaged filters or loss of collected particles from the filters. Use of a filter cartridge or cassette may reduce the magnitude of these errors. Filters must also meet the integrity specification in section 7.2.3.
6.5 Flow Rate Variation. Variations in the sampler's operating flow rate may alter the particle size discrimination characteristics of the sampler inlet. The magnitude of this error will depend on the sensitivity of the inlet to variations in flow rate and on the particle distribution in the atmosphere during the sampling period. The use of a flow control device (section 7.1.3) is required to minimize this error.
6.6 Air Volume Determination. Errors in the air volume determination may result from errors in the flow rate and/or sampling time measurements. The flow control device serves to minimize errors in the flow rate determination, and an elapsed time meter (section 7.1.5) is required to minimize the error in the sampling time measurement.
7.0 Apparatus.
7.1 PM10Sampler.
7.1.1 The sampler shall be designed to:
a. Draw the air sample into the sampler inlet and through the particle collection filter at a uniform face velocity.
b. Hold and seal the filter in a horizontal position so that sample air is drawn downward through the filter.
c. Allow the filter to be installed and removed conveniently.
d. Protect the filter and sampler from precipitation and prevent insects and other debris from being sampled.
e. Minimize air leaks that would cause error in the measurement of the air volume passing through the filter.
f. Discharge exhaust air at a sufficient distance from the sampler inlet to minimize the sampling of exhaust air.
g. Minimize the collection of dust from the supporting surface.
7.1.2 The sampler shall have a sample air inlet system that, when operated within a specified flow rate range, provides particle size discrimination characteristics meeting all of the applicable performance specifications prescribed in §53.11 of this chapter. The sampler inlet shall show no significant wind direction dependence. The latter requirement can generally be satisfied by an inlet shape that is circularly symmetrical about a vertical axis.
7.1.3 The sampler shall have a flow control device capable of maintaining the sampler's operating flow rate within the flow rate limits specified for the sampler inlet over normal variations in line voltage and filter pressure drop.
7.1.4 The sampler shall provide a means to measure the total flow rate during the sampling period. A continuous flow recorder is recommended but not required. The flow measurement device shall be accurate to ±2 percent.
7.1.5 A timing/control device capable of starting and stopping the sampler shall be used to obtain a sample collection period of 24 ±1 hr (1,440 ±60 min). An elapsed time meter, accurate to within ±15 minutes, shall be used to measure sampling time. This meter is optional for samplers with continuous flow recorders if the sampling time measurement obtained by means of the recorder meets the ±15 minute accuracy specification.
7.1.6 The sampler shall have an associated operation or instruction manual as required by §53.11 of this chapter which includes detailed instructions on the calibration, operation, and maintenance of the sampler.
7.2 Filters.
7.2.1 Filter Medium. No commercially available filter medium is ideal in all respects for all samplers. The user's goals in sampling determine the relative importance of various filter characteristics (e.g., cost, ease of handling, physical and chemical characteristics, etc.) and, consequently, determine the choice among acceptable filters. Furthermore, certain types of filters may not be suitable for use with some samplers, particularly under heavy loading conditions (high mass concentrations), because of high or rapid increase in the filter flow resistance that would exceed the capability of the sampler's flow control device. However, samplers equipped with automatic filter-changing mechanisms may allow use of these types of filters. The specifications given below are minimum requirements to ensure acceptability of the filter medium for measurement of PM10 mass concentrations. Other filter evaluation criteria should be considered to meet individual sampling and analysis objectives.
7.2.2 Collection Efficiency. ≥99 percent, as measured by the DOP test (ASTM-2986) with 0.3 µm particles at the sampler's operating face velocity.
7.2.3 Integrity. ±5 µg/m 3 (assuming sampler's nominal 24-hour air sample volume). Integrity is measured as the PM10 concentration equivalent corresponding to the average difference between the initial and the final weights of a random sample of test filters that are weighed and handled under actual or simulated sampling conditions, but have no air sample passed through them (i.e., filter blanks). As a minimum, the test procedure must include initial equilibration and weighing, installation on an inoperative sampler, removal from the sampler, and final equilibration and weighing.
7.2.4 Alkalinity. <25 microequivalents/gram of filter, as measured by the procedure given in Reference 13 following at least two months storage in a clean environment (free from contamination by acidic gases) at room temperature and humidity.
7.3 Flow Rate Transfer Standard. The flow rate transfer standard must be suitable for the sampler's operating flow rate and must be calibrated against a primary flow or volume standard that is traceable to the National Bureau of Standards (NBS). The flow rate transfer standard must be capable of measuring the sampler's operating flow rate with an accuracy of ±2 percent.
7.4 Filter Conditioning Environment.
7.4.1 Temperature range: 15 to 30 C.
7.4.2 Temperature control: ±3 C.
7.4.3 Humidity range: 20% to 45% RH.
7.4.4 Humidity control: ±5% RH.
7.5 Analytical Balance. The analytical balance must be suitable for weighing the type and size of filters required by the sampler. The range and sensitivity required will depend on the filter tare weights and mass loadings. Typically, an analytical balance with a sensitivity of 0.1 mg is required for high volume samplers (flow rates >0.5 m 3/min). Lower volume samplers (flow rates <0.5 m 3/min) will require a more sensitive balance.
8.0 Calibration.
8.1 General Requirements.
8.1.1 Calibration of the sampler's flow measurement device is required to establish traceability of subsequent flow measurements to a primary standard. A flow rate transfer standard calibrated against a primary flow or volume standard shall be used to calibrate or verify the accuracy of the sampler's flow measurement device.
8.1.2 Particle size discrimination by inertial separation requires that specific air velocities be maintained in the sampler's air inlet system. Therefore, the flow rate through the sampler's inlet must be maintained throughout the sampling period within the design flow rate range specified by the manufacturer. Design flow rates are specified as actual volumetric flow rates, measured at existing conditions of temperature and pressure (Qa). In contrast, mass concentrations of PM10 are computed using flow rates corrected to EPA reference conditions of temperature and pressure (Qstd).
8.2 Flow Rate Calibration Procedure.
8.2.1 PM10 samplers employ various types of flow control and flow measurement devices. The specific procedure used for flow rate calibration or verification will vary depending on the type of flow controller and flow indicator employed. Calibration in terms of actual volumetric flow rates (Qa) is generally recommended, but other measures of flow rate (e.g., Qstd) may be used provided the requirements of section 8.1 are met. The general procedure given here is based on actual volumetric flow units (Qa) and serves to illustrate the steps involved in the calibration of a PM10 sampler. Consult the sampler manufacturer's instruction manual and Reference 2 for specific guidance on calibration. Reference 14 provides additional information on the use of the commonly used measures of flow rate and their interrelationships.
8.2.2 Calibrate the flow rate transfer standard against a primary flow or volume standard traceable to NBS. Establish a calibration relationship (e.g., an equation or family of curves) such that traceability to the primary standard is accurate to within 2 percent over the expected range of ambient conditions (i.e., temperatures and pressures) under which the transfer standard will be used. Recalibrate the transfer standard periodically.
8.2.3 Following the sampler manufacturer's instruction manual, remove the sampler inlet and connect the flow rate transfer standard to the sampler such that the transfer standard accurately measures the sampler's flow rate. Make sure there are no leaks between the transfer standard and the sampler.
8.2.4 Choose a minimum of three flow rates (actual m 3/min), spaced over the acceptable flow rate range specified for the inlet (see 7.1.2) that can be obtained by suitable adjustment of the sampler flow rate. In accordance with the sampler manufacturer's instruction manual, obtain or verify the calibration relationship between the flow rate (actual m 3/min) as indicated by the transfer standard and the sampler's flow indicator response. Record the ambient temperature and barometric pressure. Temperature and pressure corrections to subsequent flow indicator readings may be required for certain types of flow measurement devices. When such corrections are necessary, correction on an individual or daily basis is preferable. However, seasonal average temperature and average barometric pressure for the sampling site may be incorporated into the sampler calibration to avoid daily corrections. Consult the sampler manufacturer's instruction manual and Reference 2 for additional guidance.
8.2.5 Following calibration, verify that the sampler is operating at its design flow rate (actual m 3/min) with a clean filter in place.
8.2.6 Replace the sampler inlet.
9.0 Procedure.
9.1 The sampler shall be operated in accordance with the specific guidance provided in the sampler manufacturer's instruction manual and in Reference 2. The general procedure given here assumes that the sampler's flow rate calibration is based on flow rates at ambient conditions (Qa) and serves to illustrate the steps involved in the operation of a PM10 sampler.
9.2 Inspect each filter for pinholes, particles, and other imperfections. Establish a filter information record and assign an identification number to each filter.
9.3 Equilibrate each filter in the conditioning environment (see 7.4) for at least 24 hours.
9.4 Following equilibration, weigh each filter and record the presampling weight with the filter identification number.
9.5 Install a preweighed filter in the sampler following the instructions provided in the sampler manufacturer's instruction manual.
9.6 Turn on the sampler and allow it to establish run-temperature conditions. Record the flow indicator reading and, if needed, the ambient temperature and barometric pressure. Determine the sampler flow rate (actual m 3/min) in accordance with the instructions provided in the sampler manufacturer's instruction manual. NOTE. - No onsite temperature or pressure measurements are necessary if the sampler's flow indicator does not require temperature or pressure corrections or if seasonal average temperature and average barometric pressure for the sampling site are incorporated into the sampler calibration (see step 8.2.4). If individual or daily temperature and pressure corrections are required, ambient temperature and barometric pressure can be obtained by on-site measurements or from a nearby weather station. Barometric pressure readings obtained from airports must be station pressure, not corrected to sea level, and may need to be corrected for differences in elevation between the sampling site and the airport.
9.7 If the flow rate is outside the acceptable range specified by the manufacturer, check for leaks, and if necessary, adjust the flow rate to the specified setpoint. Stop the sampler.
9.8 Set the timer to start and stop the sampler at appropriate times. Set the elapsed time meter to zero or record the initial meter reading.
9.9 Record the sample information (site location or identification number, sample date, filter identification number, and sampler model and serial number).
9.10 Sample for 24 ±1 hours.
9.11 Determine and record the average flow rate (Q͞ a) in actual m 3/min for the sampling period in accordance with the instructions provided in the sampler manufacturer's instruction manual. Record the elapsed time meter final reading and, if needed, the average ambient temperature and barometric pressure for the sampling period (see note following step 9.6).
9.12 Carefully remove the filter from the sampler, following the sampler manufacturer's instruction manual. Touch only the outer edges of the filter.
9.13 Place the filter in a protective holder or container (e.g., petri dish, glassine envelope, or manila folder).
9.14 Record any factors such as meteorological conditions, construction activity, fires or dust storms, etc., that might be pertinent to the measurement on the filter information record.
9.15 Transport the exposed sample filter to the filter conditioning environment as soon as possible for equilibration and subsequent weighing.
9.16 Equilibrate the exposed filter in the conditioning environment for at least 24 hours under the same temperature and humidity conditions used for presampling filter equilibration (see 9.3).
9.17 Immediately after equilibration, reweigh the filter and record the postsampling weight with the filter identification number.
10.0 Sampler Maintenance.
10.1 The PM10 sampler shall be maintained in strict accordance with the maintenance procedures specified in the sampler manufacturer's instruction manual.
11.0 Calculations.
11.1 Calculate the average flow rate over the sampling period corrected to EPA reference conditions as Q͞ std. When the sampler's flow indicator is calibrated in actual volumetric units (Qa), Q͞ std is calculated as:
Q͞ std = Q͞ a × (Pav/Tav)(Tstd/Pstd)
where
Q͞ std = average flow rate at EPA reference conditions, std m 3/min;
Q͞ a = average flow rate at ambient conditions, m 3/min;
Pav = average barometric pressure during the sampling period or average barometric pressure for the sampling site, kPa (or mm Hg);
Tav = average ambient temperature during the sampling period or seasonal average ambient temperature for the sampling site, K;
Tstd = standard temperature, defined as 298 K;
Pstd = standard pressure, defined as 101.3 kPa (or 760 mm Hg).
11.2 Calculate the total volume of air sampled as:
Vstd = Q͞ std × t
where
Vstd = total air sampled in standard volume units, std m 3;
t = sampling time, min.
11.3 Calculate the PM10 concentration as:
PM10 = (Wf−Wi) × 10 6/Vstd
where
PM10 = mass concentration of PM10, µg/std m 3;
Wf, Wi = final and initial weights of filter collecting PM1O particles, g;
10 6 = conversion of g to µg.
Note:
If more than one size fraction in the PM10 size range is collected by the sampler, the sum of the net weight gain by each collection filter [Σ(Wf−Wi)] is used to calculate the PM10 mass concentration.
12.0 References.
1. Quality Assurance Handbook for Air Pollution Measurement Systems, Volume I, Principles. EPA-600/9-76-005, March 1976. Available from CERI, ORD Publications, U.S. Environmental Protection Agency, 26 West St. Clair Street, Cincinnati, OH 45268.
2. Quality Assurance Handbook for Air Pollution Measurement Systems, Volume II, Ambient Air Specific Methods. EPA-600/4-77-027a, May 1977. Available from CERI, ORD Publications, U.S. Environmental Protection Agency, 26 West St. Clair Street, Cincinnati, OH 45268.
3. Clement, R.E., and F.W. Karasek. Sample Composition Changes in Sampling and Analysis of Organic Compounds in Aerosols. Int. J. Environ. Analyt. Chem., 7:109, 1979.
4. Lee, R.E., Jr., and J. Wagman. A Sampling Anomaly in the Determination of Atmospheric Sulfate Concentration. Amer. Ind. Hyg. Assoc. J., 27:266, 1966.
5. Appel, B.R., S.M. Wall, Y. Tokiwa, and M. Haik. Interference Effects in Sampling Particulate Nitrate in Ambient Air. Atmos. Environ., 13:319, 1979.
6. Coutant, R.W. Effect of Environmental Variables on Collection of Atmospheric Sulfate. Environ. Sci. Technol., 11:873, 1977.
7. Spicer, C.W., and P. Schumacher. Interference in Sampling Atmospheric Particulate Nitrate. Atmos. Environ., 11:873, 1977.
8. Appel, B.R., Y. Tokiwa, and M. Haik. Sampling of Nitrates in Ambient Air. Atmos. Environ., 15:283, 1981.
9. Spicer, C.W., and P.M. Schumacher. Particulate Nitrate: Laboratory and Field Studies of Major Sampling Interferences. Atmos. Environ., 13:543, 1979.
10. Appel, B.R. Letter to Larry Purdue, U.S. EPA, Environmental Monitoring and Support Laboratory. March 18, 1982, Docket No. A-82-37, II-I-1.
11. Pierson, W.R., W.W. Brachaczek, T.J. Korniski, T.J. Truex, and J.W. Butler. Artifact Formation of Sulfate, Nitrate, and Hydrogen Ion on Backup Filters: Allegheny Mountain Experiment. J. Air Pollut. Control Assoc., 30:30, 1980.
12. Dunwoody, C.L. Rapid Nitrate Loss From PM10 Filters. J. Air Pollut. Control Assoc., 36:817, 1986.
13. Harrell, R.M. Measuring the Alkalinity of Hi-Vol Air Filters. EMSL/RTP-SOP-QAD-534, October 1985. Available from the U.S. Environmental Protection Agency, EMSL/QAD, Research Triangle Park, NC 27711.
14. Smith, F., P.S. Wohlschlegel, R.S.C. Rogers, and D.J. Mulligan. Investigation of Flow Rate Calibration Procedures Associated With the High Volume Method for Determination of Suspended Particulates. EPA-600/4-78-047, U.S. Environmental Protection Agency, Research Triangle Park, NC 27711, 1978.
[52 FR 24664, July 1, 1987; 52 FR 29467, Aug. 7, 1987]
Appendix K to Part 50 - Interpretation of the National Ambient Air Quality Standards for Particulate Matter
1.0 General
(a) This appendix explains the computations necessary for analyzing particulate matter data to determine attainment of the 24-hour standards specified in 40 CFR 50.6. For the primary and secondary standards, particulate matter is measured in the ambient air as PM10 (particles with an aerodynamic diameter less than or equal to a nominal 10 micrometers) by a reference method based on appendix J of this part and designated in accordance with §53.11 of this chapter, or by an equivalent method designated in accordance with §53.11 of this chapter. The required frequency of measurements is specified in §53.11 of this chapter.
(b) The terms used in this appendix are defined as follows:
Average refers to the arithmetic mean of the estimated number of exceedances per year, as per section 3.1 of this appendix.
Collocated monitors refer to two or more air measurement instruments for the same parameter (e.g., PM 10 mass) operated at the same site location, and whose placement is consistent with part 53 of this chapter. For purposes of considering a combined site record in this appendix, when two or more monitors are operated at the same site, one monitor is designated as the “primary” monitor with any additional monitors designated as “collocated.” It is implicit in these appendix procedures that the primary monitor and collocated monitor(s) are all reference or equivalent methods; however, it is not a requirement that the primary and collocated monitors utilize the same specific sampling and analysis method.
Combined site data record is the data set used for performing computations in this appendix and represents data for the primary monitors augmented with data from collocated monitors according to the procedure specified in section 3.0(a) of this appendix.
Daily value for PM 10 refers to the 24-hour average concentration of PM 10 calculated or measured from midnight to midnight (local time).
Exceedance means a daily value that is above the level of the 24-hour standard after rounding to the nearest 10 µg/m 3 i.e., values ending in 5 or greater are to be rounded up).
Expected annual value is the number approached when the annual values from an increasing number of years are averaged, in the absence of long-term trends in emissions or meteorological conditions.
Primary monitors are suitable monitors designated by a State or local agency in their annual network plan as the default data source for creating a combined site data record. If there is only one suitable monitor at a particular site location, then it is presumed to be a primary monitor.
Year refers to a calendar year.
(c) Although the discussion in this appendix focuses on monitored data, the same principles apply to modeling data, subject to EPA modeling guidelines.
2.0 Attainment Determinations
2.1 24-Hour Primary and Secondary Standards
(a) Under 40 CFR 50.6(a) the 24-hour primary and secondary standards are attained when the expected number of exceedances per year at each monitoring site is less than or equal to one. In the simplest case, the number of expected exceedances at a site is determined by recording the number of exceedances in each calendar year and then averaging them over the past 3 calendar years. Situations in which 3 years of data are not available and possible adjustments for unusual events or trends are discussed in sections 2.3 and 2.4 of this appendix. Further, when data for a year are incomplete, it is necessary to compute an estimated number of exceedances for that year by adjusting the observed number of exceedances. This procedure, performed by calendar quarter, is described in section 3.0 of this appendix. The expected number of exceedances is then estimated by averaging the individual annual estimates for the past 3 years.
(b) The comparison with the allowable expected exceedance rate of one per year is made in terms of a number rounded to the nearest tenth (fractional values equal to or greater than 0.05 are to be rounded up; e.g., an exceedance rate of 1.05 would be rounded to 1.1, which is the lowest rate for nonattainment).
2.2 Reserved
2.3 Data Requirements
(a) 40 CFR 58.12 specifies the required minimum frequency of sampling for PM10. For the purposes of making comparisons with the particulate matter standards, all data produced by State and Local Air Monitoring Stations (SLAMS) and other sites submitted to EPA in accordance with the §53.11 requirements must be used, and a minimum of 75 percent of the scheduled PM10 samples per quarter are required.
(b) To demonstrate attainment of the 24-hour standards at a monitoring site, the monitor must provide sufficient data to perform the required calculations of sections 3.0 and 4.0 of this appendix. The amount of data required varies with the sampling frequency, data capture rate and the number of years of record. In all cases, 3 years of representative monitoring data that meet the 75 percent criterion of the previous paragraph should be utilized, if available, and would suffice. More than 3 years may be considered, if all additional representative years of data meeting the 75 percent criterion are utilized. Data not meeting these criteria may also suffice to show attainment; however, such exceptions will have to be approved by the appropriate Regional Administrator in accordance with EPA guidance.
(c) There are less stringent data requirements for showing that a monitor has failed an attainment test and thus has recorded a violation of the particulate matter standards. Although it is generally necessary to meet the minimum 75 percent data capture requirement per quarter to use the computational equations described in section 3.0 of this appendix, this criterion does not apply when less data is sufficient to unambiguously establish nonattainment. The following examples illustrate how nonattainment can be demonstrated when a site fails to meet the completeness criteria. Nonattainment of the 24-hour primary standards can be established by the observed annual number of exceedances (e.g., four observed exceedances in a single year), or by the estimated number of exceedances derived from the observed number of exceedances and the required number of scheduled samples (e.g., two observed exceedances with every other day sampling). In both cases, expected annual values must exceed the levels allowed by the standards.
(d) 24-hour average concentrations will be computed from submitted hourly PM 10 concentration data for each corresponding day of the year and the result will be stored in the first, or start, hour ( i.e., midnight, hour `0') of the 24-hour period. A 24-hour average concentration shall be considered valid if at least 75 percent of the hourly averages ( i.e., 18 hourly values) for the 24-hour period are available. In the event that fewer than all 24 hourly average concentrations are available ( i.e., fewer than 24 but at least 18), the 24-hour average concentration shall be computed on the basis of the hours available using the number of available hours within the 24-hour period as the divisor ( e.g., the divisor is 19 if 19 hourly values are available). 24-hour periods with 7 or more missing hours shall also be considered for computations in this appendix if, after substituting zero for all missing hourly concentrations, the resulting 24-hour average daily value exceeds the level of the 24-hour standard specified in §50.6 after rounding to the nearest 10 µg/m 3 .
2.4 Adjustment for Exceptional Events and Trends
(a) An exceptional event is an uncontrollable event caused by natural sources of particulate matter or an event that is not expected to recur at a given location. Inclusion of such a value in the computation of exceedances or averages could result in inappropriate estimates of their respective expected annual values. To reduce the effect of unusual events, more than 3 years of representative data may be used. Alternatively, other techniques, such as the use of statistical models or the use of historical data could be considered so that the event may be discounted or weighted according to the likelihood that it will recur. The use of such techniques is subject to the approval of the appropriate Regional Administrator in accordance with EPA guidance.
(b) In cases where long-term trends in emissions and air quality are evident, mathematical techniques should be applied to account for the trends to ensure that the expected annual values are not inappropriately biased by unrepresentative data. In the simplest case, if 3 years of data are available under stable emission conditions, this data should be used. In the event of a trend or shift in emission patterns, either the most recent representative year(s) could be used or statistical techniques or models could be used in conjunction with previous years of data to adjust for trends. The use of less than 3 years of data, and any adjustments are subject to the approval of the appropriate Regional Administrator in accordance with EPA guidance.
3.0 Computational Equations for the 24-Hour Standards
(a) All computations shown in this appendix shall be implemented on a site-level basis. Site level concentration data shall be processed as follows:
(1) The default dataset for PM 10 mass concentrations for a site shall consist of the measured concentrations recorded from the designated primary monitor(s). All daily values produced by the primary monitor are considered part of the site record.
(2) If a daily value is not produced by the primary monitor for a particular day, but a value is available from a single collocated monitor, then that collocated monitor value shall be considered part of the combined site data record. If daily value data is available from two or more collocated monitors, the average of those collocated values shall be used as the daily value. The data record resulting from this procedure is referred to as the “combined site data record.”
(b) In certain circumstances, including but not limited to site closures or relocations, data from two nearby sites may be combined into a single site data record for the purpose of calculating a valid design value. The appropriate Regional Administrator may approve such combinations if the Regional Administrator determines that the measured concentrations do not differ substantially between the two sites, taking into consideration factors such as distance between sites, spatial and temporal patterns in air quality, local emissions and meteorology, jurisdictional boundaries, and terrain features.
3.1 Estimating Exceedances for a Year
(a) If PM10 sampling is scheduled less frequently than every day, or if some scheduled samples are missed, a PM10 value will not be available for each day of the year. To account for the possible effect of incomplete data, an adjustment must be made to the data collected at each monitoring location to estimate the number of exceedances in a calendar year. In this adjustment, the assumption is made that the fraction of missing values that would have exceeded the standard level is identical to the fraction of measured values above this level. This computation is to be made for all sites that are scheduled to monitor throughout the entire year and meet the minimum data requirements of section 2.3 of this appendix. Because of possible seasonal imbalance, this adjustment shall be applied on a quarterly basis. The estimate of the expected number of exceedances for the quarter is equal to the observed number of exceedances plus an increment associated with the missing data. The following equation must be used for these computations:
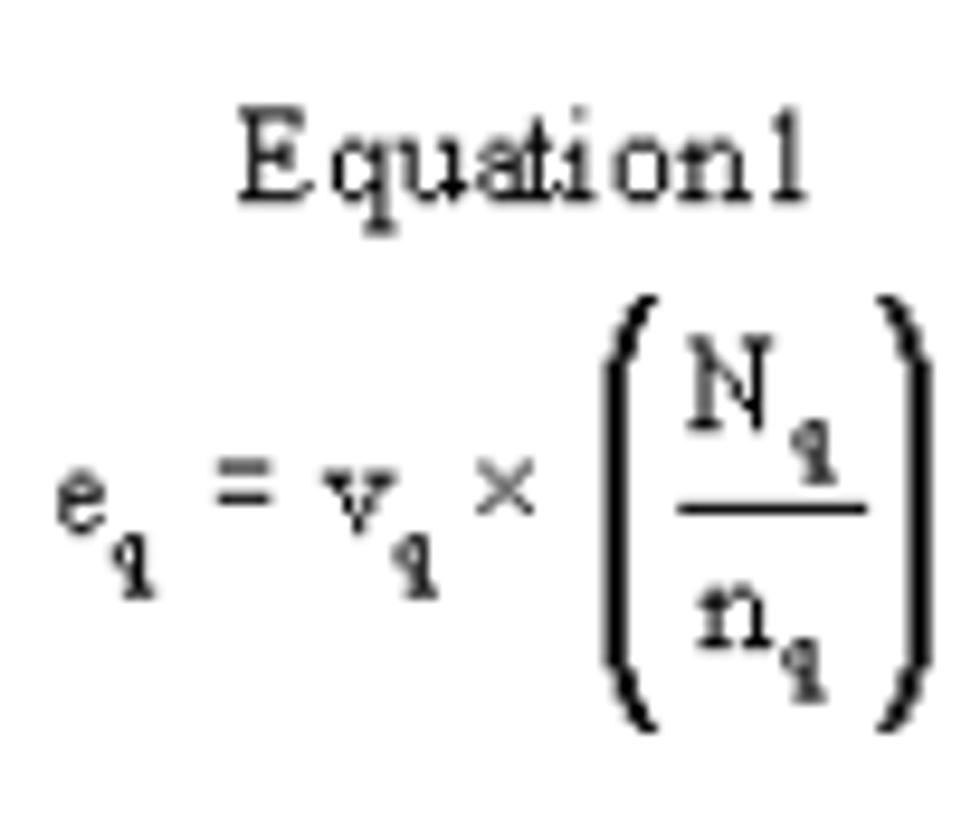
Where:
eq = the estimated number of exceedances for calendar quarter q;
vq = the observed number of exceedances for calendar quarter q;
Nq = the number of days in calendar quarter q;
nq = the number of days in calendar quarter q with PM10 data; and
q = the index for calendar quarter, q = 1, 2, 3 or 4.
(b) The estimated number of exceedances for a calendar quarter must be rounded to the nearest hundredth (fractional values equal to or greater than 0.005 must be rounded up).
(c) The estimated number of exceedances for the year, e, is the sum of the estimates for each calendar quarter.
(d) The estimated number of exceedances for a single year must be rounded to one decimal place (fractional values equal to or greater than 0.05 are to be rounded up). The expected number of exceedances is then estimated by averaging the individual annual estimates for the most recent 3 or more representative years of data. The expected number of exceedances must be rounded to one decimal place (fractional values equal to or greater than 0.05 are to be rounded up).
(e) The adjustment for incomplete data will not be necessary for monitoring or modeling data which constitutes a complete record, i.e., 365 days per year.
(f) To reduce the potential for overestimating the number of expected exceedances, the correction for missing data will not be required for a calendar quarter in which the first observed exceedance has occurred if:
(1) There was only one exceedance in the calendar quarter;
(2) Everyday sampling is subsequently initiated and maintained for 4 calendar quarters in accordance with 40 CFR 58.12; and
(3) Data capture of 75 percent is achieved during the required period of everyday sampling. In addition, if the first exceedance is observed in a calendar quarter in which the monitor is already sampling every day, no adjustment for missing data will be made to the first exceedance if a 75 percent data capture rate was achieved in the quarter in which it was observed.
Example 1
a. During a particular calendar quarter, 39 out of a possible 92 samples were recorded, with one observed exceedance of the 24-hour standard. Using Equation 1, the estimated number of exceedances for the quarter is:
eq = 1 × 92/39 = 2.359 or 2.36.
b. If the estimated exceedances for the other 3 calendar quarters in the year were 2.30, 0.0 and 0.0, then, using Equation 2, the estimated number of exceedances for the year is 2.36 + 2.30 + 0.0 + 0.0 which equals 4.66 or 4.7. If no exceedances were observed for the 2 previous years, then the expected number of exceedances is estimated by: ( 1/3) × (4.7 + 0 + 0) = 1.57 or 1.6. Since 1.6 exceeds the allowable number of expected exceedances, this monitoring site would fail the attainment test.
Example 2
In this example, everyday sampling was initiated following the first observed exceedance as required by 40 CFR 58.12. Accordingly, the first observed exceedance would not be adjusted for incomplete sampling. During the next three quarters, 1.2 exceedances were estimated. In this case, the estimated exceedances for the year would be 1.0 + 1.2 + 0.0 + 0.0 which equals 2.2. If, as before, no exceedances were observed for the two previous years, then the estimated exceedances for the 3-year period would then be ( 1/3) × (2.2 + 0.0 + 0.0) = 0.7, and the monitoring site would not fail the attainment test.
3.2 Adjustments for Non-Scheduled Sampling Days
(a) If a systematic sampling schedule is used and sampling is performed on days in addition to the days specified by the systematic sampling schedule, e.g., during episodes of high pollution, then an adjustment must be made in the equation for the estimation of exceedances. Such an adjustment is needed to eliminate the bias in the estimate of the quarterly and annual number of exceedances that would occur if the chance of an exceedance is different for scheduled than for non-scheduled days, as would be the case with episode sampling.
(b) The required adjustment treats the systematic sampling schedule as a stratified sampling plan. If the period from one scheduled sample until the day preceding the next scheduled sample is defined as a sampling stratum, then there is one stratum for each scheduled sampling day. An average number of observed exceedances is computed for each of these sampling strata. With nonscheduled sampling days, the estimated number of exceedances is defined as:
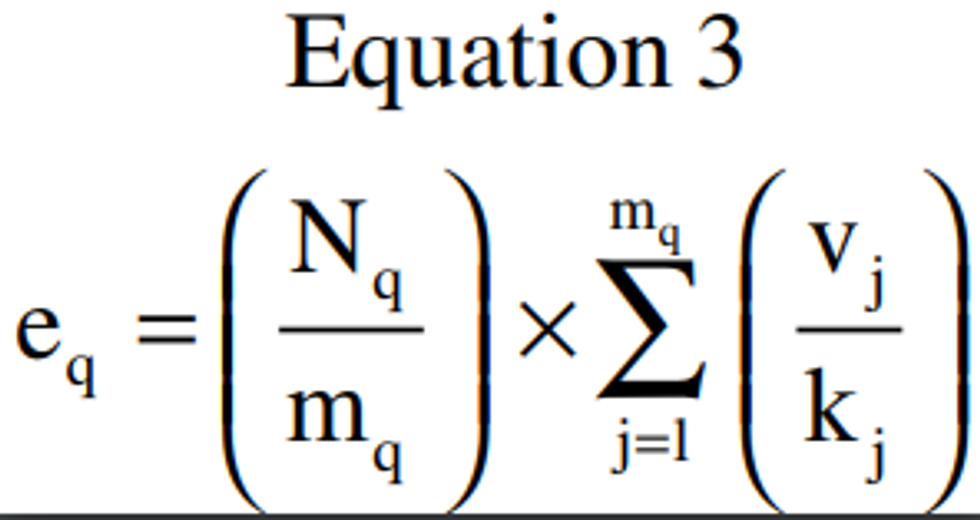
Where:
eq = the estimated number of exceedances for the quarter;
Nq = the number of days in the quarter;
mq = the number of strata with samples during the quarter;
vj = the number of observed exceedances in stratum j; and
kj = the number of actual samples in stratum j.
(c) Note that if only one sample value is recorded in each stratum, then Equation 3 reduces to Equation 1.
Example 3
A monitoring site samples according to a systematic sampling schedule of one sample every 6 days, for a total of 15 scheduled samples in a quarter out of a total of 92 possible samples. During one 6-day period, potential episode levels of PM10 were suspected, so 5 additional samples were taken. One of the regular scheduled samples was missed, so a total of 19 samples in 14 sampling strata were measured. The one 6-day sampling stratum with 6 samples recorded 2 exceedances. The remainder of the quarter with one sample per stratum recorded zero exceedances. Using Equation 3, the estimated number of exceedances for the quarter is:
Eq = (92/14) × (2/6 + 0 + . . . + 0) = 2.19.
[71 FR 61224, Oct. 17, 2006; 89 FR 16380, March 6, 2024]
Appendix L to Part 50 - Reference Method for the Determination of Fine Particulate Matter as PM2.5 in the Atmosphere
1.0 Applicability.
1.1 This method provides for the measurement of the mass concentration of fine particulate matter having an aerodynamic diameter less than or equal to a nominal 2.5 micrometers (PM2.5) in ambient air over a 24-hour period for purposes of determining whether the primary and secondary national ambient air quality standards for fine particulate matter specified in §50.7 and §50.13 of this part are met. The measurement process is considered to be nondestructive, and the PM2.5 sample obtained can be subjected to subsequent physical or chemical analyses. Quality assessment procedures are provided in part 58, appendix A of this chapter, and quality assurance guidance are provided in references 1, 2, and 3 in section 13.0 of this appendix.
1.2 This method will be considered a reference method for purposes of §53.11 of this chapter only if:
(a) The associated sampler meets the requirements specified in this appendix and the applicable requirements in §53.11 of this chapter, and
(b) The method and associated sampler have been designated as a reference method in accordance with §53.11 of this chapter.
1.3 PM2.5 samplers that meet nearly all specifications set forth in this method but have minor deviations and/or modifications of the reference method sampler will be designated as “Class I” equivalent methods for PM2.5 in accordance with §53.11 of this chapter.
2.0 Principle.
2.1 An electrically powered air sampler draws ambient air at a constant volumetric flow rate into a specially shaped inlet and through an inertial particle size separator (impactor) where the suspended particulate matter in the PM2.5 size range is separated for collection on a polytetrafluoroethylene (PTFE) filter over the specified sampling period. The air sampler and other aspects of this reference method are specified either explicitly in this appendix or generally with reference to other applicable regulations or quality assurance guidance.
2.2 Each filter is weighed (after moisture and temperature conditioning) before and after sample collection to determine the net gain due to collected PM2.5. The total volume of air sampled is determined by the sampler from the measured flow rate at actual ambient temperature and pressure and the sampling time. The mass concentration of PM2.5 in the ambient air is computed as the total mass of collected particles in the PM2.5 size range divided by the actual volume of air sampled, and is expressed in micrograms per cubic meter of air (µg/m 3).
3.0 PM 2.5 Measurement Range.
3.1 Lower concentration limit. The lower detection limit of the mass concentration measurement range is estimated to be approximately 2 µg/m 3, based on noted mass changes in field blanks in conjunction with the 24 m 3 nominal total air sample volume specified for the 24-hour sample.
3.2 Upper concentration limit. The upper limit of the mass concentration range is determined by the filter mass loading beyond which the sampler can no longer maintain the operating flow rate within specified limits due to increased pressure drop across the loaded filter. This upper limit cannot be specified precisely because it is a complex function of the ambient particle size distribution and type, humidity, the individual filter used, the capacity of the sampler flow rate control system, and perhaps other factors. Nevertheless, all samplers are estimated to be capable of measuring 24-hour PM2.5 mass concentrations of at least 200 µg/m 3 while maintaining the operating flow rate within the specified limits.
3.3 Sample period. The required sample period for PM2.5 concentration measurements by this method shall be 1,380 to 1500 minutes (23 to 25 hours). However, when a sample period is less than 1,380 minutes, the measured concentration (as determined by the collected PM2.5 mass divided by the actual sampled air volume), multiplied by the actual number of minutes in the sample period and divided by 1,440, may be used as if it were a valid concentration measurement for the specific purpose of determining a violation of the NAAQS. This value assumes that the PM2.5 concentration is zero for the remaining portion of the sample period and therefore represents the minimum concentration that could have been measured for the full 24-hour sample period. Accordingly, if the value thus calculated is high enough to be an exceedance, such an exceedance would be a valid exceedance for the sample period. When reported to AIRS, this data value should receive a special code to identify it as not to be commingled with normal concentration measurements or used for other purposes.
4.0 Accuracy.
4.1 Because the size and volatility of the particles making up ambient particulate matter vary over a wide range and the mass concentration of particles varies with particle size, it is difficult to define the accuracy of PM2.5 measurements in an absolute sense. The accuracy of PM2.5 measurements is therefore defined in a relative sense, referenced to measurements provided by this reference method. Accordingly, accuracy shall be defined as the degree of agreement between a subject field PM2.5 sampler and a collocated PM2.5 reference method audit sampler operating simultaneously at the monitoring site location of the subject sampler and includes both random (precision) and systematic (bias) errors. The requirements for this field sampler audit procedure are set forth in part 58, appendix A of this chapter.
4.2 Measurement system bias. Results of collocated measurements where the duplicate sampler is a reference method sampler are used to assess a portion of the measurement system bias according to the schedule and procedure specified in part 58, appendix A of this chapter.
4.3 Audits with reference method samplers to determine system accuracy and bias. According to the schedule and procedure specified in part 58, appendix A of this chapter, a reference method sampler is required to be located at each of selected PM2.5 SLAMS sites as a duplicate sampler. The results from the primary sampler and the duplicate reference method sampler are used to calculate accuracy of the primary sampler on a quarterly basis, bias of the primary sampler on an annual basis, and bias of a single reporting organization on an annual basis. Reference 2 in section 13.0 of this appendix provides additional information and guidance on these reference method audits.
4.4 Flow rate accuracy and bias. Part 58, appendix A of this chapter requires that the flow rate accuracy and bias of individual PM2.5 samplers used in SLAMS monitoring networks be assessed periodically via audits of each sampler's operational flow rate. In addition, part 58, appendix A of this chapter requires that flow rate bias for each reference and equivalent method operated by each reporting organization be assessed quarterly and annually. Reference 2 in section 13.0 of this appendix provides additional information and guidance on flow rate accuracy audits and calculations for accuracy and bias.
5.0 Precision. A data quality objective of 10 percent coefficient of variation or better has been established for the operational precision of PM2.5 monitoring data.
5.1 Tests to establish initial operational precision for each reference method sampler are specified as a part of the requirements for designation as a reference method under §53.58 of this chapter.
5.2 Measurement System Precision. Collocated sampler results, where the duplicate sampler is not a reference method sampler but is a sampler of the same designated method as the primary sampler, are used to assess measurement system precision according to the schedule and procedure specified in part 58, appendix A of this chapter. Part 58, appendix A of this chapter requires that these collocated sampler measurements be used to calculate quarterly and annual precision estimates for each primary sampler and for each designated method employed by each reporting organization. Reference 2 in section 13.0 of this appendix provides additional information and guidance on this requirement.
6.0 Filter for PM 2.5 Sample Collection. Any filter manufacturer or vendor who sells or offers to sell filters specifically identified for use with this PM2.5 reference method shall certify that the required number of filters from each lot of filters offered for sale as such have been tested as specified in this section 6.0 and meet all of the following design and performance specifications.
6.1 Size. Circular, 46.2 mm diameter ±0.25 mm.
6.2 Medium. Polytetrafluoroethylene (PTFE Teflon), with integral support ring.
6.3 Support ring. Polymethylpentene (PMP) or equivalent inert material, 0.38 ±0.04 mm thick, outer diameter 46.2 mm ±0.25 mm, and width of 3.68 mm (±0.00, −0.51 mm).
6.4 Pore size. 2 µm as measured by ASTM F 316-94.
6.5 Filter thickness. 30 to 50 µm.
6.6 Maximum pressure drop (clean filter). 30 cm H2O column @ 16.67 L/min clean air flow.
6.7 Maximum moisture pickup. Not more than 10 µg weight increase after 24-hour exposure to air of 40 percent relative humidity, relative to weight after 24-hour exposure to air of 35 percent relative humidity.
6.8 Collection efficiency. Greater than 99.7 percent, as measured by the DOP test (ASTM D 2986-91) with 0.3 µm particles at the sampler's operating face velocity.
6.9 Filter weight stability. Filter weight loss shall be less than 20 µg, as measured in each of the following two tests specified in sections 6.9.1 and 6.9.2 of this appendix. The following conditions apply to both of these tests: Filter weight loss shall be the average difference between the initial and the final filter weights of a random sample of test filters selected from each lot prior to sale. The number of filters tested shall be not less than 0.1 percent of the filters of each manufacturing lot, or 10 filters, whichever is greater. The filters shall be weighed under laboratory conditions and shall have had no air sample passed through them, i.e., filter blanks. Each test procedure must include initial conditioning and weighing, the test, and final conditioning and weighing. Conditioning and weighing shall be in accordance with sections 8.0 through 8.2 of this appendix and general guidance provided in reference 2 of section 13.0 of this appendix.
6.9.1 Test for loose, surface particle contamination. After the initial weighing, install each test filter, in turn, in a filter cassette (Figures L-27, L-28, and L-29 of this appendix) and drop the cassette from a height of 25 cm to a flat hard surface, such as a particle-free wood bench. Repeat two times, for a total of three drop tests for each test filter. Remove the test filter from the cassette and weigh the filter. The average change in weight must be less than 20 µg.
6.9.2 Test for temperature stability. After weighing each filter, place the test filters in a drying oven set at 40°C ±2°C for not less than 48 hours. Remove, condition, and reweigh each test filter. The average change in weight must be less than 20 µg.
6.10 Alkalinity. Less than 25 microequivalents/gram of filter, as measured by the guidance given in reference 2 in section 13.0 of this appendix.
6.11 Supplemental requirements. Although not required for determination of PM2.5 mass concentration under this reference method, additional specifications for the filter must be developed by users who intend to subject PM2.5 filter samples to subsequent chemical analysis. These supplemental specifications include background chemical contamination of the filter and any other filter parameters that may be required by the method of chemical analysis. All such supplemental filter specifications must be compatible with and secondary to the primary filter specifications given in this section 6.0 of this appendix.
7.0 PM 2.5 Sampler.
7.1 Configuration. The sampler shall consist of a sample air inlet, downtube, particle size separator (impactor), filter holder assembly, air pump and flow rate control system, flow rate measurement device, ambient and filter temperature monitoring system, barometric pressure measurement system, timer, outdoor environmental enclosure, and suitable mechanical, electrical, or electronic control capability to meet or exceed the design and functional performance as specified in this section 7.0 of this appendix. The performance specifications require that the sampler:
(a) Provide automatic control of sample volumetric flow rate and other operational parameters.
(b) Monitor these operational parameters as well as ambient temperature and pressure.
(c) Provide this information to the sampler operator at the end of each sample period in digital form, as specified in table L-1 of section 7.4.19 of this appendix.
7.2 Nature of specifications. The PM2.5 sampler is specified by a combination of design and performance requirements. The sample inlet, downtube, particle size discriminator, filter cassette, and the internal configuration of the filter holder assembly are specified explicitly by design figures and associated mechanical dimensions, tolerances, materials, surface finishes, assembly instructions, and other necessary specifications. All other aspects of the sampler are specified by required operational function and performance, and the design of these other aspects (including the design of the lower portion of the filter holder assembly) is optional, subject to acceptable operational performance. Test procedures to demonstrate compliance with both the design and performance requirements are set forth in subpart E of §53.11 of this chapter.
7.3 Design specifications. Except as indicated in this section 7.3 of this appendix, these components must be manufactured or reproduced exactly as specified, in an ISO 9001-registered facility, with registration initially approved and subsequently maintained during the period of manufacture. See §53.1(t) of this chapter for the definition of an ISO-registered facility. Minor modifications or variances to one or more components that clearly would not affect the aerodynamic performance of the inlet, downtube, impactor, or filter cassette will be considered for specific approval. Any such proposed modifications shall be described and submitted to the EPA for specific individual acceptability either as part of a reference or equivalent method application under §53.11 of this chapter or in writing in advance of such an intended application under §53.11 of this chapter.
7.3.1 Sample inlet assembly. The sample inlet assembly, consisting of the inlet, downtube, and impactor shall be configured and assembled as indicated in Figure L-1 of this appendix and shall meet all associated requirements. A portion of this assembly shall also be subject to the maximum overall sampler leak rate specification under section 7.4.6 of this appendix.
7.3.2 Inlet. The sample inlet shall be fabricated as indicated in Figures L-2 through L-18 of this appendix and shall meet all associated requirements.
7.3.3 Downtube. The downtube shall be fabricated as indicated in Figure L-19 of this appendix and shall meet all associated requirements.
7.3.4 Particle size separator. The sampler shall be configured with one of the three alternative particle size separators described in this section. One separator is an impactor-type separator (WINS impactor) described in sections 7.3.4.1, 7.3.4.2, and 7.3.4.3 of this appendix. One alternative separator is a cyclone-type separator (VSCC TM) described in section 7.3.4.4 of this appendix. The other alternative separator is also a cyclone-type separator (TE–PM 2.5 C) described in section 7.3.4.5 of this appendix.
7.3.4.1 The impactor (particle size separator) shall be fabricated as indicated in Figures L-20 through L-24 of this appendix and shall meet all associated requirements. Following the manufacture and finishing of each upper impactor housing (Figure L-21 of this appendix), the dimension of the impaction jet must be verified by the manufacturer using Class ZZ go/no-go plug gauges that are traceable to NIST.
7.3.4.2 Impactor filter specifications:
(a) Size. Circular, 35 to 37 mm diameter.
(b) Medium. Borosilicate glass fiber, without binder.
(c) Pore size. 1 to 1.5 micrometer, as measured by ASTM F 316-80.
(d) Thickness. 300 to 500 micrometers.
7.3.4.3 Impactor oil specifications:
(a) Composition. Dioctyl sebacate (DOS), single-compound diffusion oil.
(b) Vapor pressure. Maximum 2 × 10−8 mm Hg at 25°C.
(c) Viscosity. 36 to 40 centistokes at 25°C.
(d) Density. 1.06 to 1.07 g/cm 3 at 25°C.
(e) Quantity. 1 mL ±0.1 mL.
7.3.4.4 The cyclone-type separator is identified as a BGI VSCC TM Very Sharp Cut Cyclone particle size separator specified as part of EPA-designated equivalent method EQPM-0202-142 (67 FR 15567, April 2, 2002) and as manufactured by BGI Incorporated, 58 Guinan Street, Waltham, Massachusetts 20451.
7.3.4.5 A second cyclone-type separator is identified as a Tisch TE–PM 2.5 C Cyclone particle size separator specified as part of EPA-designated reference method RFPS–1014–219 and as manufactured by Tisch Environmental Incorporated, 145 S. Miami Avenue, Village of Cleves, Ohio 45002.
7.3.5 Filter holder assembly. The sampler shall have a sample filter holder assembly to adapt and seal to the down tube and to hold and seal the specified filter, under section 6.0 of this appendix, in the sample air stream in a horizontal position below the downtube such that the sample air passes downward through the filter at a uniform face velocity. The upper portion of this assembly shall be fabricated as indicated in Figures L-25 and L-26 of this appendix and shall accept and seal with the filter cassette, which shall be fabricated as indicated in Figures L-27 through L-29 of this appendix.
(a) The lower portion of the filter holder assembly shall be of a design and construction that:
(1) Mates with the upper portion of the assembly to complete the filter holder assembly,
(2) Completes both the external air seal and the internal filter cassette seal such that all seals are reliable over repeated filter changings, and
(3) Facilitates repeated changing of the filter cassette by the sampler operator.
(b) Leak-test performance requirements for the filter holder assembly are included in section 7.4.6 of this appendix.
(c) If additional or multiple filters are stored in the sampler as part of an automatic sequential sample capability, all such filters, unless they are currently and directly installed in a sampling channel or sampling configuration (either active or inactive), shall be covered or (preferably) sealed in such a way as to:
(1) Preclude significant exposure of the filter to possible contamination or accumulation of dust, insects, or other material that may be present in the ambient air, sampler, or sampler ventilation air during storage periods either before or after sampling; and
(2) To minimize loss of volatile or semi-volatile PM sample components during storage of the filter following the sample period.
7.3.6 Flow rate measurement adapter. A flow rate measurement adapter as specified in Figure L-30 of this appendix shall be furnished with each sampler.
7.3.7 Surface finish. All internal surfaces exposed to sample air prior to the filter shall be treated electrolytically in a sulfuric acid bath to produce a clear, uniform anodized surface finish of not less than 1000 mg/ft 2 (1.08 mg/cm 2) in accordance with military standard specification (mil. spec.) 8625F, Type II, Class 1 in reference 4 of section 13.0 of this appendix. This anodic surface coating shall not be dyed or pigmented. Following anodization, the surfaces shall be sealed by immersion in boiling deionized water for not less than 15 minutes. Section 53.51(d)(2) of this chapter should also be consulted.
7.3.8 Sampling height. The sampler shall be equipped with legs, a stand, or other means to maintain the sampler in a stable, upright position and such that the center of the sample air entrance to the inlet, during sample collection, is maintained in a horizontal plane and is 2.0 ±0.2 meters above the floor or other horizontal supporting surface. Suitable bolt holes, brackets, tie-downs, or other means should be provided to facilitate mechanically securing the sample to the supporting surface to prevent toppling of the sampler due to wind.
7.4 Performance specifications.
7.4.1 Sample flow rate. Proper operation of the impactor requires that specific air velocities be maintained through the device. Therefore, the design sample air flow rate through the inlet shall be 16.67 L/min (1.000 m 3/hour) measured as actual volumetric flow rate at the temperature and pressure of the sample air entering the inlet.
7.4.2 Sample air flow rate control system. The sampler shall have a sample air flow rate control system which shall be capable of providing a sample air volumetric flow rate within the specified range, under section 7.4.1 of this appendix, for the specified filter, under section 6.0 of this appendix, at any atmospheric conditions specified, under section 7.4.7 of this appendix, at a filter pressure drop equal to that of a clean filter plus up to 75 cm water column (55 mm Hg), and over the specified range of supply line voltage, under section 7.4.15.1 of this appendix. This flow control system shall allow for operator adjustment of the operational flow rate of the sampler over a range of at least ±15 percent of the flow rate specified in section 7.4.1 of this appendix.
7.4.3 Sample flow rate regulation. The sample flow rate shall be regulated such that for the specified filter, under section 6.0 of this appendix, at any atmospheric conditions specified, under section 7.4.7 of this appendix, at a filter pressure drop equal to that of a clean filter plus up to 75 cm water column (55 mm Hg), and over the specified range of supply line voltage, under section 7.4.15.1 of this appendix, the flow rate is regulated as follows:
7.4.3.1 The volumetric flow rate, measured or averaged over intervals of not more than 5 minutes over a 24-hour period, shall not vary more than ±5 percent from the specified 16.67 L/min flow rate over the entire sample period.
7.4.3.2 The coefficient of variation (sample standard deviation divided by the mean) of the flow rate, measured over a 24-hour period, shall not be greater than 2 percent.
7.4.3.3 The amplitude of short-term flow rate pulsations, such as may originate from some types of vacuum pumps, shall be attenuated such that they do not cause significant flow measurement error or affect the collection of particles on the particle collection filter.
7.4.4 Flow rate cut off. The sampler's sample air flow rate control system shall terminate sample collection and stop all sample flow for the remainder of the sample period in the event that the sample flow rate deviates by more than 10 percent from the sampler design flow rate specified in section 7.4.1 of this appendix for more than 60 seconds. However, this sampler cut-off provision shall not apply during periods when the sampler is inoperative due to a temporary power interruption, and the elapsed time of the inoperative period shall not be included in the total sample time measured and reported by the sampler, under section 7.4.13 of this appendix.
7.4.5 Flow rate measurement.
7.4.5.1 The sampler shall provide a means to measure and indicate the instantaneous sample air flow rate, which shall be measured as volumetric flow rate at the temperature and pressure of the sample air entering the inlet, with an accuracy of ±2 percent. The measured flow rate shall be available for display to the sampler operator at any time in either sampling or standby modes, and the measurement shall be updated at least every 30 seconds. The sampler shall also provide a simple means by which the sampler operator can manually start the sample flow temporarily during non-sampling modes of operation, for the purpose of checking the sample flow rate or the flow rate measurement system.
7.4.5.2 During each sample period, the sampler's flow rate measurement system shall automatically monitor the sample volumetric flow rate, obtaining flow rate measurements at intervals of not greater than 30 seconds.
(a) Using these interval flow rate measurements, the sampler shall determine or calculate the following flow-related parameters, scaled in the specified engineering units:
(1) The instantaneous or interval-average flow rate, in L/min.
(2) The value of the average sample flow rate for the sample period, in L/min.
(3) The value of the coefficient of variation (sample standard deviation divided by the average) of the sample flow rate for the sample period, in percent.
(4) The occurrence of any time interval during the sample period in which the measured sample flow rate exceeds a range of ±5 percent of the average flow rate for the sample period for more than 5 minutes, in which case a warning flag indicator shall be set.
(5) The value of the integrated total sample volume for the sample period, in m 3.
(b) Determination or calculation of these values shall properly exclude periods when the sampler is inoperative due to temporary interruption of electrical power, under section 7.4.13 of this appendix, or flow rate cut off, under section 7.4.4 of this appendix.
(c) These parameters shall be accessible to the sampler operator as specified in table L-1 of section 7.4.19 of this appendix. In addition, it is strongly encouraged that the flow rate for each 5-minute interval during the sample period be available to the operator following the end of the sample period.
7.4.6 Leak test capability.
7.4.6.1 External leakage. The sampler shall include an external air leak-test capability consisting of components, accessory hardware, operator interface controls, a written procedure in the associated Operation/Instruction Manual, under section 7.4.18 of this appendix, and all other necessary functional capability to permit and facilitate the sampler operator to conveniently carry out a leak test of the sampler at a field monitoring site without additional equipment. The sampler components to be subjected to this leak test include all components and their interconnections in which external air leakage would or could cause an error in the sampler's measurement of the total volume of sample air that passes through the sample filter.
(a) The suggested technique for the operator to use for this leak test is as follows:
(1) Remove the sampler inlet and installs the flow rate measurement adapter supplied with the sampler, under section 7.3.6 of this appendix.
(2) Close the valve on the flow rate measurement adapter and use the sampler air pump to draw a partial vacuum in the sampler, including (at least) the impactor, filter holder assembly (filter in place), flow measurement device, and interconnections between these devices, of at least 55 mm Hg (75 cm water column), measured at a location downstream of the filter holder assembly.
(3) Plug the flow system downstream of these components to isolate the components under vacuum from the pump, such as with a built-in valve.
(4) Stop the pump.
(5) Measure the trapped vacuum in the sampler with a built-in pressure measuring device.
(6) (i) Measure the vacuum in the sampler with the built-in pressure measuring device again at a later time at least 10 minutes after the first pressure measurement.
(ii) Caution: Following completion of the test, the adaptor valve should be opened slowly to limit the flow rate of air into the sampler. Excessive air flow rate may blow oil out of the impactor.
(7) Upon completion of the test, open the adaptor valve, remove the adaptor and plugs, and restore the sampler to the normal operating configuration.
(b) The associated leak test procedure shall require that for successful passage of this test, the difference between the two pressure measurements shall not be greater than the number of mm of Hg specified for the sampler by the manufacturer, based on the actual internal volume of the sampler, that indicates a leak of less than 80 mL/min.
(c) Variations of the suggested technique or an alternative external leak test technique may be required for samplers whose design or configuration would make the suggested technique impossible or impractical. The specific proposed external leak test procedure, or particularly an alternative leak test technique, proposed for a particular candidate sampler may be described and submitted to the EPA for specific individual acceptability either as part of a reference or equivalent method application under §53.11 of this chapter or in writing in advance of such an intended application under §53.11 of this chapter.
7.4.6.2 Internal, filter bypass leakage. The sampler shall include an internal, filter bypass leak-check capability consisting of components, accessory hardware, operator interface controls, a written procedure in the Operation/Instruction Manual, and all other necessary functional capability to permit and facilitate the sampler operator to conveniently carry out a test for internal filter bypass leakage in the sampler at a field monitoring site without additional equipment. The purpose of the test is to determine that any portion of the sample flow rate that leaks past the sample filter without passing through the filter is insignificant relative to the design flow rate for the sampler.
(a) The suggested technique for the operator to use for this leak test is as follows:
(1) Carry out an external leak test as provided under section 7.4.6.1 of this appendix which indicates successful passage of the prescribed external leak test.
(2) Install a flow-impervious membrane material in the filter cassette, either with or without a filter, as appropriate, which effectively prevents air flow through the filter.
(3) Use the sampler air pump to draw a partial vacuum in the sampler, downstream of the filter holder assembly, of at least 55 mm Hg (75 cm water column).
(4) Plug the flow system downstream of the filter holder to isolate the components under vacuum from the pump, such as with a built-in valve.
(5) Stop the pump.
(6) Measure the trapped vacuum in the sampler with a built-in pressure measuring device.
(7) Measure the vacuum in the sampler with the built-in pressure measuring device again at a later time at least 10 minutes after the first pressure measurement.
(8) Remove the flow plug and membrane and restore the sampler to the normal operating configuration.
(b) The associated leak test procedure shall require that for successful passage of this test, the difference between the two pressure measurements shall not be greater than the number of mm of Hg specified for the sampler by the manufacturer, based on the actual internal volume of the portion of the sampler under vacuum, that indicates a leak of less than 80 mL/min.
(c) Variations of the suggested technique or an alternative internal, filter bypass leak test technique may be required for samplers whose design or configuration would make the suggested technique impossible or impractical. The specific proposed internal leak test procedure, or particularly an alternative internal leak test technique proposed for a particular candidate sampler may be described and submitted to the EPA for specific individual acceptability either as part of a reference or equivalent method application under §53.11 of this chapter or in writing in advance of such intended application under §53.11 of this chapter.
7.4.7 Range of operational conditions. The sampler is required to operate properly and meet all requirements specified in this appendix over the following operational ranges.
7.4.7.1 Ambient temperature. −30 to = 45°C (Note: Although for practical reasons, the temperature range over which samplers are required to be tested under §53.11 of this chapter is −20 to = 40°C, the sampler shall be designed to operate properly over this wider temperature range.).
7.4.7.2 Ambient relative humidity. 0 to 100 percent.
7.4.7.3 Barometric pressure range. 600 to 800 mm Hg.
7.4.8 Ambient temperature sensor. The sampler shall have capability to measure the temperature of the ambient air surrounding the sampler over the range of −30 to = 45°C, with a resolution of 0.1°C and accuracy of ±2.0°C, referenced as described in reference 3 in section 13.0 of this appendix, with and without maximum solar insolation.
7.4.8.1 The ambient temperature sensor shall be mounted external to the sampler enclosure and shall have a passive, naturally ventilated sun shield. The sensor shall be located such that the entire sun shield is at least 5 cm above the horizontal plane of the sampler case or enclosure (disregarding the inlet and downtube) and external to the vertical plane of the nearest side or protuberance of the sampler case or enclosure. The maximum temperature measurement error of the ambient temperature measurement system shall be less than 1.6°C at 1 m/s wind speed and 1000 W/m2 solar radiation intensity.
7.4.8.2 The ambient temperature sensor shall be of such a design and mounted in such a way as to facilitate its convenient dismounting and immersion in a liquid for calibration and comparison to the filter temperature sensor, under section 7.4.11 of this appendix.
7.4.8.3 This ambient temperature measurement shall be updated at least every 30 seconds during both sampling and standby (non-sampling) modes of operation. A visual indication of the current (most recent) value of the ambient temperature measurement, updated at least every 30 seconds, shall be available to the sampler operator during both sampling and standby (non-sampling) modes of operation, as specified in table L-1 of section 7.4.19 of this appendix.
7.4.8.4 This ambient temperature measurement shall be used for the purpose of monitoring filter temperature deviation from ambient temperature, as required by section 7.4.11 of this appendix, and may be used for purposes of effecting filter temperature control, under section 7.4.10 of this appendix, or computation of volumetric flow rate, under sections 7.4.1 to 7.4.5 of this appendix, if appropriate.
7.4.8.5 Following the end of each sample period, the sampler shall report the maximum, minimum, and average temperature for the sample period, as specified in table L-1 of section 7.4.19 of this appendix.
7.4.9 Ambient barometric sensor. The sampler shall have capability to measure the barometric pressure of the air surrounding the sampler over a range of 600 to 800 mm Hg referenced as described in reference 3 in section 13.0 of this appendix; also see part 53, subpart E of this chapter. This barometric pressure measurement shall have a resolution of 5 mm Hg and an accuracy of ±10 mm Hg and shall be updated at least every 30 seconds. A visual indication of the value of the current (most recent) barometric pressure measurement, updated at least every 30 seconds, shall be available to the sampler operator during both sampling and standby (non-sampling) modes of operation, as specified in table L-1 of section 7.4.19 of this appendix. This barometric pressure measurement may be used for purposes of computation of volumetric flow rate, under sections 7.4.1 to 7.4.5 of this appendix, if appropriate. Following the end of a sample period, the sampler shall report the maximum, minimum, and mean barometric pressures for the sample period, as specified in table L-1 of section 7.4.19 of this appendix.
7.4.10 Filter temperature control (sampling and post-sampling). The sampler shall provide a means to limit the temperature rise of the sample filter (all sample filters for sequential samplers), from insolation and other sources, to no more 5°C above the temperature of the ambient air surrounding the sampler, during both sampling and post-sampling periods of operation. The post-sampling period is the non-sampling period between the end of the active sampling period and the time of retrieval of the sample filter by the sampler operator.
7.4.11 Filter temperature sensor(s).
7.4.11.1 The sampler shall have the capability to monitor the temperature of the sample filter (all sample filters for sequential samplers) over the range of −30 to = 45°C during both sampling and non-sampling periods. While the exact location of this temperature sensor is not explicitly specified, the filter temperature measurement system must demonstrate agreement, within 1°C, with a test temperature sensor located within 1 cm of the center of the filter downstream of the filter during both sampling and non-sampling modes, as specified in the filter temperature measurement test described in part 53, subpart E of this chapter. This filter temperature measurement shall have a resolution of 0.1°C and accuracy of ±1.0°C, referenced as described in reference 3 in section 13.0 of this appendix. This temperature sensor shall be of such a design and mounted in such a way as to facilitate its reasonably convenient dismounting and immersion in a liquid for calibration and comparison to the ambient temperature sensor under section 7.4.8 of this appendix.
7.4.11.2 The filter temperature measurement shall be updated at least every 30 seconds during both sampling and standby (non-sampling) modes of operation. A visual indication of the current (most recent) value of the filter temperature measurement, updated at least every 30 seconds, shall be available to the sampler operator during both sampling and standby (non-sampling) modes of operation, as specified in table L-1 of section 7.4.19 of this appendix.
7.4.11.3 For sequential samplers, the temperature of each filter shall be measured individually unless it can be shown, as specified in the filter temperature measurement test described in §53.57 of this chapter, that the temperature of each filter can be represented by fewer temperature sensors.
7.4.11.4 The sampler shall also provide a warning flag indicator following any occurrence in which the filter temperature (any filter temperature for sequential samplers) exceeds the ambient temperature by more than 5°C for more than 30 consecutive minutes during either the sampling or post-sampling periods of operation, as specified in table L-1 of section 7.4.19 of this appendix, under section 10.12 of this appendix, regarding sample validity when a warning flag occurs. It is further recommended (not required) that the sampler be capable of recording the maximum differential between the measured filter temperature and the ambient temperature and its time and date of occurrence during both sampling and post-sampling (non-sampling) modes of operation and providing for those data to be accessible to the sampler operator following the end of the sample period, as suggested in table L-1 of section 7.4.19 of this appendix.
7.4.12 Clock/timer system.
(a) The sampler shall have a programmable real-time clock timing/control system that:
(1) Is capable of maintaining local time and date, including year, month, day-of-month, hour, minute, and second to an accuracy of ±1.0 minute per month.
(2) Provides a visual indication of the current system time, including year, month, day-of-month, hour, and minute, updated at least each minute, for operator verification.
(3) Provides appropriate operator controls for setting the correct local time and date.
(4) Is capable of starting the sample collection period and sample air flow at a specific, operator-settable time and date, and stopping the sample air flow and terminating the sampler collection period 24 hours (1440 minutes) later, or at a specific, operator-settable time and date.
(b) These start and stop times shall be readily settable by the sampler operator to within ±1.0 minute. The system shall provide a visual indication of the current start and stop time settings, readable to ±1.0 minute, for verification by the operator, and the start and stop times shall also be available via the data output port, as specified in table L-1 of section 7.4.19 of this appendix. Upon execution of a programmed sample period start, the sampler shall automatically reset all sample period information and warning flag indications pertaining to a previous sample period. Refer also to section 7.4.15.4 of this appendix regarding retention of current date and time and programmed start and stop times during a temporary electrical power interruption.
7.4.13 Sample time determination. The sampler shall be capable of determining the elapsed sample collection time for each PM2.5 sample, accurate to within ±1.0 minute, measured as the time between the start of the sampling period, under section 7.4.12 of this appendix and the termination of the sample period, under section 7.4.12 of this appendix or section 7.4.4 of this appendix. This elapsed sample time shall not include periods when the sampler is inoperative due to a temporary interruption of electrical power, under section 7.4.15.4 of this appendix. In the event that the elapsed sample time determined for the sample period is not within the range specified for the required sample period in section 3.3 of this appendix, the sampler shall set a warning flag indicator. The date and time of the start of the sample period, the value of the elapsed sample time for the sample period, and the flag indicator status shall be available to the sampler operator following the end of the sample period, as specified in table L-1 of section 7.4.19 of this appendix.
7.4.14 Outdoor environmental enclosure. The sampler shall have an outdoor enclosure (or enclosures) suitable to protect the filter and other non-weatherproof components of the sampler from precipitation, wind, dust, extremes of temperature and humidity; to help maintain temperature control of the filter (or filters, for sequential samplers); and to provide reasonable security for sampler components and settings.
7.4.15 Electrical power supply.
7.4.15.1 The sampler shall be operable and function as specified herein when operated on an electrical power supply voltage of 105 to 125 volts AC (RMS) at a frequency of 59 to 61 Hz. Optional operation as specified at additional power supply voltages and/or frequencies shall not be precluded by this requirement.
7.4.15.2 The design and construction of the sampler shall comply with all applicable National Electrical Code and Underwriters Laboratories electrical safety requirements.
7.4.15.3 The design of all electrical and electronic controls shall be such as to provide reasonable resistance to interference or malfunction from ordinary or typical levels of stray electromagnetic fields (EMF) as may be found at various monitoring sites and from typical levels of electrical transients or electronic noise as may often or occasionally be present on various electrical power lines.
7.4.15.4 In the event of temporary loss of electrical supply power to the sampler, the sampler shall not be required to sample or provide other specified functions during such loss of power, except that the internal clock/timer system shall maintain its local time and date setting within ±1 minute per week, and the sampler shall retain all other time and programmable settings and all data required to be available to the sampler operator following each sample period for at least 7 days without electrical supply power. When electrical power is absent at the operator-set time for starting a sample period or is interrupted during a sample period, the sampler shall automatically start or resume sampling when electrical power is restored, if such restoration of power occurs before the operator-set stop time for the sample period.
7.4.15.5 The sampler shall have the capability to record and retain a record of the year, month, day-of-month, hour, and minute of the start of each power interruption of more than 1 minute duration, up to 10 such power interruptions per sample period. (More than 10 such power interruptions shall invalidate the sample, except where an exceedance is measured, under section 3.3 of this appendix.) The sampler shall provide for these power interruption data to be available to the sampler operator following the end of the sample period, as specified in table L-1 of section 7.4.19 of this appendix.
7.4.16 Control devices and operator interface. The sampler shall have mechanical, electrical, or electronic controls, control devices, electrical or electronic circuits as necessary to provide the timing, flow rate measurement and control, temperature control, data storage and computation, operator interface, and other functions specified. Operator-accessible controls, data displays, and interface devices shall be designed to be simple, straightforward, reliable, and easy to learn, read, and operate under field conditions. The sampler shall have provision for operator input and storage of up to 64 characters of numeric (or alphanumeric) data for purposes of site, sampler, and sample identification. This information shall be available to the sampler operator for verification and change and for output via the data output port along with other data following the end of a sample period, as specified in table L-1 of section 7.4.19 of this appendix. All data required to be available to the operator following a sample collection period or obtained during standby mode in a post-sampling period shall be retained by the sampler until reset, either manually by the operator or automatically by the sampler upon initiation of a new sample collection period.
7.4.17 Data output port requirement. The sampler shall have a standard RS-232C data output connection through which digital data may be exported to an external data storage or transmission device. All information which is required to be available at the end of each sample period shall be accessible through this data output connection. The information that shall be accessible though this output port is summarized in table L-1 of section 7.4.19 of this appendix. Since no specific format for the output data is provided, the sampler manufacturer or vendor shall make available to sampler purchasers appropriate computer software capable of receiving exported sampler data and correctly translating the data into a standard spreadsheet format and optionally any other formats as may be useful to sampler users. This requirement shall not preclude the sampler from offering other types of output connections in addition to the required RS-232C port.
7.4.18 Operation/instruction manual. The sampler shall include an associated comprehensive operation or instruction manual, as required by §53.11 of this chapter, which includes detailed operating instructions on the setup, operation, calibration, and maintenance of the sampler. This manual shall provide complete and detailed descriptions of the operational and calibration procedures prescribed for field use of the sampler and all instruments utilized as part of this reference method. The manual shall include adequate warning of potential safety hazards that may result from normal use or malfunction of the method and a description of necessary safety precautions. The manual shall also include a clear description of all procedures pertaining to installation, operation, periodic and corrective maintenance, and troubleshooting, and shall include parts identification diagrams.
7.4.19 Data reporting requirements. The various information that the sampler is required to provide and how it is to be provided is summarized in the following table L-1.
| Information to be provided | Appendix L section
reference | Availability | Format | ||||
|---|---|---|---|---|---|---|---|
| Anytime 1 | End of
period 2 | Visual
display 3 | Data
output 4 | Digital
reading 5 | Units | ||
| ✓ Provision of this information is required. * Provision of this information is optional. If information related to the entire sample period is optionally provided prior to the end of the sample period, the value provided should be the value calculated for the portion of the sampler period completed up to the time the information is provided. ▪ Indicates that this information is also required to be provided to the Air Quality System (AQS) data bank; see §58.16 of this chapter. For ambient temperature and barometric pressure, only the average for the sample period must be reported. 1. Information is required to be available to the operator at any time the sampler is operating, whether sampling or not. 2. Information relates to the entire sampler period and must be provided following the end of the sample period until reset manually by the operator or automatically by the sampler upon the start of a new sample period. 3. Information shall be available to the operator visually. 4. Information is to be available as digital data at the sampler's data output port specified in section 7.4.16 of this appendix following the end of the sample period until reset manually by the operator or automatically by the sampler upon the start of a new sample period. 5. Digital readings, both visual and data output, shall have not less than the number of significant digits and resolution specified. 6. Flag warnings may be displayed to the operator by a single flag indicator or each flag may be displayed individually. Only a set (on) flag warning must be indicated; an off (unset) flag may be indicated by the absence of a flag warning. Sampler users should refer to section 10.12 of this appendix regarding the validity of samples for which the sampler provided an associated flag warning. | |||||||
| Flow rate, 30-second maximum interval | 7.4.5.1 | ✓ | ✓ | * | XX.X | L/min | |
| Flow rate, average for the sample period | 7.4.5.2 | * | ✓ | * | ✓ | XX.X | L/min |
| Flow rate, CV, for sample period | 7.4.5.2 | * | ✓ | * | ✓ | XX.X | % |
| Flow rate, 5-min. average out of spec. (FLAG 6) | 7.4.5.2 | ✓ | ✓ | ✓ | ✓▪ | On/Off | |
| Sample volume, total | 7.4.5.2 | * | ✓ | ✓ | ✓ | XX.X | m 3 |
| Temperature, ambient, 30-second interval | 7.4.8 | ✓ | ✓ | XX.X | °C | ||
| Temperature, ambient, min., max., average for the sample period | 7.4.8 | * | ✓ | ✓ | ✓▪ | XX.X | °C |
| Baro. pressure, ambient, 30-second interval | 7.4.9 | ✓ | ✓ | XXX | mm Hg | ||
| Baro. pressure, ambient, min., max., average for the sample period | 7.4.9 | * | ✓ | ✓ | ✓▪ | XXX | mm Hg |
| Filter temperature, 30-second interval | 7.4.11 | ✓ | ✓ | XX.X | °C | ||
| Filter temp. differential, 30-second interval, out of spec. (FLAG 6) | 7.4.11 | * | ✓ | ✓ | ✓▪ | On/Off | |
| Filter temp., maximum differential from ambient, date, time of occurrence | 7.4.11 | * | * | * | * | X.X, YY/MM/DD HH.mm | °C, Yr/Mon/Day Hrs. min |
| Date and Time | 7.4.12 | ✓ | ✓ | YY/MM/DD HH.mm | Yr/Mon/Day Hrs. min | ||
| Sample start and stop time settings | 7.4.12 | ✓ | ✓ | ✓ | ✓ | YY/MM/DD HH.mm | Yr/Mon/Day Hrs. min |
| Sample period start time | 7.4.12 | ✓ | ✓ | ✓ | YY/MM/DD HH.mm | Yr/Mon/Day Hrs. min | |
| Elapsed sample time | 7.4.13 | * | ✓ | ✓ | ✓ | HH.mm | Hrs. min |
| Elapsed sample time, out of spec. (FLAG 6) | 7.4.13 | ✓ | ✓ | ✓▪ | On/Off | ||
| Power interruptions ≤1 min., start time of first 10 | 7.4.15.5 | * | ✓ | * | ✓ | 1HH.mm, 2HH.mm, etc. | Hrs. min |
| User-entered information, such as sampler and site identification | 7.4.16 | ✓ | ✓ | ✓ | ✓▪ | As entered | |
8.0 Filter Weighing. See reference 2 in section 13.0 of this appendix, for additional, more detailed guidance.
8.1 Analytical balance. The analytical balance used to weigh filters must be suitable for weighing the type and size of filters specified, under section 6.0 of this appendix, and have a readability of ±1 µg. The balance shall be calibrated as specified by the manufacturer at installation and recalibrated immediately prior to each weighing session. See reference 2 in section 13.0 of this appendix for additional guidance.
8.2 Filter conditioning. All sample filters used shall be conditioned immediately before both the pre- and post-sampling weighings as specified below. See reference 2 in section 13.0 of this appendix for additional guidance.
8.2.1 Mean temperature. 20 - 23°C.
8.2.2 Temperature control. ±2°C over 24 hours.
8.2.3 Mean humidity. Generally, 30-40 percent relative humidity; however, where it can be shown that the mean ambient relative humidity during sampling is less than 30 percent, conditioning is permissible at a mean relative humidity within ±5 relative humidity percent of the mean ambient relative humidity during sampling, but not less than 20 percent.
8.2.4 Humidity control. ±5 relative humidity percent over 24 hours.
8.2.5 Conditioning time. Not less than 24 hours.
8.3 Weighing procedure.
8.3.1 New filters should be placed in the conditioning environment immediately upon arrival and stored there until the pre-sampling weighing. See reference 2 in section 13.0 of this appendix for additional guidance.
8.3.2 The analytical balance shall be located in the same controlled environment in which the filters are conditioned. The filters shall be weighed immediately following the conditioning period without intermediate or transient exposure to other conditions or environments.
8.3.3 Filters must be conditioned at the same conditions (humidity within ±5 relative humidity percent) before both the pre- and post-sampling weighings.
8.3.4 Both the pre- and post-sampling weighings should be carried out on the same analytical balance, using an effective technique to neutralize static charges on the filter, under reference 2 in section 13.0 of this appendix. If possible, both weighings should be carried out by the same analyst.
8.3.5 The pre-sampling (tare) weighing shall be within 30 days of the sampling period.
8.3.6 The post-sampling conditioning and weighing shall be completed within 240 hours (10 days) after the end of the sample period, unless the filter sample is maintained at temperatures below the average ambient temperature during sampling (or 4°C or below for average sampling temperatures less than 4°C) during the time between retrieval from the sampler and the start of the conditioning, in which case the period shall not exceed 30 days. Reference 2 in section 13.0 of this appendix has additional guidance on transport of cooled filters.
8.3.7 Filter blanks.
8.3.7.1 New field blank filters shall be weighed along with the pre-sampling (tare) weighing of each lot of PM2.5 filters. These blank filters shall be transported to the sampling site, installed in the sampler, retrieved from the sampler without sampling, and reweighed as a quality control check.
8.3.7.2 New laboratory blank filters shall be weighed along with the pre-sampling (tare) weighing of each set of PM2.5 filters. These laboratory blank filters should remain in the laboratory in protective containers during the field sampling and should be reweighed as a quality control check.
8.3.8 Additional guidance for proper filter weighing and related quality assurance activities is provided in reference 2 in section 13.0 of this appendix.
9.0 Calibration. Reference 2 in section 13.0 of this appendix contains additional guidance.
9.1 General requirements.
9.1.1 Multipoint calibration and single-point verification of the sampler's flow rate measurement device must be performed periodically to establish and maintain traceability of subsequent flow measurements to a flow rate standard.
9.1.2 An authoritative flow rate standard shall be used for calibrating or verifying the sampler's flow rate measurement device with an accuracy of ±2 percent. The flow rate standard shall be a separate, stand-alone device designed to connect to the flow rate measurement adapter, Figure L-30 of this appendix. This flow rate standard must have its own certification and be traceable to a National Institute of Standards and Technology (NIST) primary standard for volume or flow rate. If adjustments to the sampler's flow rate measurement system calibration are to be made in conjunction with an audit of the sampler's flow measurement system, such adjustments shall be made following the audit. Reference 2 in section 13.0 of this appendix contains additional guidance.
9.1.3 The sampler's flow rate measurement device shall be re-calibrated after electromechanical maintenance or transport of the sampler.
9.2 Flow rate calibration/verification procedure.
9.2.1 PM2.5 samplers may employ various types of flow control and flow measurement devices. The specific procedure used for calibration or verification of the flow rate measurement device will vary depending on the type of flow rate controller and flow rate measurement employed. Calibration shall be in terms of actual ambient volumetric flow rates (Q a), measured at the sampler's inlet downtube. The generic procedure given here serves to illustrate the general steps involved in the calibration of a PM2.5 sampler. The sampler operation/instruction manual required under section 7.4.18 of this appendix and the Quality Assurance Handbook in reference 2 in section 13.0 of this appendix provide more specific and detailed guidance for calibration.
9.2.2 The flow rate standard used for flow rate calibration shall have its own certification and be traceable to a NIST primary standard for volume or flow rate. A calibration relationship for the flow rate standard, e.g., an equation, curve, or family of curves relating actual flow rate (Qa) to the flow rate indicator reading, shall be established that is accurate to within 2 percent over the expected range of ambient temperatures and pressures at which the flow rate standard may be used. The flow rate standard must be re-calibrated or re-verified at least annually.
9.2.3 The sampler flow rate measurement device shall be calibrated or verified by removing the sampler inlet and connecting the flow rate standard to the sampler's downtube in accordance with the operation/instruction manual, such that the flow rate standard accurately measures the sampler's flow rate. The sampler operator shall first carry out a sampler leak check and confirm that the sampler passes the leak test and then verify that no leaks exist between the flow rate standard and the sampler.
9.2.4 The calibration relationship between the flow rate (in actual L/min) indicated by the flow rate standard and by the sampler's flow rate measurement device shall be established or verified in accordance with the sampler operation/instruction manual. Temperature and pressure corrections to the flow rate indicated by the flow rate standard may be required for certain types of flow rate standards. Calibration of the sampler's flow rate measurement device shall consist of at least three separate flow rate measurements (multipoint calibration) evenly spaced within the range of −10 percent to = 10 percent of the sampler's operational flow rate, section 7.4.1 of this appendix. Verification of the sampler's flow rate shall consist of one flow rate measurement at the sampler's operational flow rate. The sampler operation/instruction manual and reference 2 in section 13.0 of this appendix provide additional guidance.
9.2.5 If during a flow rate verification the reading of the sampler's flow rate indicator or measurement device differs by ±4 percent or more from the flow rate measured by the flow rate standard, a new multipoint calibration shall be performed and the flow rate verification must then be repeated.
9.2.6 Following the calibration or verification, the flow rate standard shall be removed from the sampler and the sampler inlet shall be reinstalled. Then the sampler's normal operating flow rate (in L/min) shall be determined with a clean filter in place. If the flow rate indicated by the sampler differs by ±2 percent or more from the required sampler flow rate, the sampler flow rate must be adjusted to the required flow rate, under section 7.4.1 of this appendix.
9.3 Periodic calibration or verification of the calibration of the sampler's ambient temperature, filter temperature, and barometric pressure measurement systems is also required. Reference 3 of section 13.0 of this appendix contains additional guidance.
10.0 PM 2.5 Measurement Procedure. The detailed procedure for obtaining valid PM2.5 measurements with each specific sampler designated as part of a reference method for PM2.5 under §53.11 of this chapter shall be provided in the sampler-specific operation or instruction manual required by section 7.4.18 of this appendix. Supplemental guidance is provided in section 2.12 of the Quality Assurance Handbook listed in reference 2 in section 13.0 of this appendix. The generic procedure given here serves to illustrate the general steps involved in the PM2.5 sample collection and measurement, using a PM2.5 reference method sampler.
10.1 The sampler shall be set up, calibrated, and operated in accordance with the specific, detailed guidance provided in the specific sampler's operation or instruction manual and in accordance with a specific quality assurance program developed and established by the user, based on applicable supplementary guidance provided in reference 2 in section 13.0 of this appendix.
10.2 Each new sample filter shall be inspected for correct type and size and for pinholes, particles, and other imperfections. Unacceptable filters should be discarded. A unique identification number shall be assigned to each filter, and an information record shall be established for each filter. If the filter identification number is not or cannot be marked directly on the filter, alternative means, such as a number-identified storage container, must be established to maintain positive filter identification.
10.3 Each filter shall be conditioned in the conditioning environment in accordance with the requirements specified in section 8.2 of this appendix.
10.4 Following conditioning, each filter shall be weighed in accordance with the requirements specified in section 8.0 of this appendix and the presampling weight recorded with the filter identification number.
10.5 A numbered and preweighed filter shall be installed in the sampler following the instructions provided in the sampler operation or instruction manual.
10.6 The sampler shall be checked and prepared for sample collection in accordance with instructions provided in the sampler operation or instruction manual and with the specific quality assurance program established for the sampler by the user.
10.7 The sampler's timer shall be set to start the sample collection at the beginning of the desired sample period and stop the sample collection 24 hours later.
10.8 Information related to the sample collection (site location or identification number, sample date, filter identification number, and sampler model and serial number) shall be recorded and, if appropriate, entered into the sampler.
10.9 The sampler shall be allowed to collect the PM2.5 sample during the set 24-hour time period.
10.10 Within 177 hours (7 days, 9 hours) of the end of the sample collection period, the filter, while still contained in the filter cassette, shall be carefully removed from the sampler, following the procedure provided in the sampler operation or instruction manual and the quality assurance program, and placed in a protective container. The protective container shall contain no loose material that could be transferred to the filter. The protective container shall hold the filter cassette securely such that the cover shall not come in contact with the filter's surfaces. Reference 2 in section 13.0 of this appendix contains additional information.
10.11 The total sample volume in actual m 3 for the sampling period and the elapsed sample time shall be obtained from the sampler and recorded in accordance with the instructions provided in the sampler operation or instruction manual. All sampler warning flag indications and other information required by the local quality assurance program shall also be recorded.
10.12 All factors related to the validity or representativeness of the sample, such as sampler tampering or malfunctions, unusual meteorological conditions, construction activity, fires or dust storms, etc. shall be recorded as required by the local quality assurance program. The occurrence of a flag warning during a sample period shall not necessarily indicate an invalid sample but rather shall indicate the need for specific review of the QC data by a quality assurance officer to determine sample validity.
10.13 After retrieval from the sampler, the exposed filter containing the PM2.5 sample should be transported to the filter conditioning environment as soon as possible, ideally to arrive at the conditioning environment within 24 hours for conditioning and subsequent weighing. During the period between filter retrieval from the sampler and the start of the conditioning, the filter shall be maintained as cool as practical and continuously protected from exposure to temperatures over 25°C to protect the integrity of the sample and minimize loss of volatile components during transport and storage. See section 8.3.6 of this appendix regarding time limits for completing the post-sampling weighing. See reference 2 in section 13.0 of this appendix for additional guidance on transporting filter samplers to the conditioning and weighing laboratory.
10.14. The exposed filter containing the PM2.5 sample shall be re-conditioned in the conditioning environment in accordance with the requirements specified in section 8.2 of this appendix.
10.15. The filter shall be reweighed immediately after conditioning in accordance with the requirements specified in section 8.0 of this appendix, and the postsampling weight shall be recorded with the filter identification number.
10.16 The PM2.5 concentration shall be calculated as specified in section 12.0 of this appendix.
11.0 Sampler Maintenance. The sampler shall be maintained as described by the sampler's manufacturer in the sampler-specific operation or instruction manual required under section 7.4.18 of this appendix and in accordance with the specific quality assurance program developed and established by the user based on applicable supplementary guidance provided in reference 2 in section 13.0 of this appendix.
12.0 Calculations
12.1 (a) The PM2.5 concentration is calculated as:
PM2.5 = (Wf − Wi)/Va
where:
PM2.5 = mass concentration of PM2.5, µg/m 3;
Wf, Wi = final and initial weights, respectively, of the filter used to collect the PM2.5 particle sample, µg;
Va = total air volume sampled in actual volume units, as provided by the sampler, m 3.
Note:
Total sample time must be between 1,380 and 1,500 minutes (23 and 25 hrs) for a fully valid PM2.5 sample; however, see also section 3.3 of this appendix.
13.0 References.
1. Quality Assurance Handbook for Air Pollution Measurement Systems, Volume I, Principles. EPA/600/R-94/038a, April 1994. Available from CERI, ORD Publications, U.S. Environmental Protection Agency, 26 West Martin Luther King Drive, Cincinnati, Ohio 45268.
2. Quality Assurance Guidance Document 2.12. Monitoring PM2.5 in Ambient Air Using Designated Reference or Class I Equivalent Methods. U.S. EPA, National Exposure Research Laboratory. Research Triangle Park, NC, November 1988 or later edition. Currently available at: http://www.epa.gov/ttn/amtic/pmqainf.html.
3. Quality Assurance Handbook for Air Pollution Measurement Systems, Volume IV: Meteorological Measurements, (Revised Edition) EPA/600/R-94/038d, March, 1995. Available from CERI, ORD Publications, U.S. Environmental Protection Agency, 26 West Martin Luther King Drive, Cincinnati, Ohio 45268.
4. Military standard specification (mil. spec.) 8625F, Type II, Class 1 as listed in Department of Defense Index of Specifications and Standards (DODISS), available from DODSSP-Customer Service, Standardization Documents Order Desk, 700 Robbins Avenue, Building 4D, Philadelphia, PA 1911-5094.
14.0 Figures L-1 through L-30 to Appendix L.
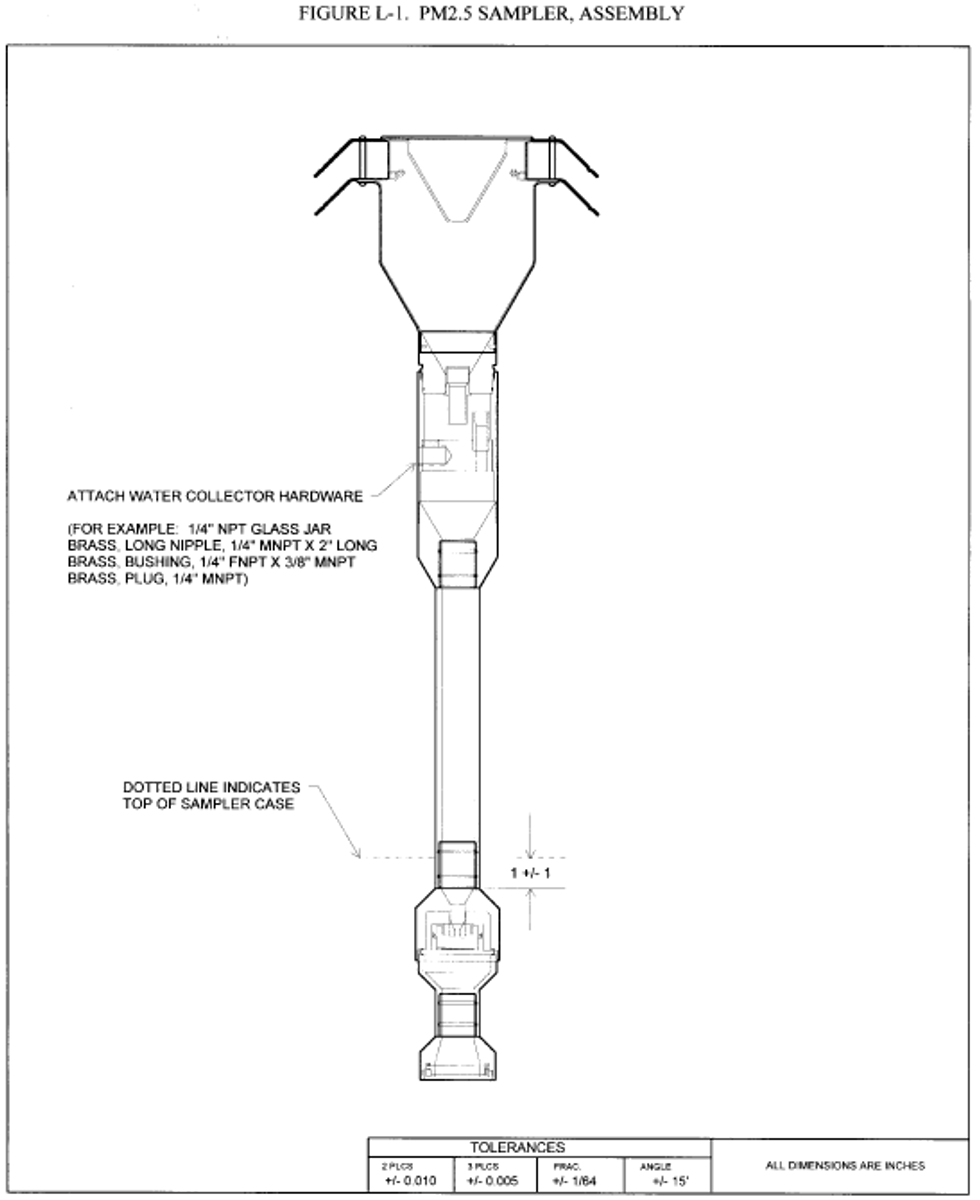
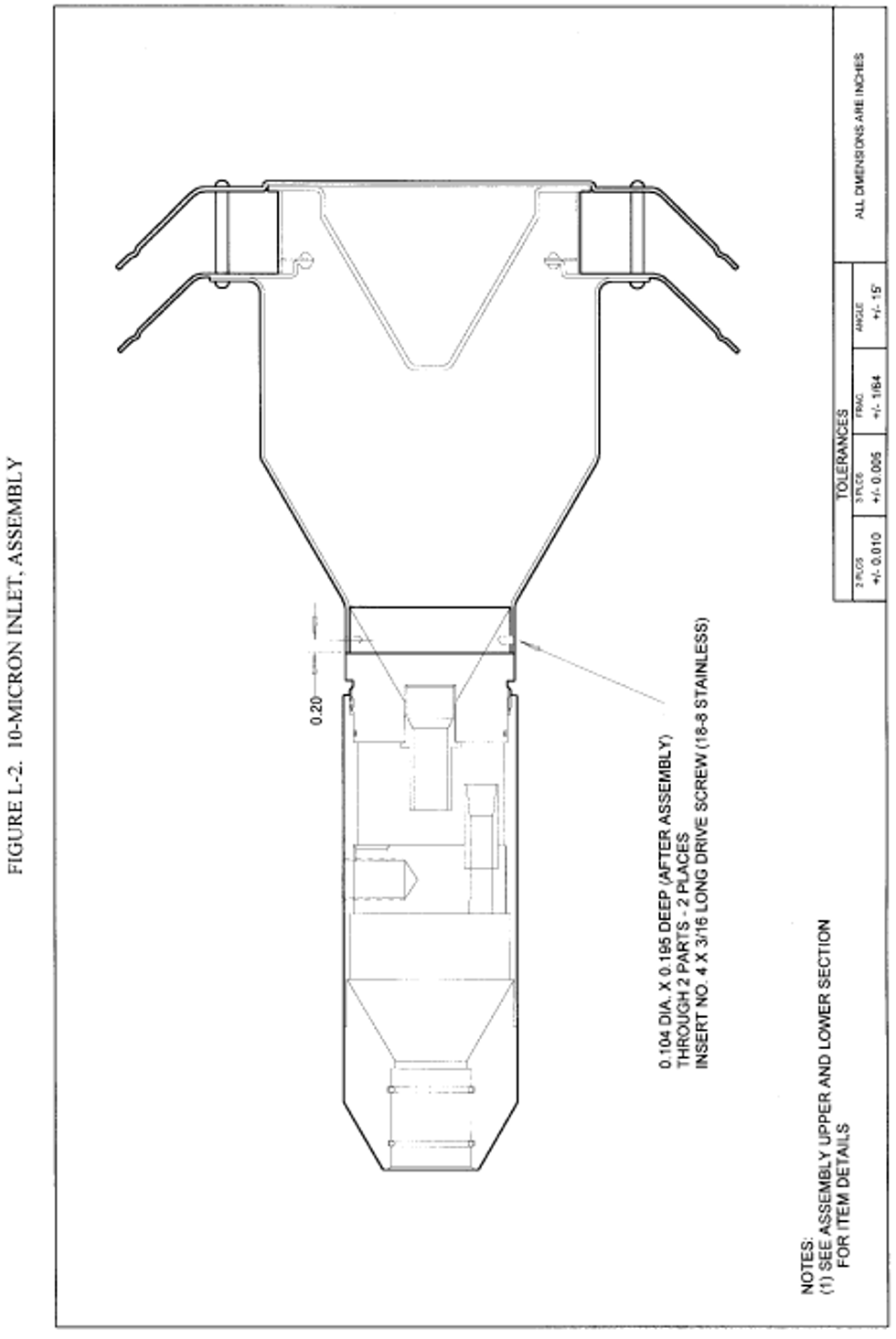
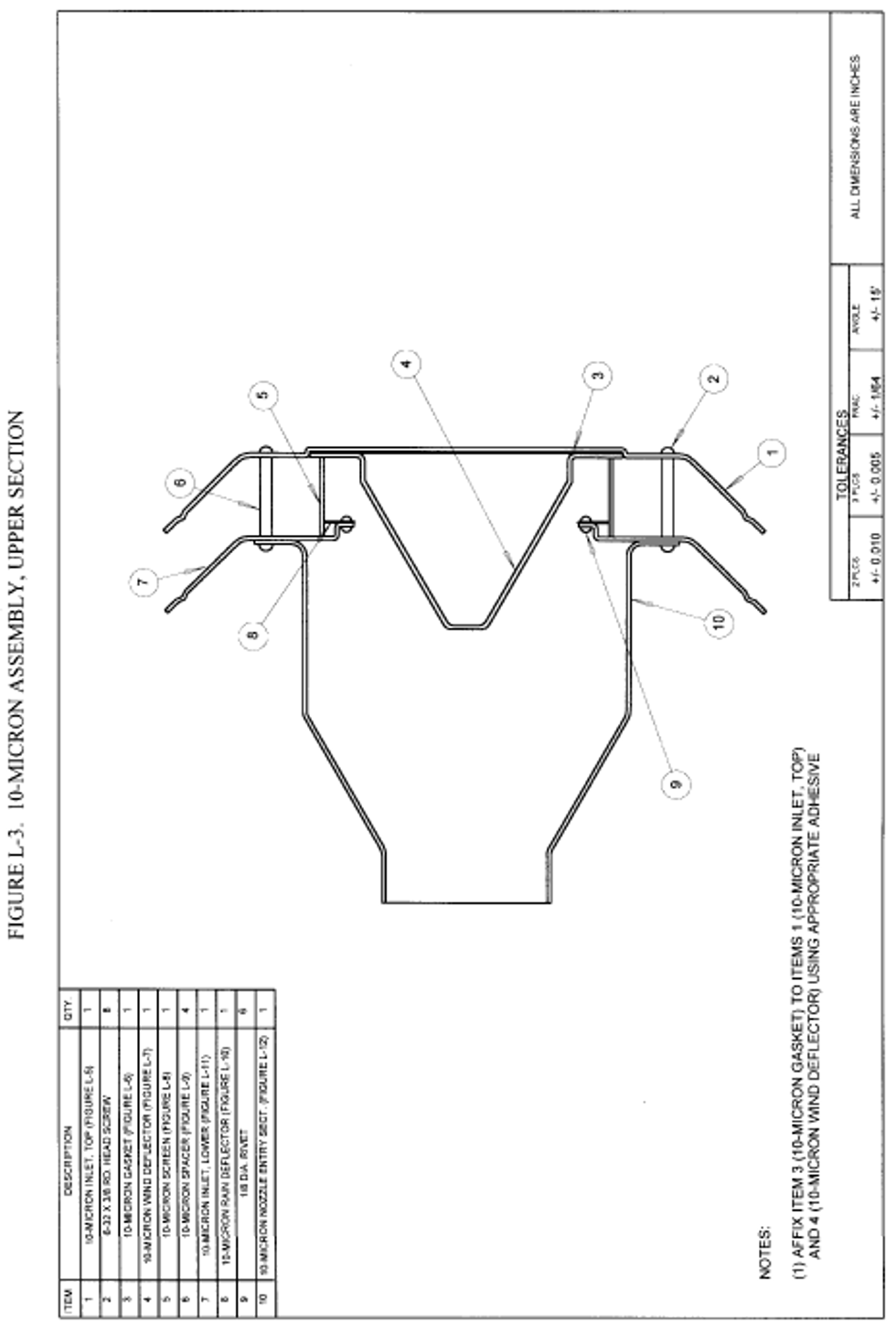
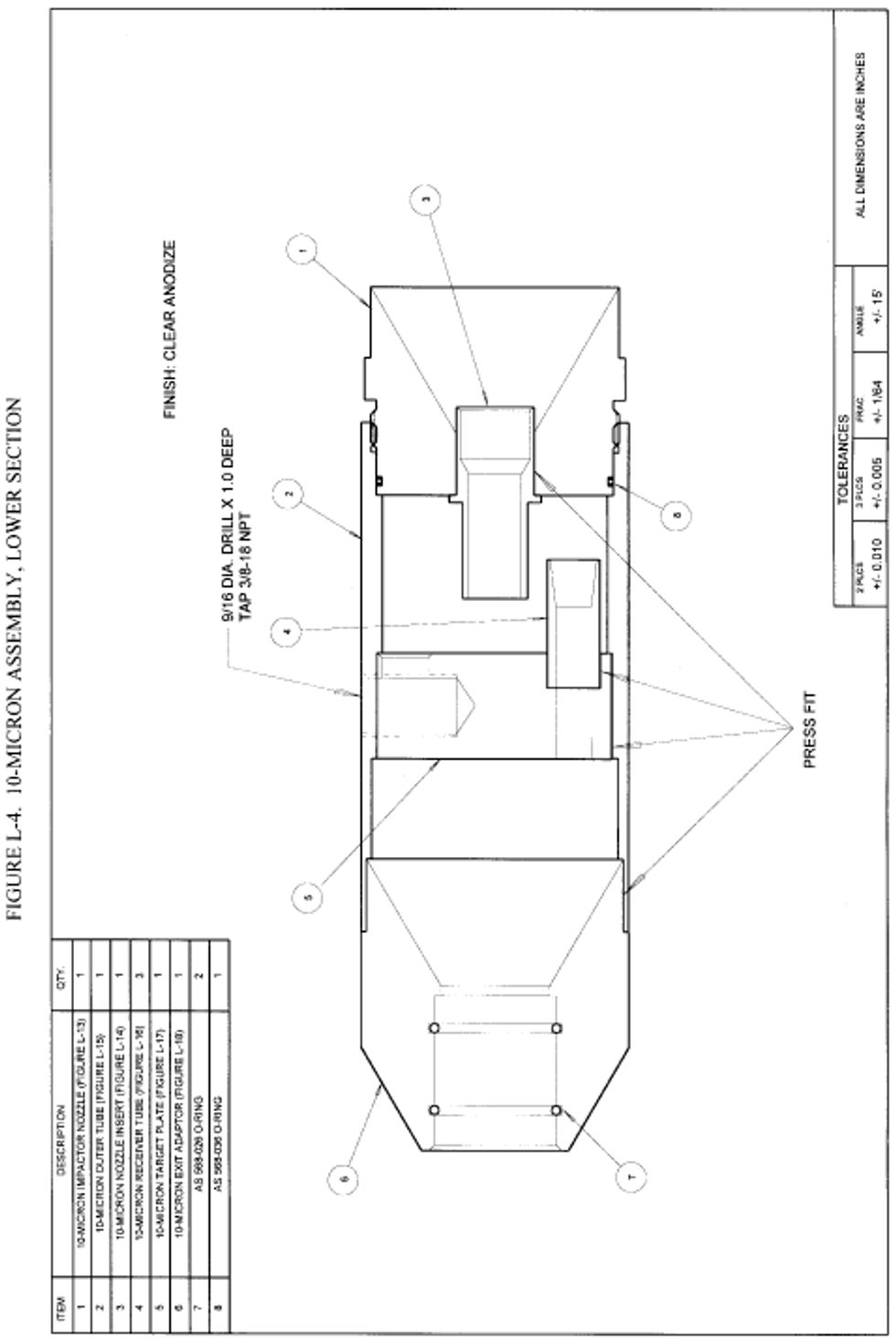
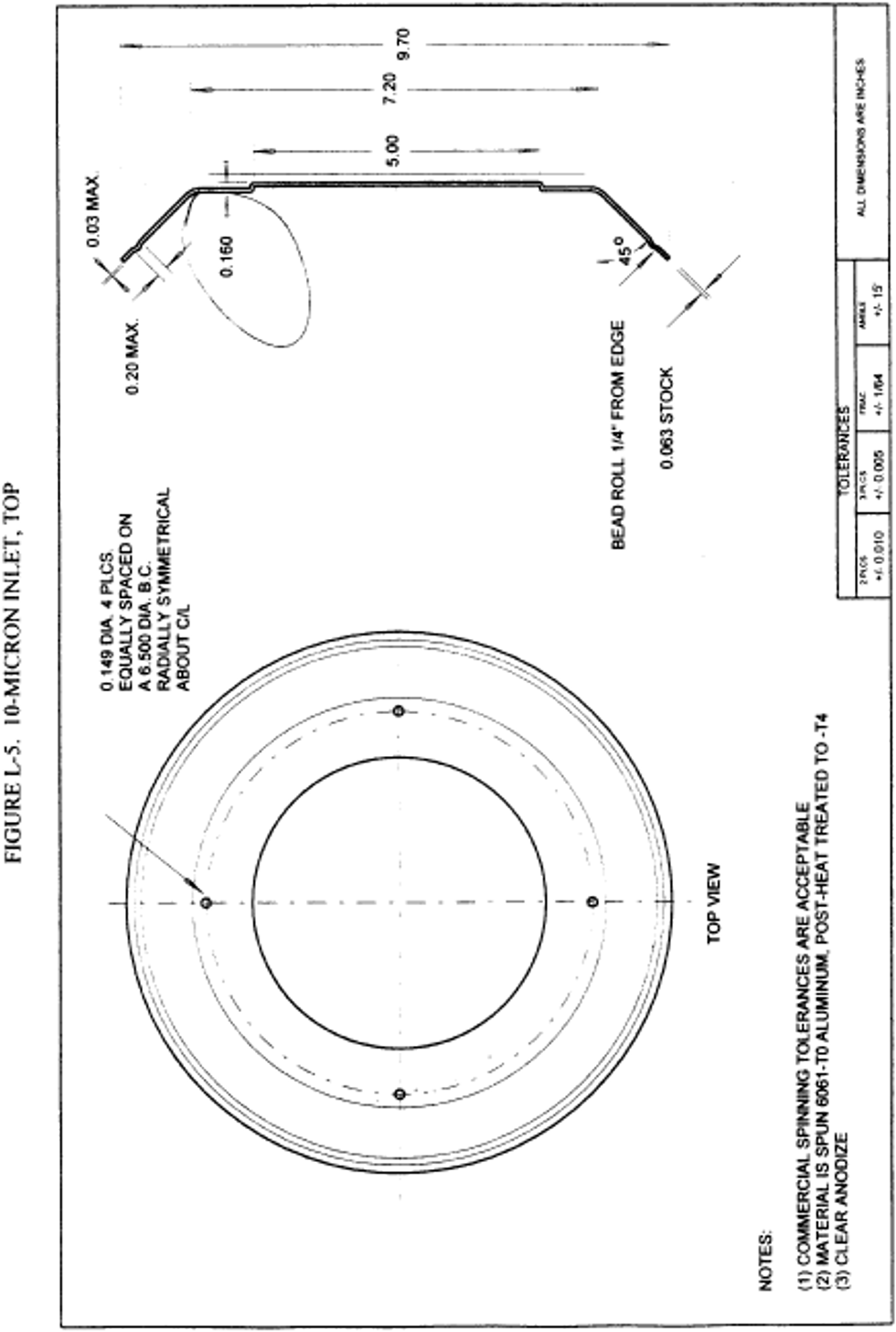
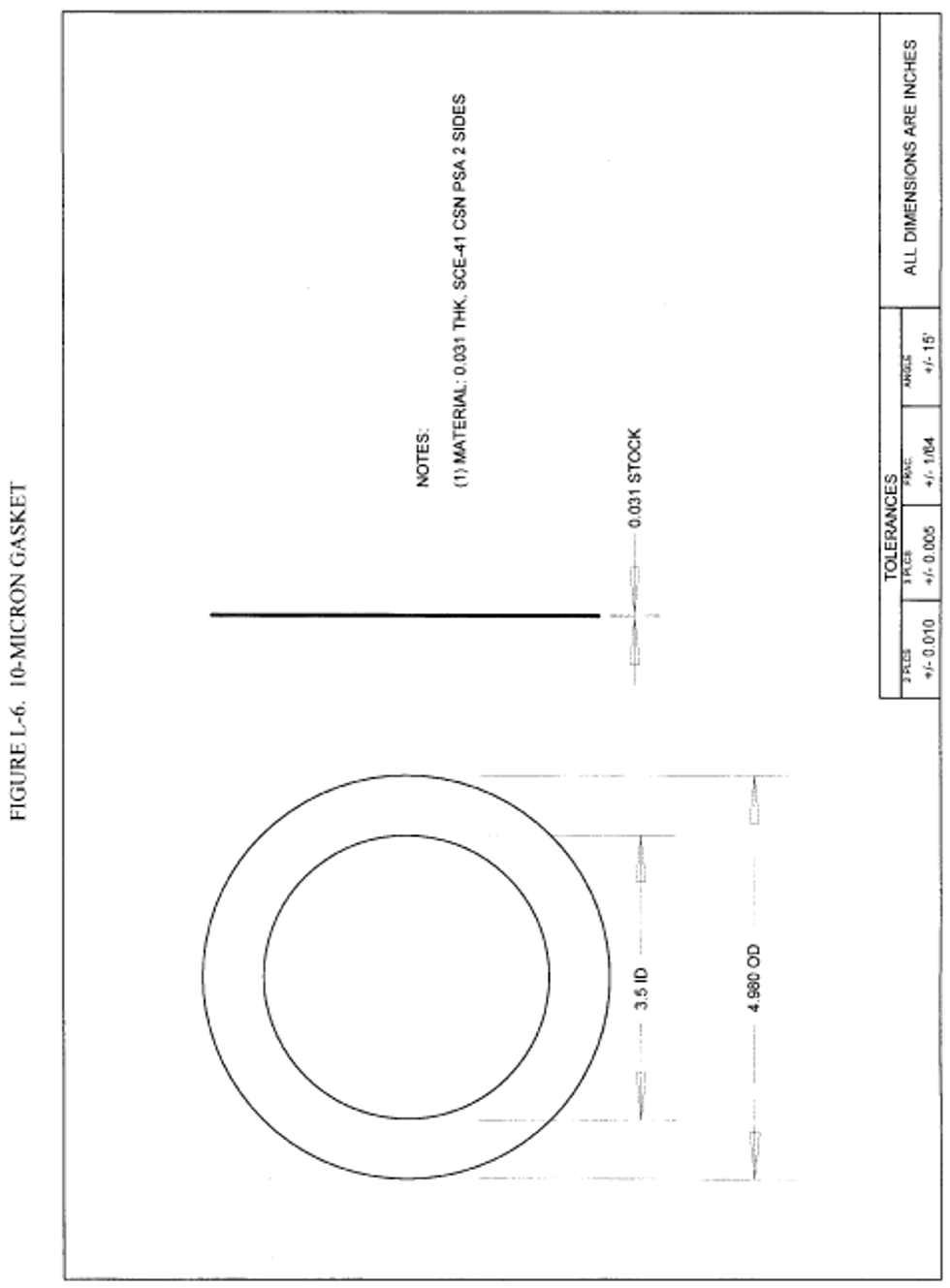
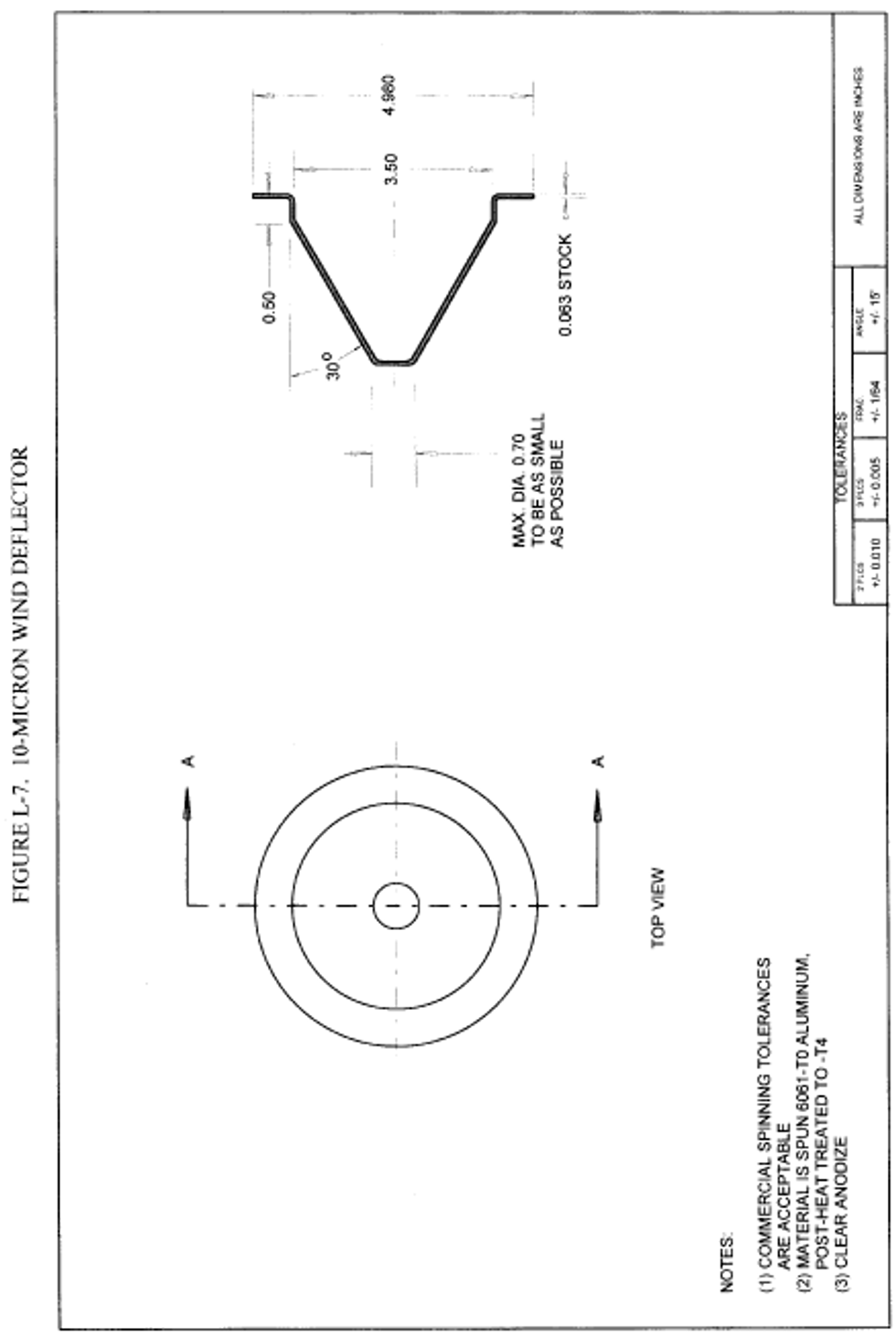
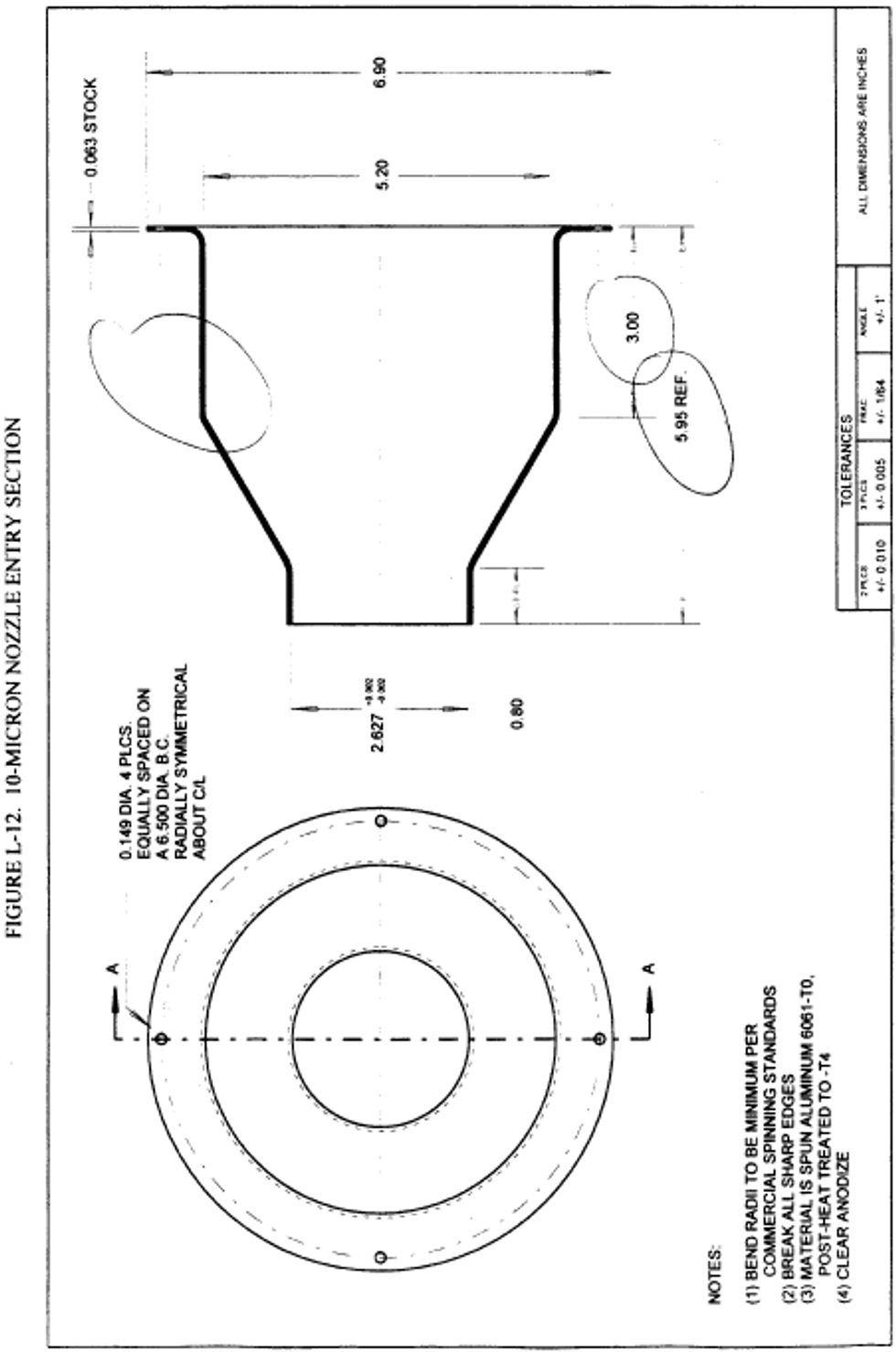
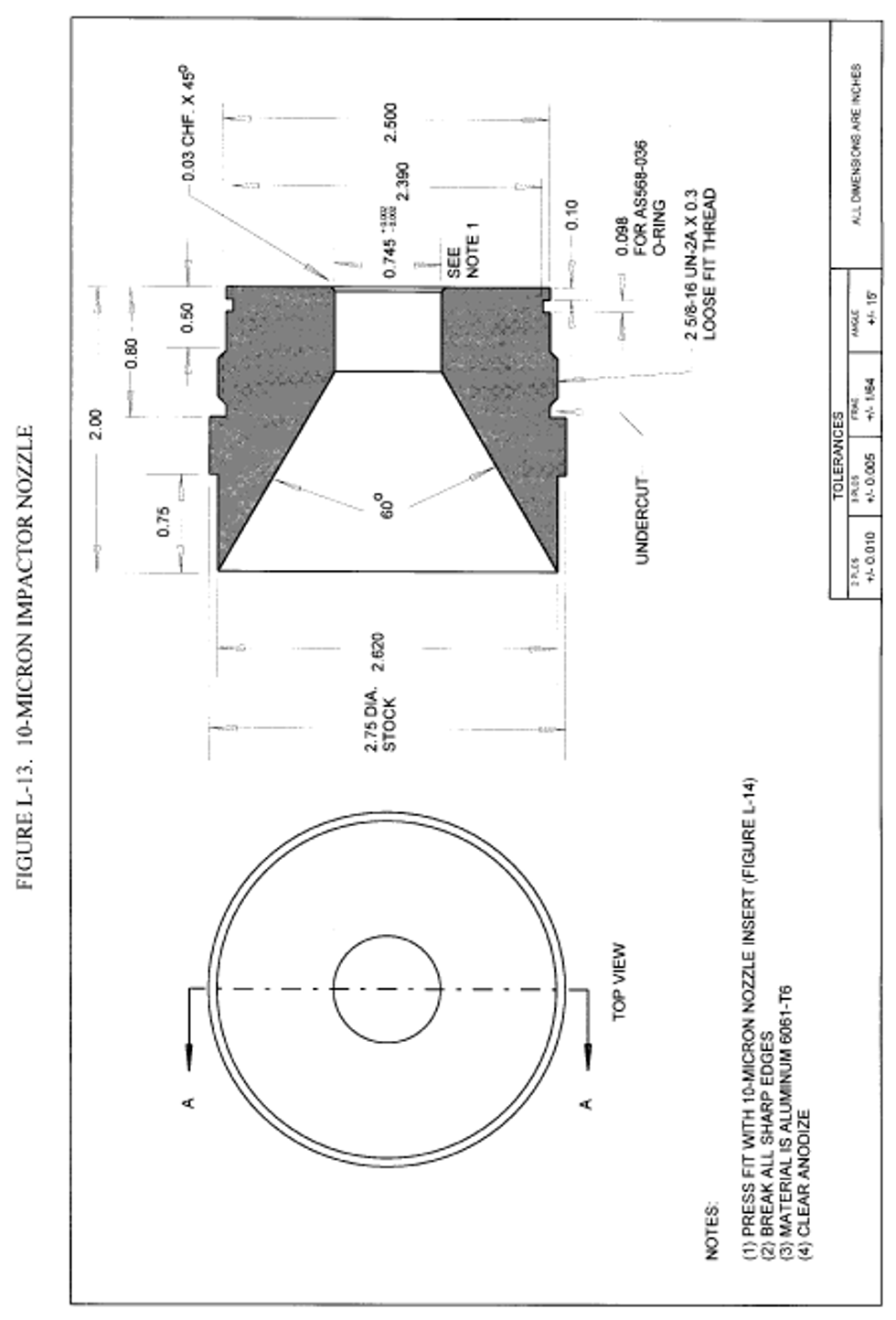
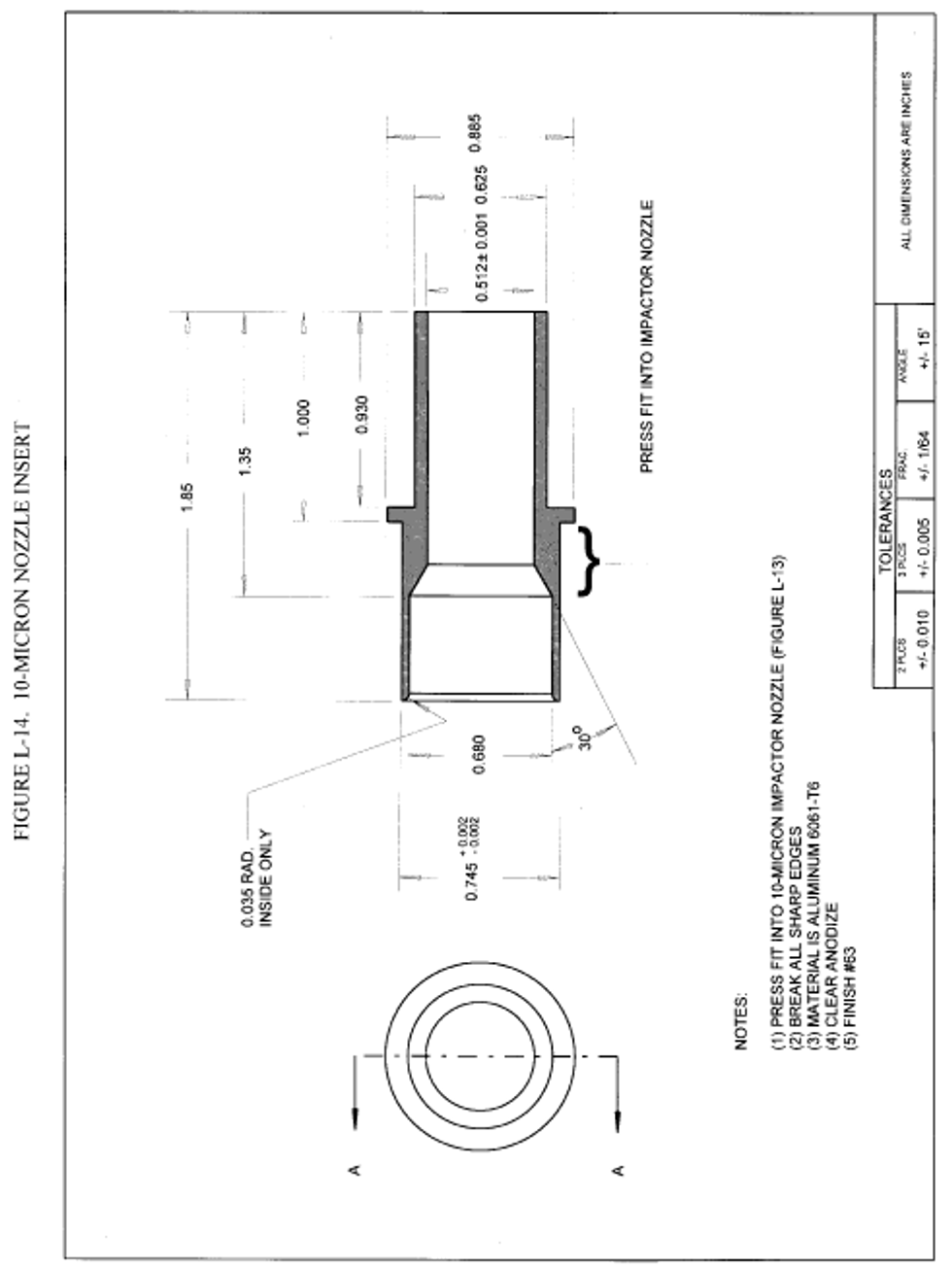
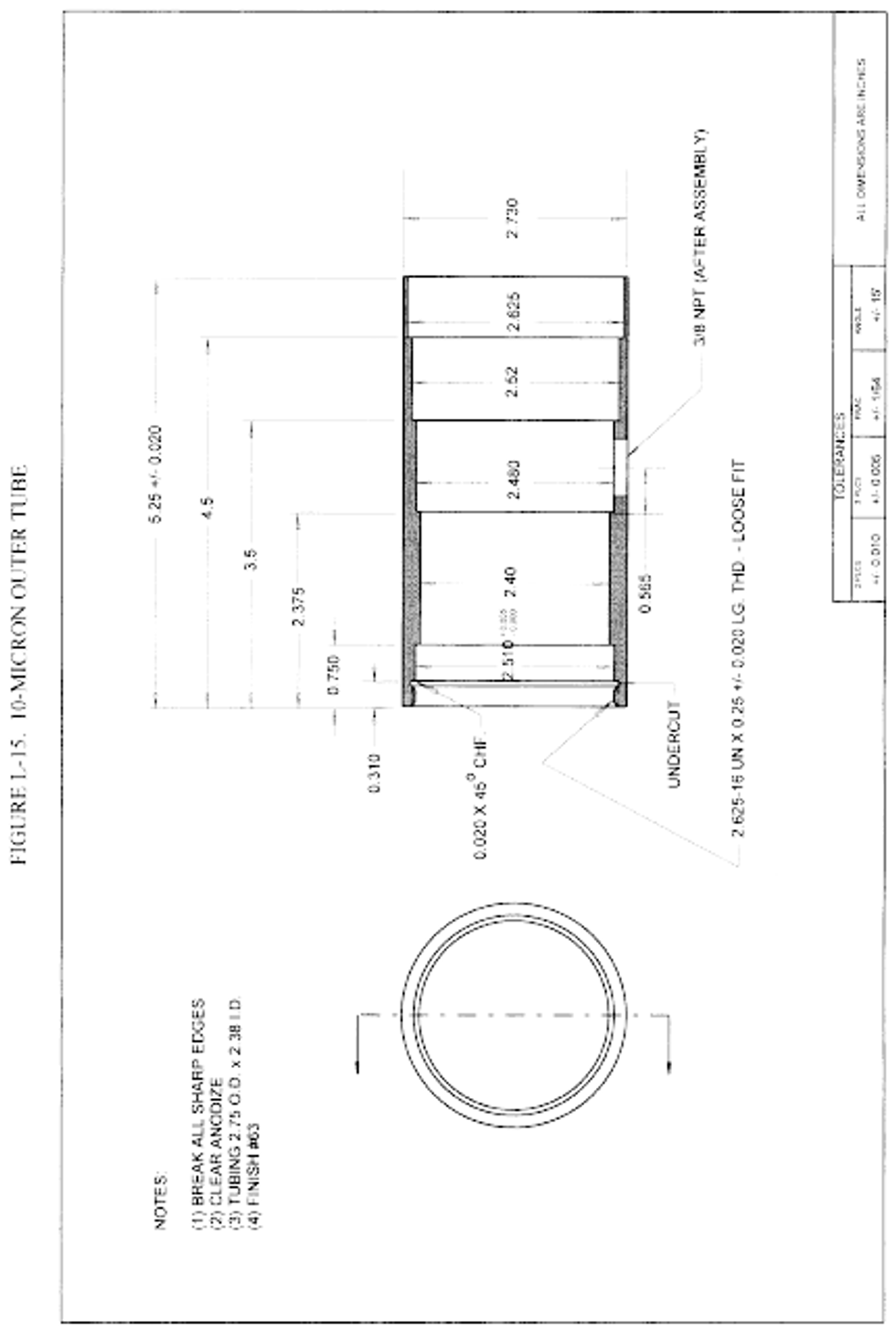
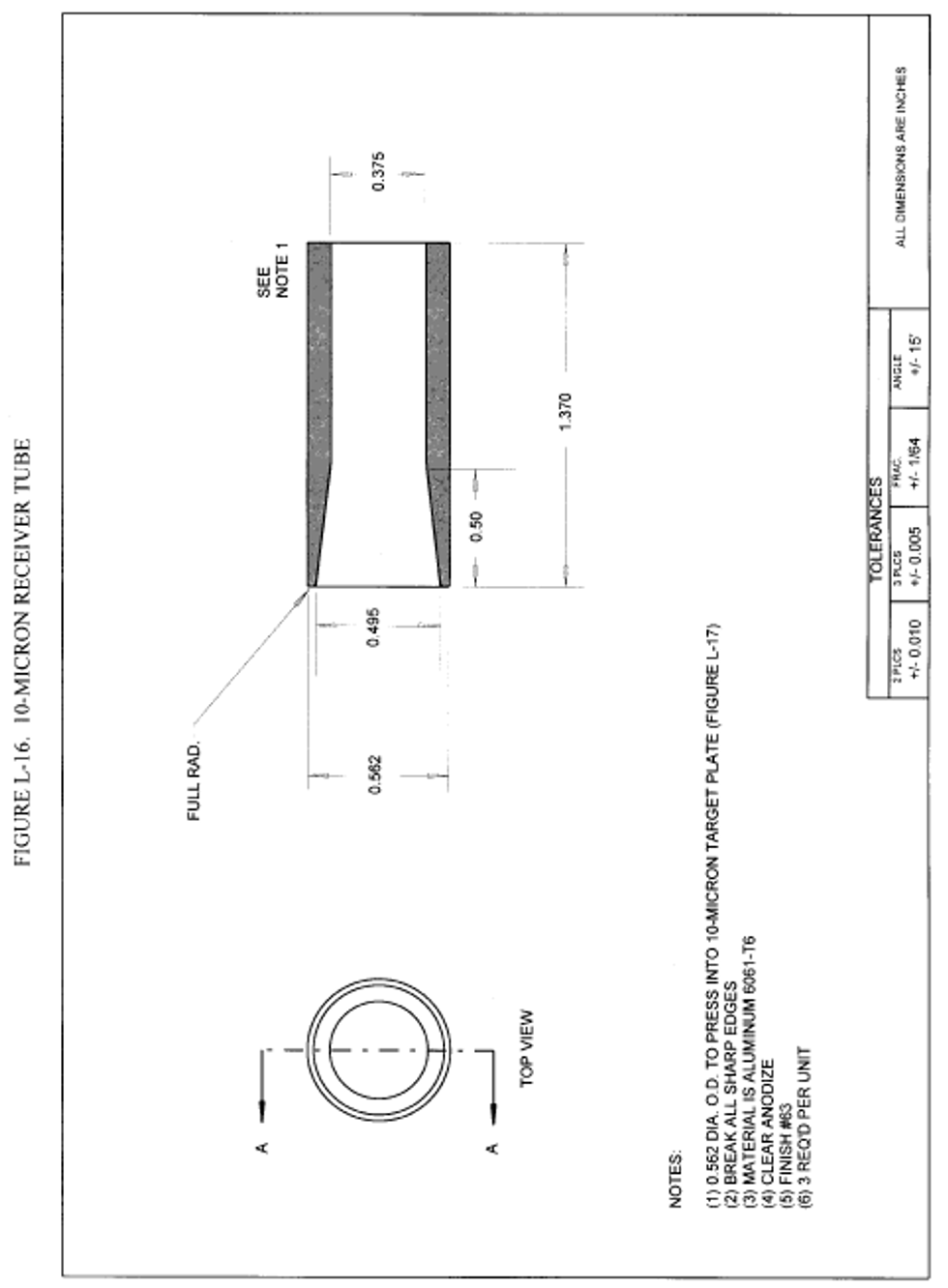
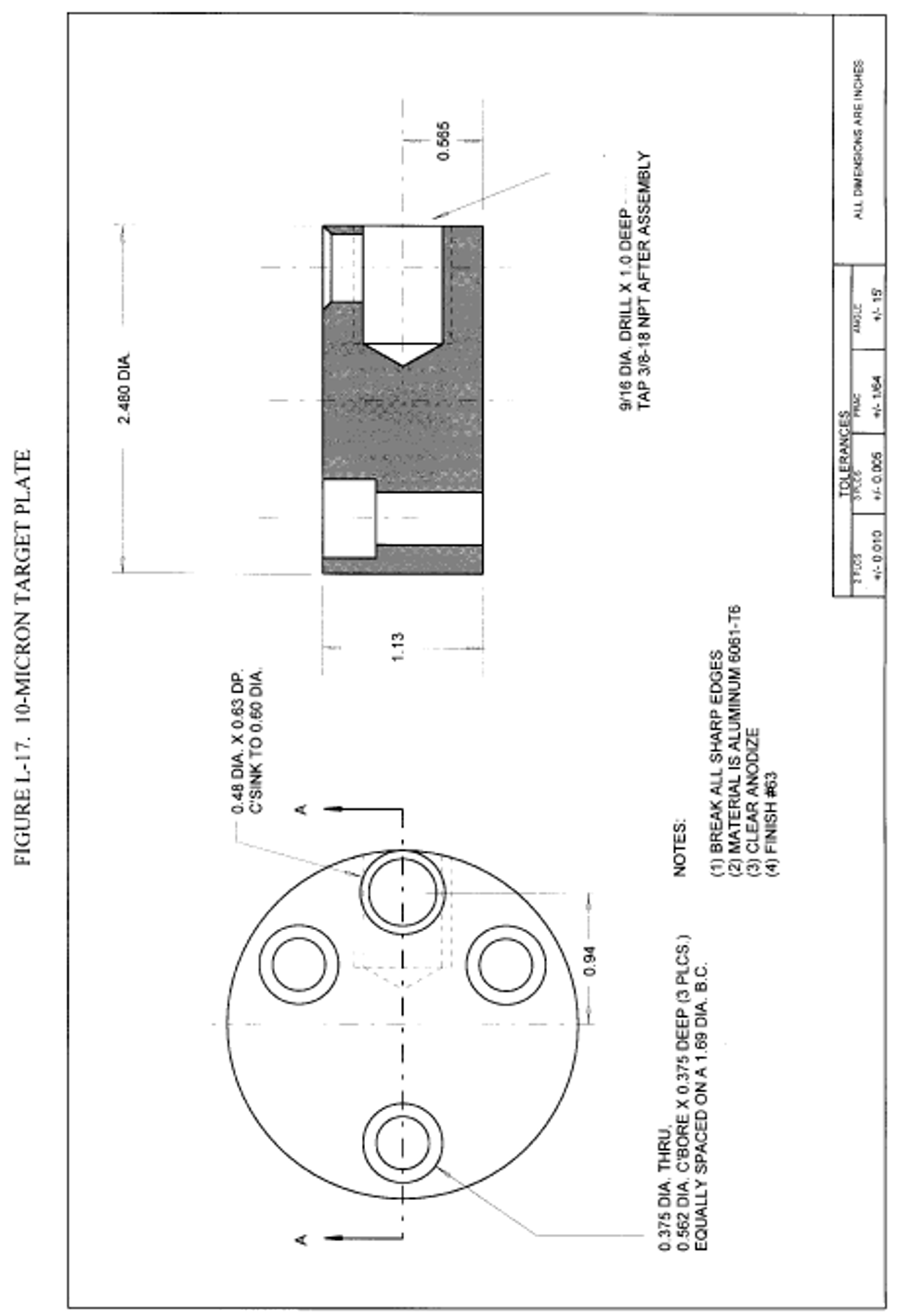
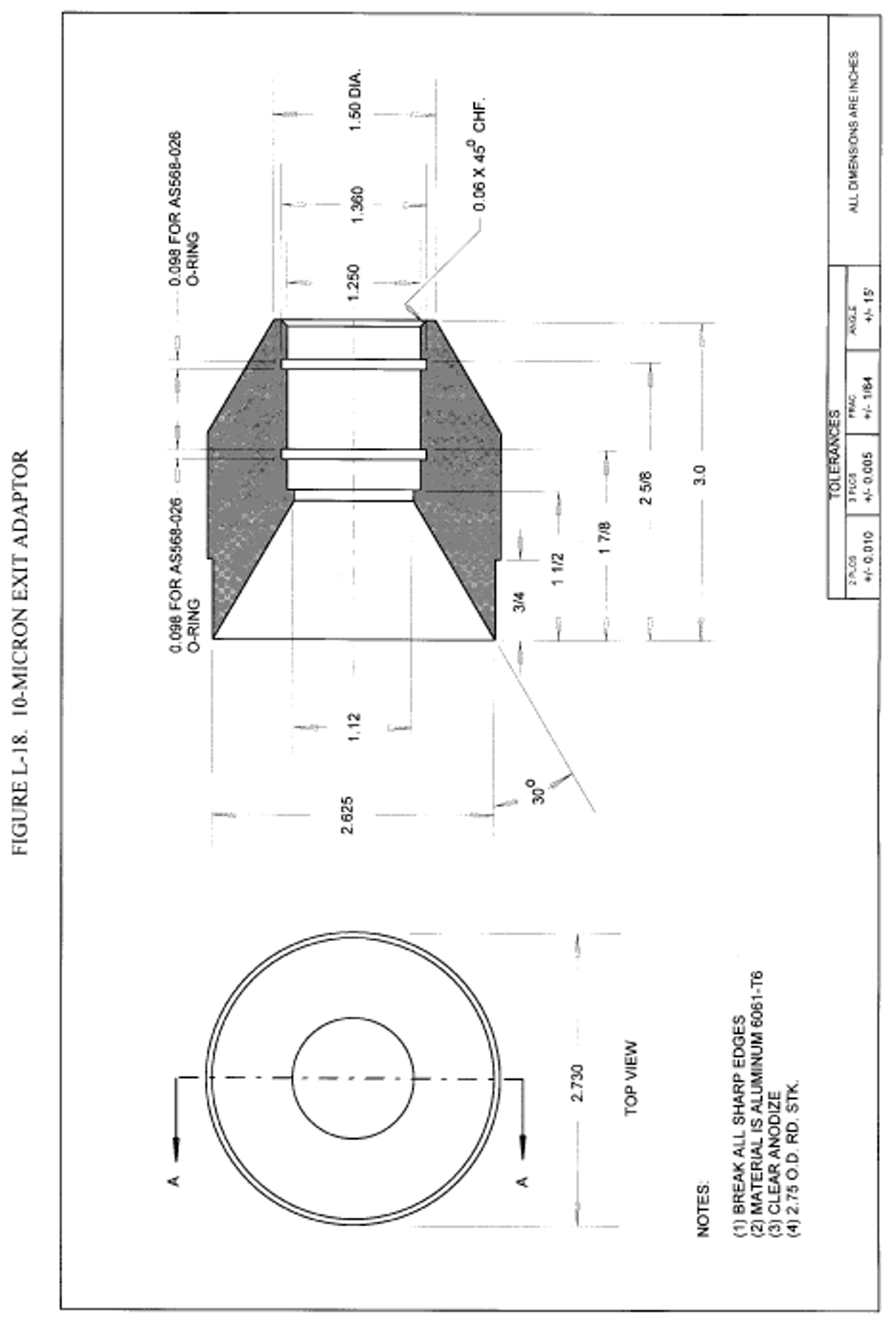
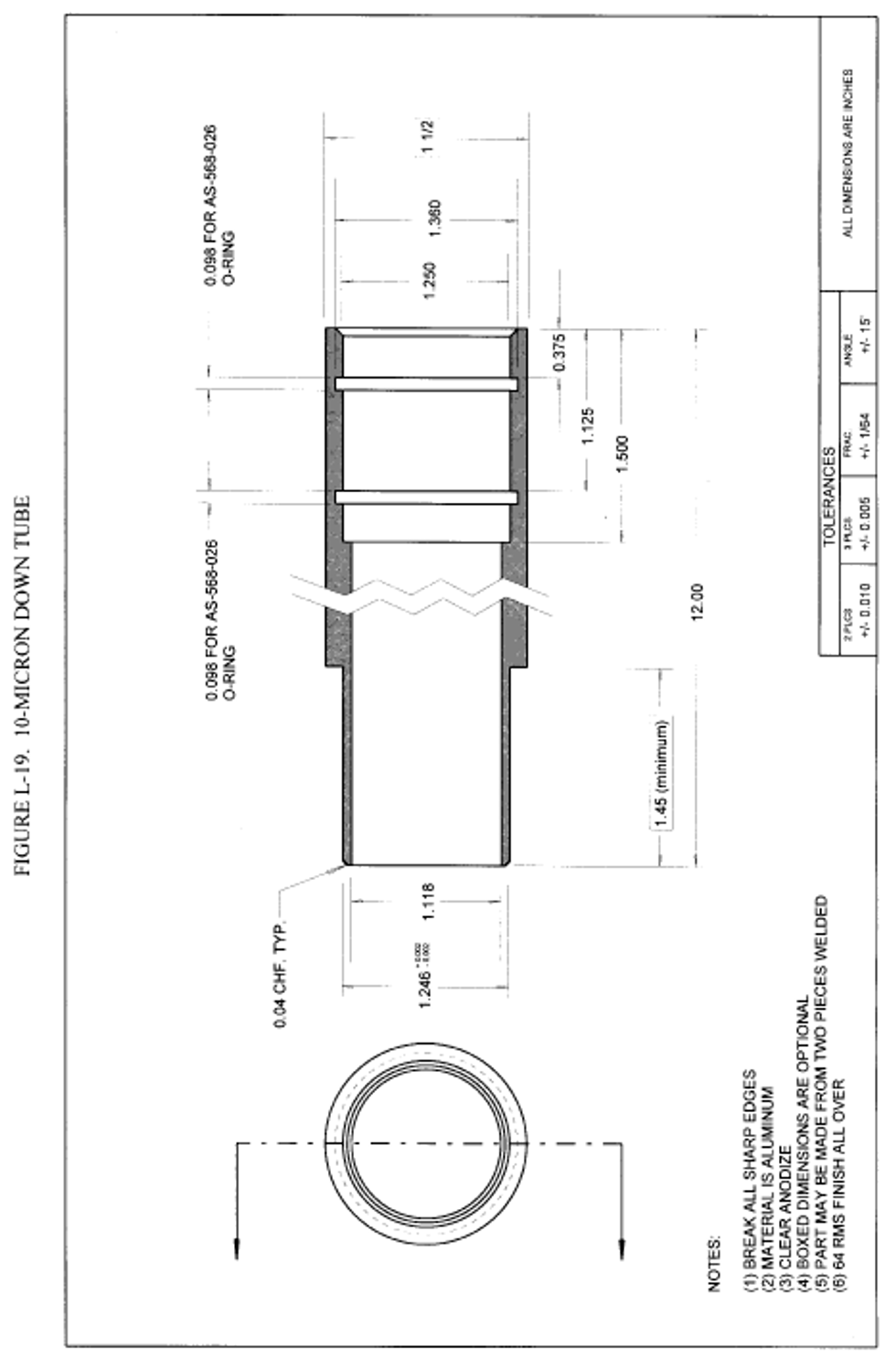
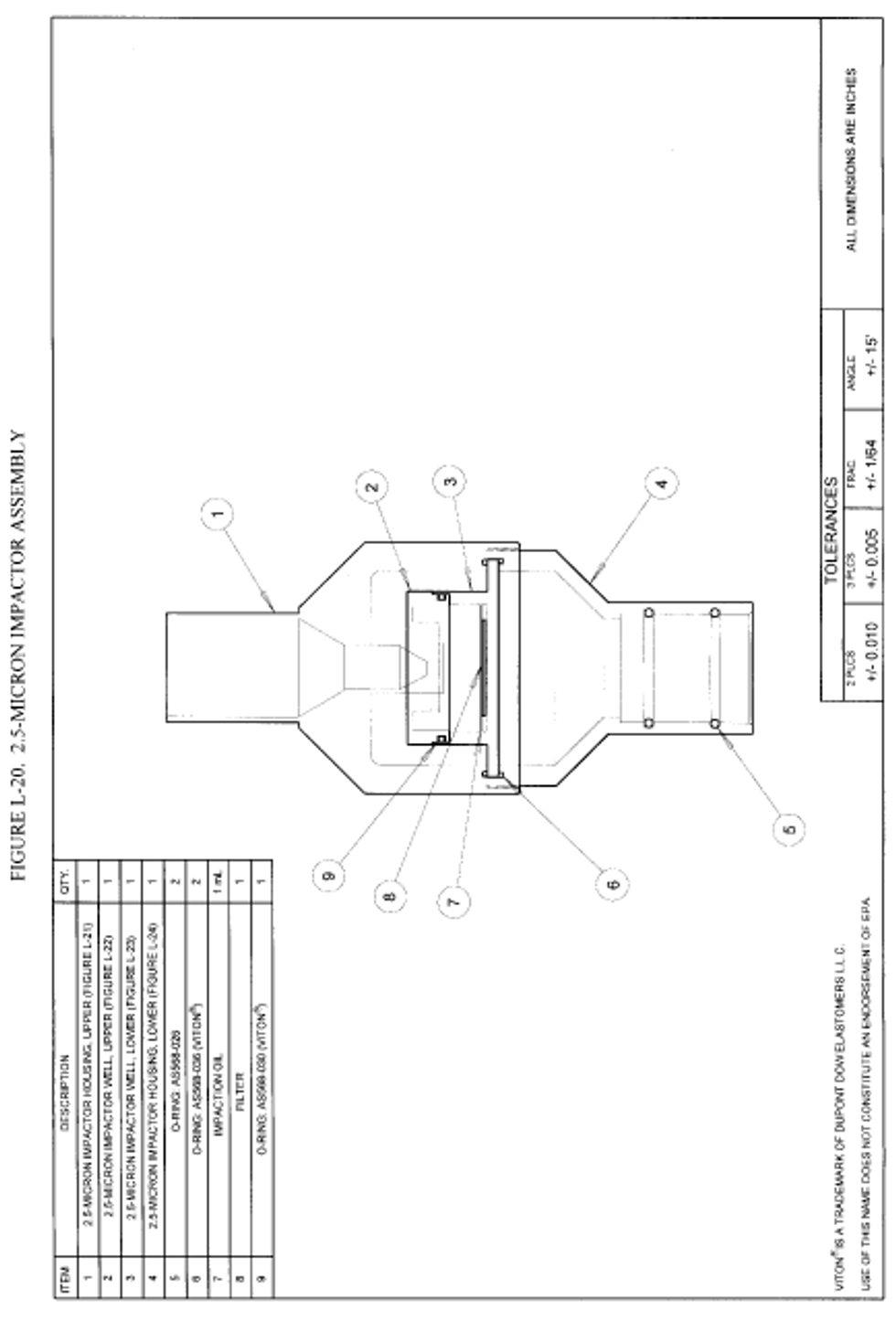
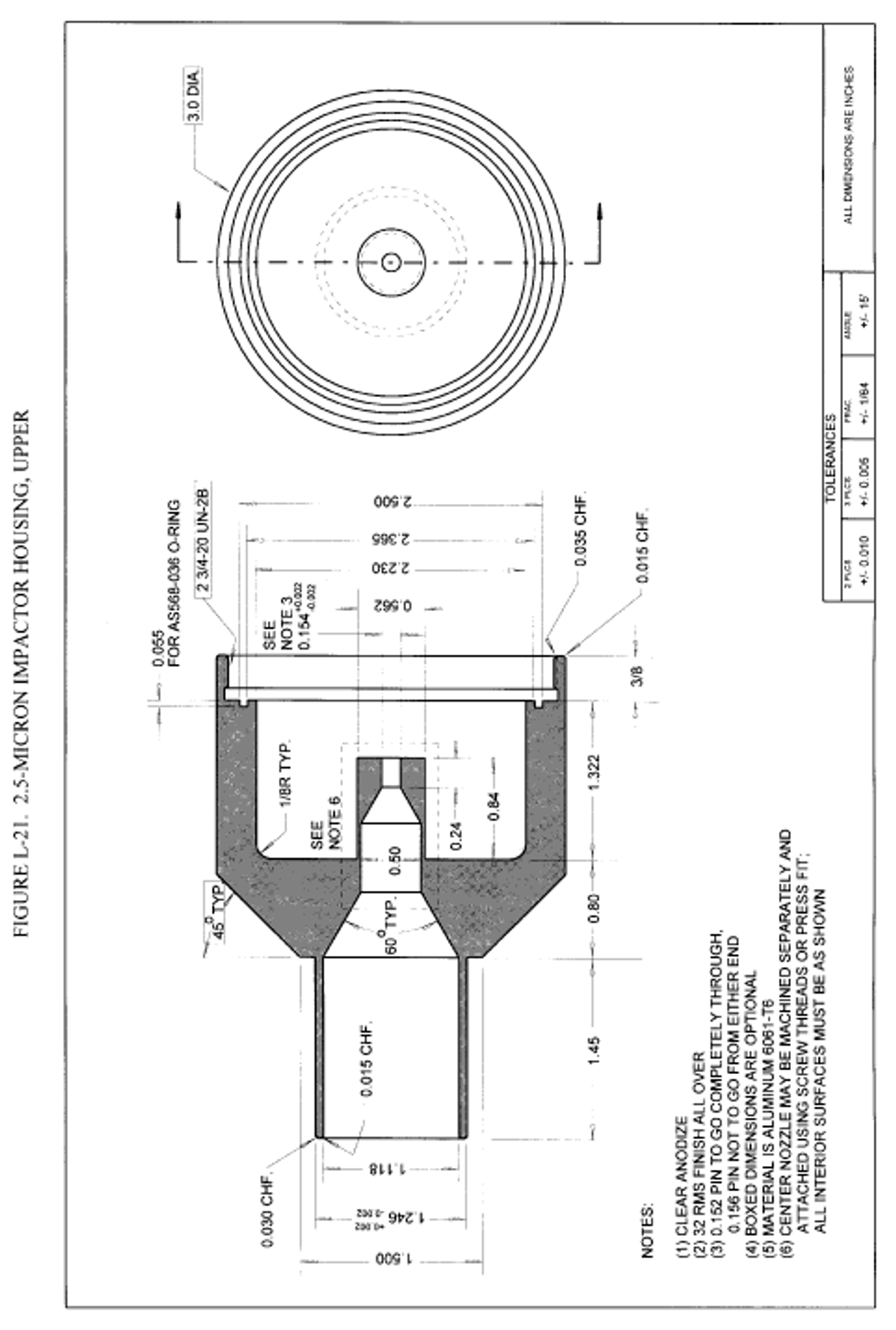
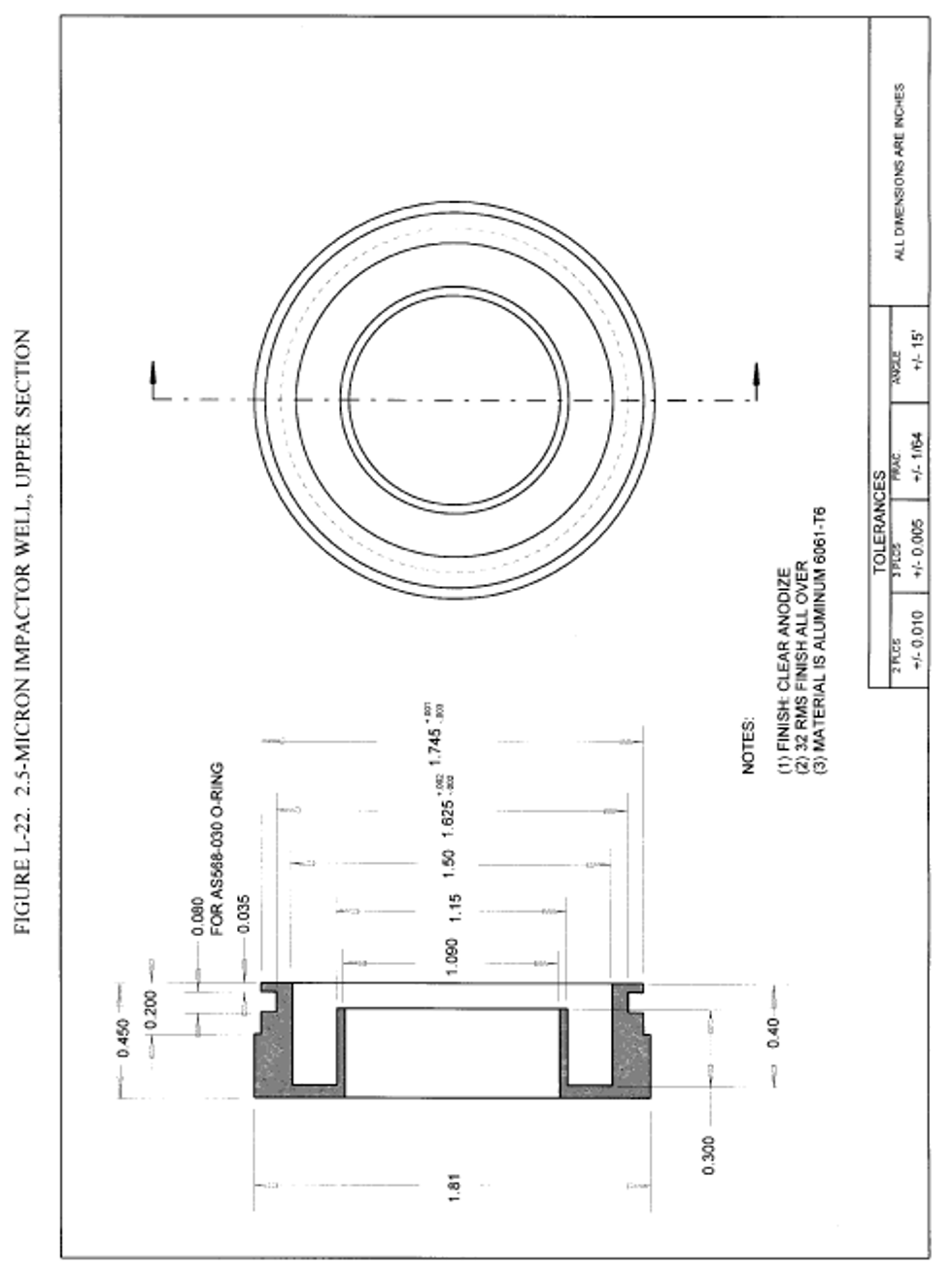
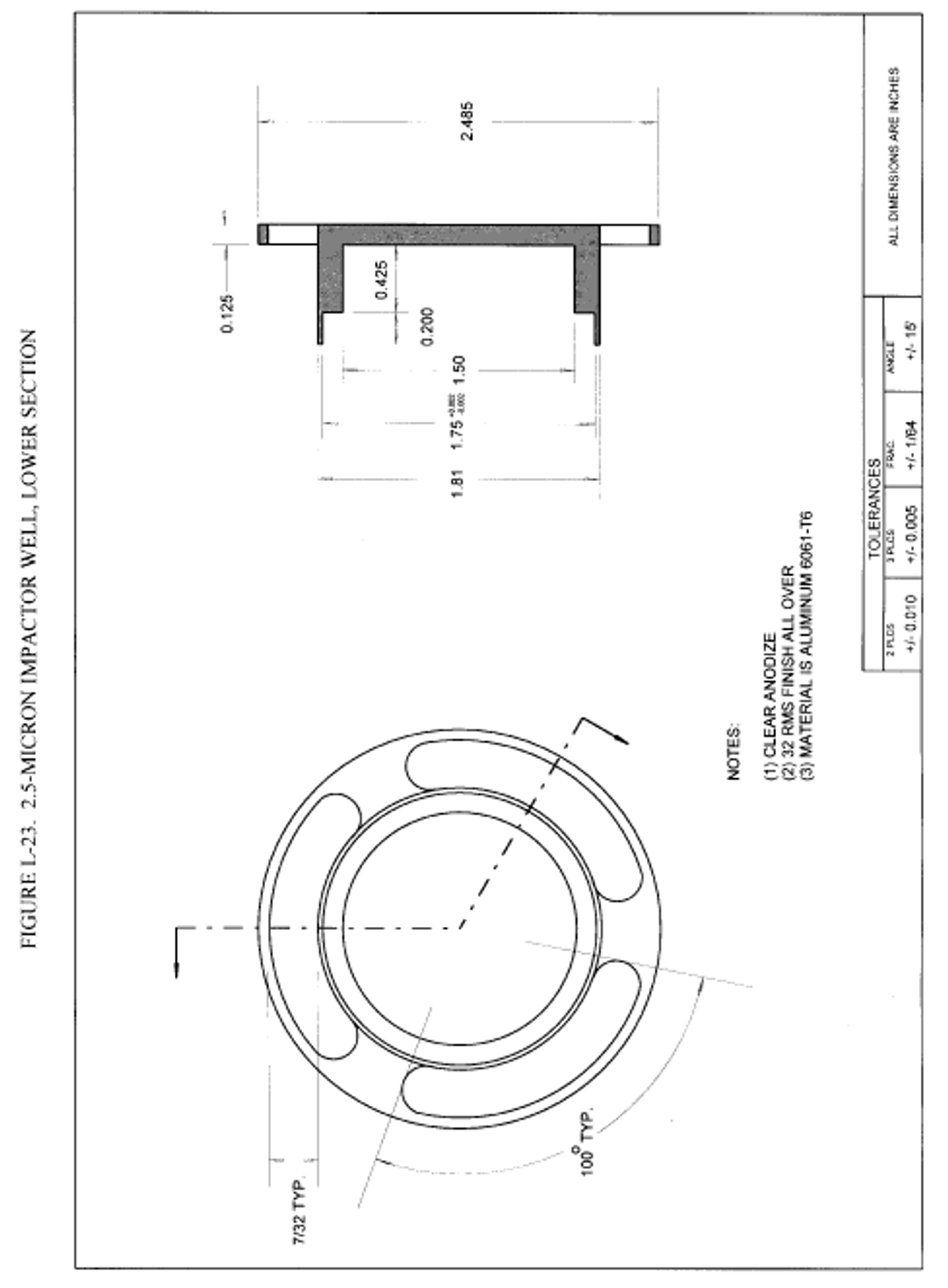
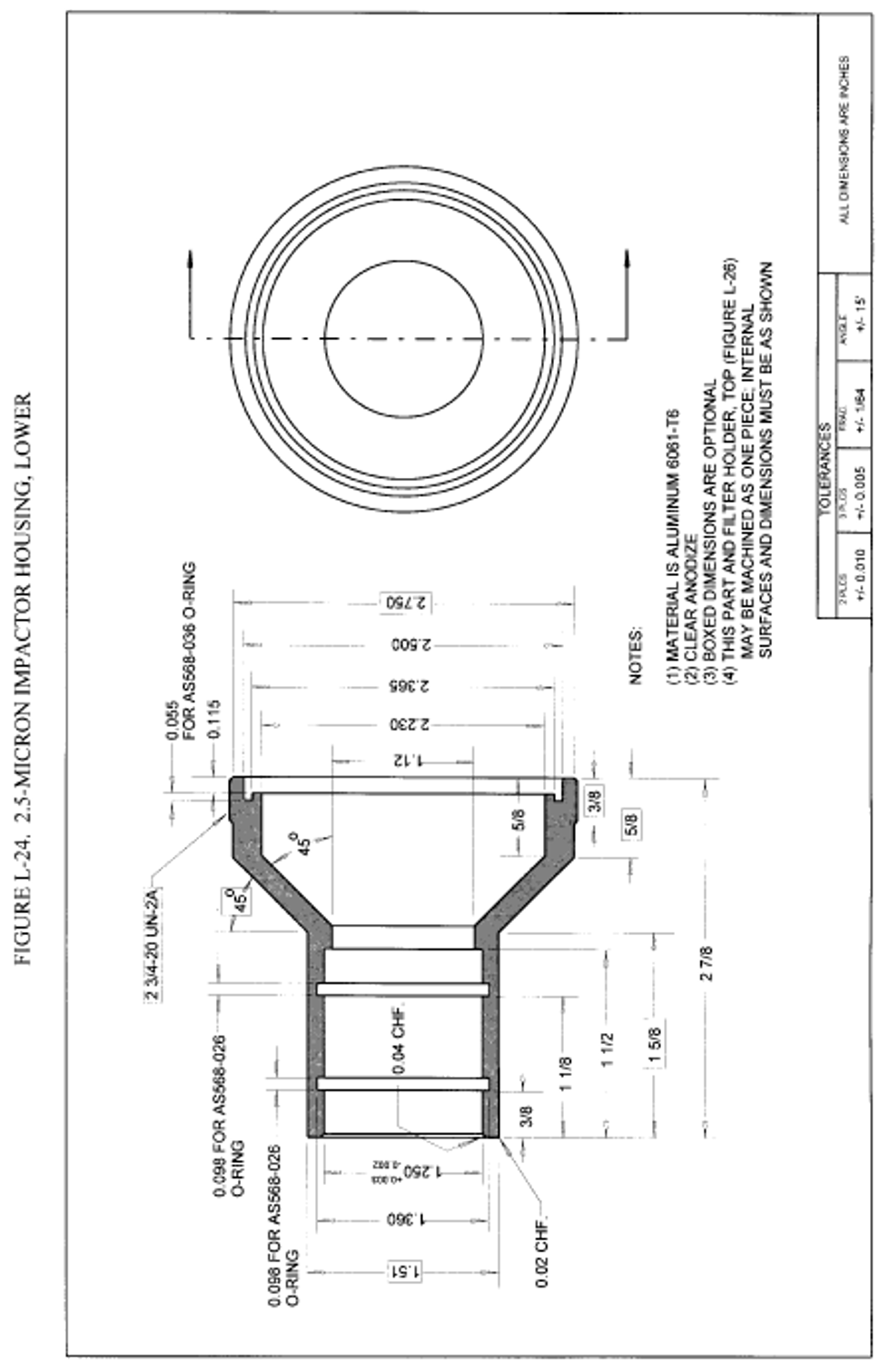
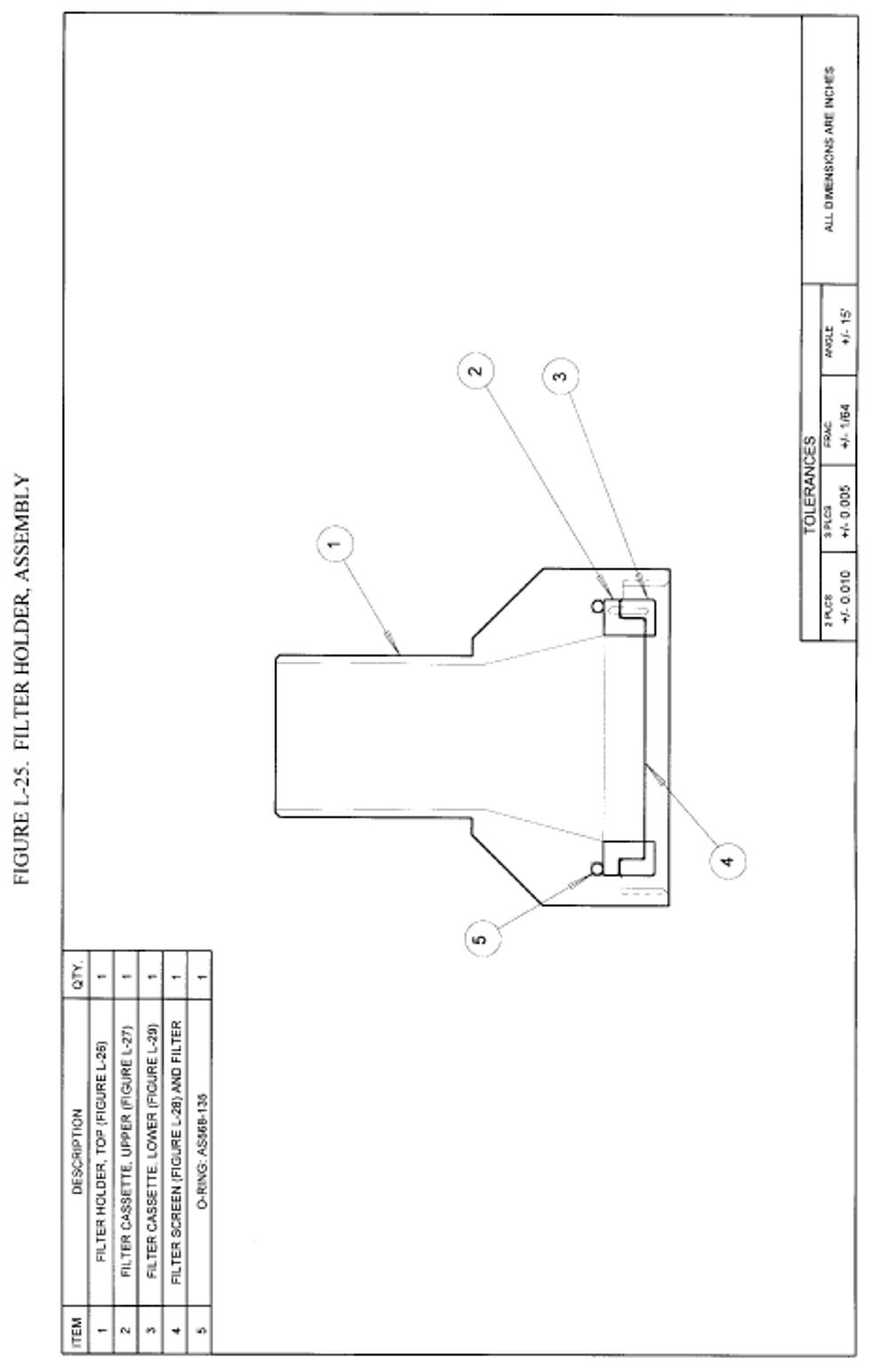
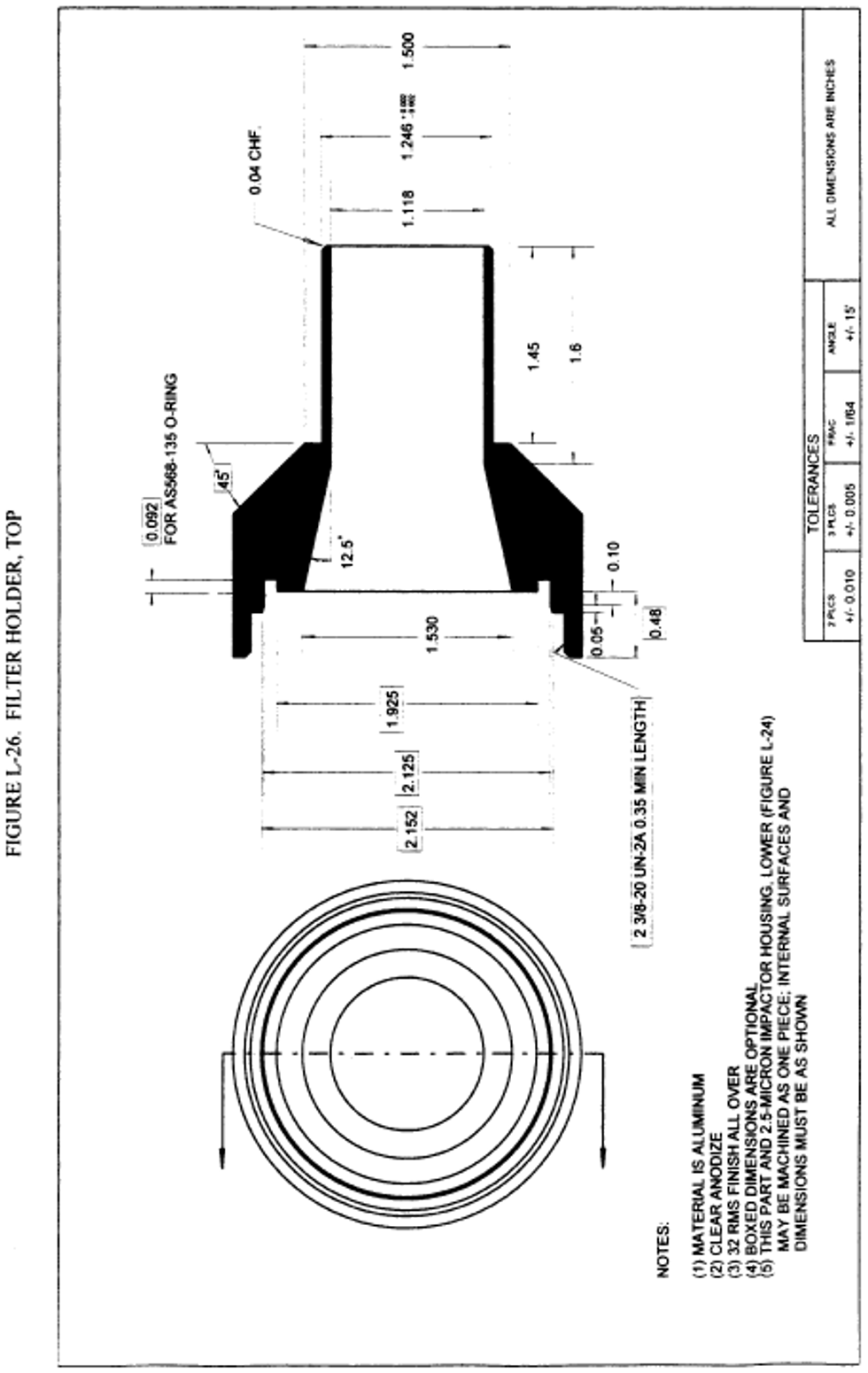
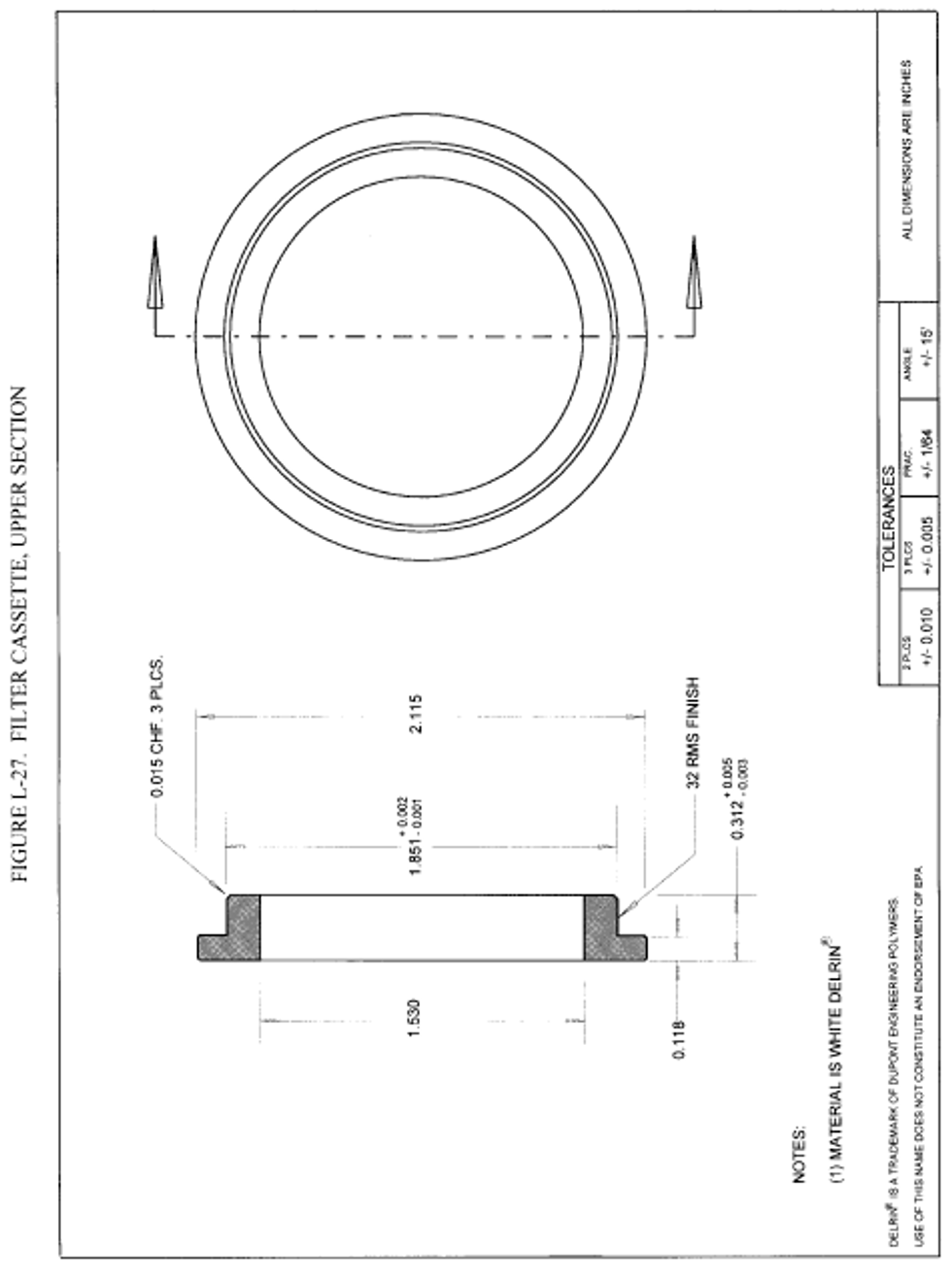
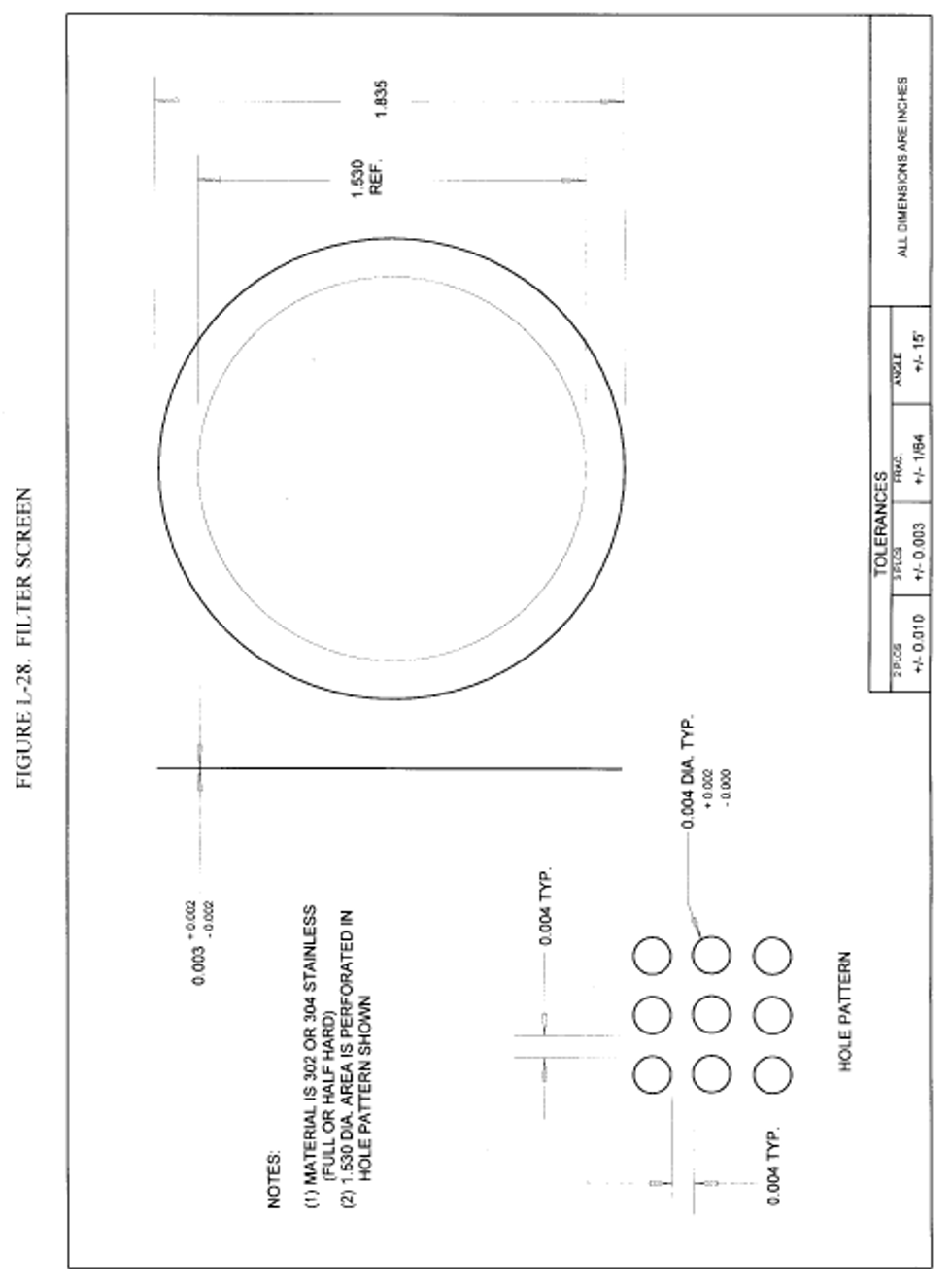
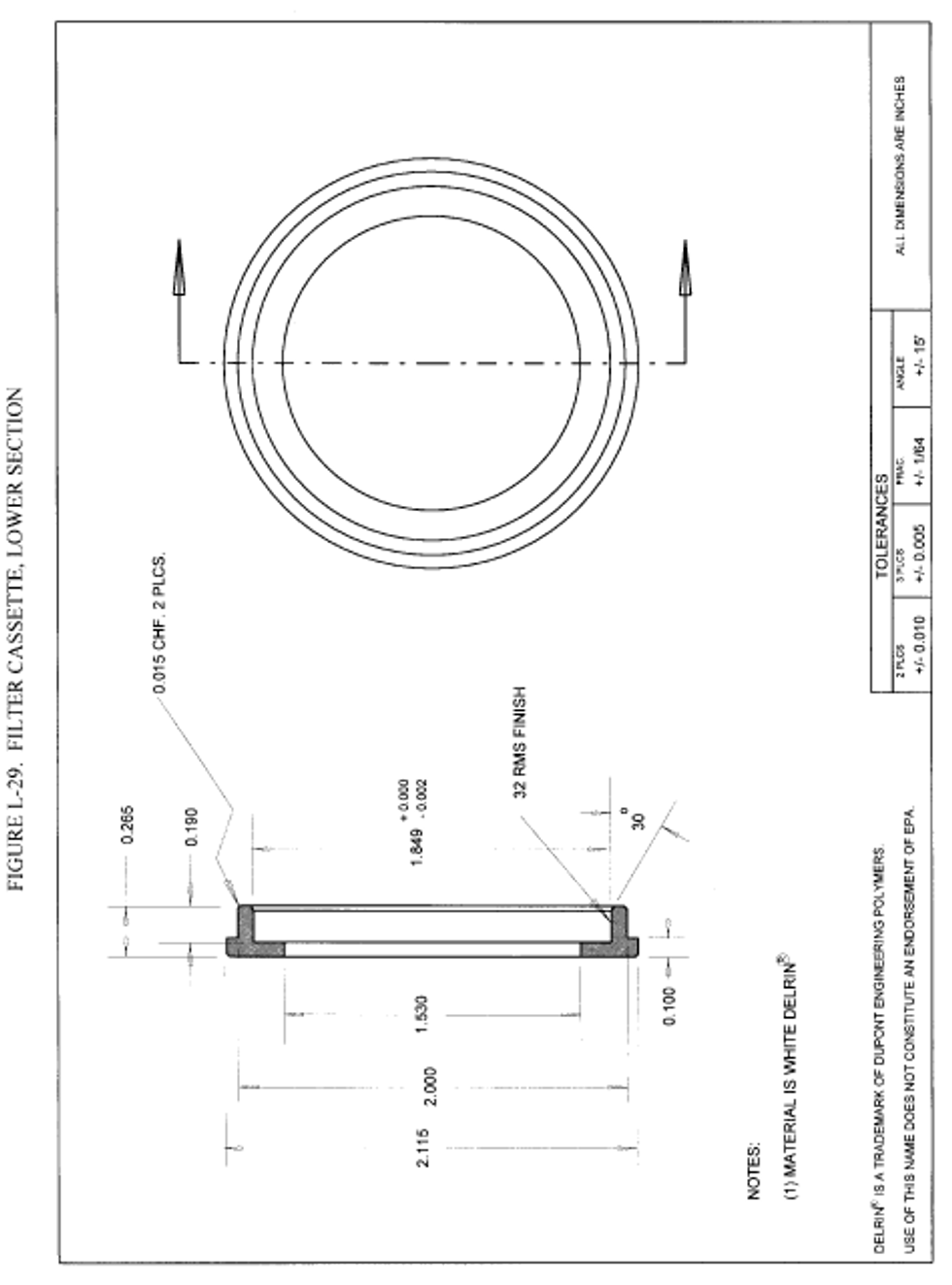
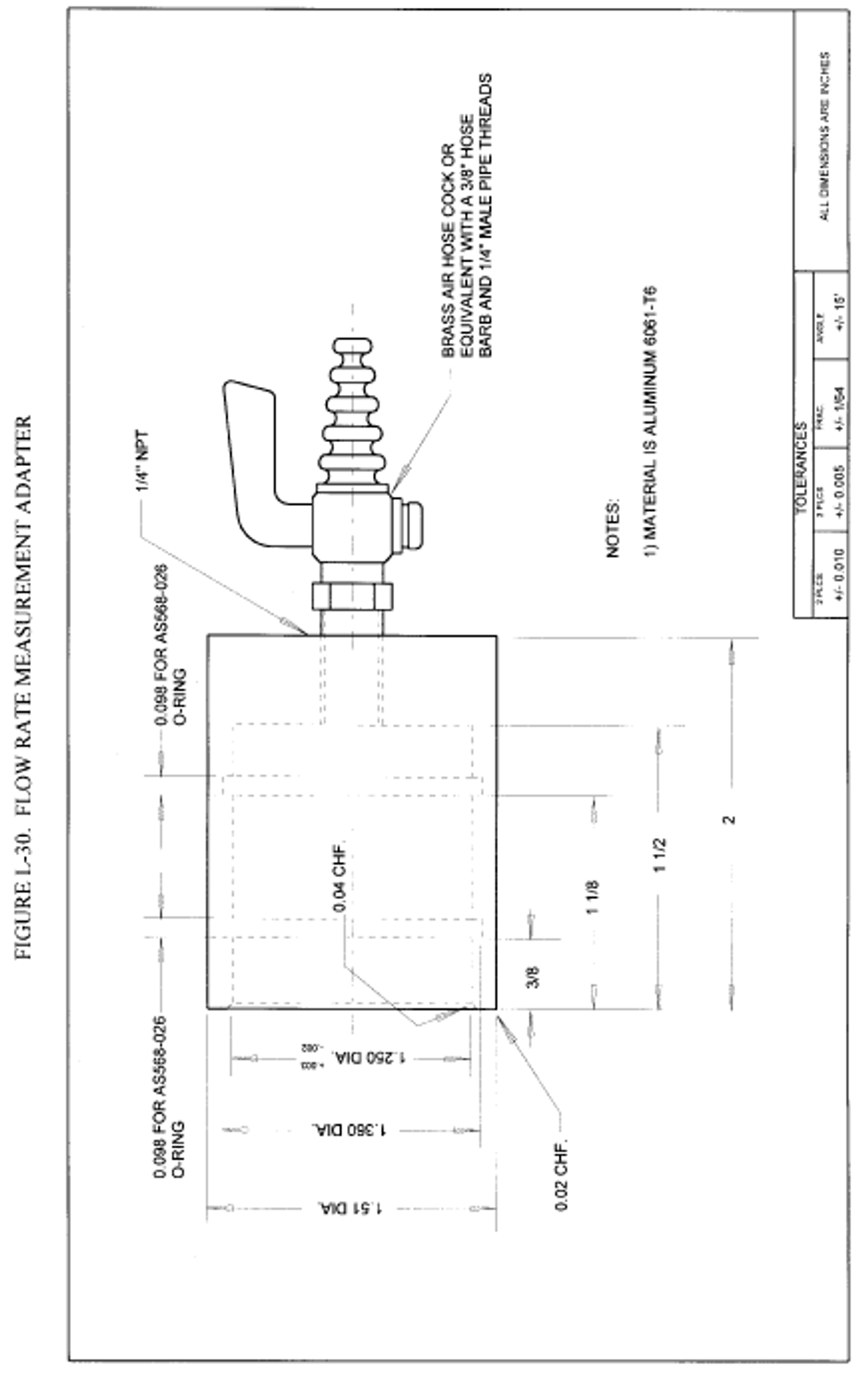
[62 FR 38714, July 18, 1997, as amended at 64 FR 19719, Apr. 22, 1999; 71 FR 61226, Oct. 17, 2006; 89 FR 16381, March 6, 2024]
Appendix M to Part 50 [Reserved]
Appendix N to Part 50 - Interpretation of the National Ambient Air Quality Standards for PM2.5
1.0 General
(a) This appendix explains the data handling conventions and computations necessary for determining when the national ambient air quality standards (NAAQS) for PM 2.5 are met, specifically the primary and secondary annual and 24-hour PM 2.5 NAAQS specified in §§50.7, 50.13, 50.18, and 50.20. PM 2.5 is defined, in general terms, as particles with an aerodynamic diameter less than or equal to a nominal 2.5 micrometers. PM 2.5 mass concentrations are measured in the ambient air by a Federal Reference Method (FRM) based on appendix L to this part, as applicable, and designated in accordance with part 53 of this chapter or by a Federal Equivalent Method (FEM) designated in accordance with part 53 of this chapter. Only those FRM and FEM measurements that are derived in accordance with part 58 of this chapter (i.e., that are deemed “suitable”) shall be used in comparisons with the PM 2.5 NAAQS. The data handling and computation procedures to be used to construct annual and 24-hour NAAQS metrics from reported PM 2.5 mass concentrations, and the associated instructions for comparing these calculated metrics to the levels of the PM 2.5 NAAQS, are specified in sections 2.0, 3.0, and 4.0 of this appendix.
(b) Decisions to exclude, retain, or make adjustments to the data affected by exceptional events, including natural events, are made according to the requirements and process deadlines specified in §§50.1, 50.14 and 51.930 of this chapter.
(c) The terms used in this appendix are defined as follows:
Annual mean refers to a weighted arithmetic mean, based on quarterly means, as defined in section 4.4 of this appendix.
The Air Quality System (AQS) is EPA's official repository of ambient air data.
Collocated monitors refers to two or more air measurement instruments for the same parameter (e.g., PM2.5 mass) operated at the same site location, and whose placement is consistent with §53.1 of this chapter. For purposes of considering a combined site record in this appendix, when two or more monitors are operated at the same site, one monitor is designated as the “primary” monitor with any additional monitors designated as “collocated.” It is implicit in these appendix procedures that the primary monitor and collocated monitor(s) are all deemed suitable for the applicable NAAQS comparison; however, it is not a requirement that the primary and monitors utilize the same specific sampling and analysis method.
Combined site data record is the data set used for performing calculations in appendix N. It represents data for the primary monitors augmented with data from collocated monitors according to the procedure specified in section 3.0(d) of this appendix.
Creditable samples are daily values in the combined site record that are given credit for data completeness. The number of creditable samples (cn) for a given year also governs which value in the sorted series of daily values represents the 98th percentile for that year. Creditable samples include daily values collected on scheduled sampling days and valid make-up samples taken for missed or invalidated samples on scheduled sampling days.
Daily values refer to the 24-hour average concentrations of PM2.5 mass measured (or averaged from hourly measurements in AQS) from midnight to midnight (local standard time) from suitable monitors.
Data substitution tests are diagnostic evaluations performed on an annual PM2.5 NAAQS design value (DV) or a 24-hour PM2.5 NAAQS DV to determine if those metrics, which are judged to be based on incomplete data in accordance with 4.1(b) or 4.2(b) of this appendix shall nevertheless be deemed valid for NAAQS comparisons, or alternatively, shall still be considered incomplete and not valid for NAAQS comparisons. There are two data substitution tests, the “minimum quarterly value” test and the “maximum quarterly value” test. Design values (DVs) are the 3-year average NAAQS metrics that are compared to the NAAQS levels to determine when a monitoring site meets or does not meet the NAAQS, calculated as shown in section 4. There are two separate DVs specified in this appendix:
(1) The 3-year average of PM2.5 annual mean mass concentrations for each eligible monitoring site is referred to as the “annual PM2.5NAAQS DV”.
(2) The 3-year average of annual 98th percentile 24-hour average PM2.5 mass concentration values recorded at each eligible monitoring site is referred to as the “24-hour (or daily) PM2.5NAAQS DV”.
Eligible sites are monitoring stations that meet the criteria specified in §58.11 and §58.30 of this chapter, and thus are approved for comparison to the annual PM2.5 NAAQS. For the 24-hour PM2.5 NAAQS, all site locations that meet the criteria specified in §58.11 are approved (i.e., eligible) for NAAQS comparisons.
Extra samples are non-creditable samples. They are daily values that do not occur on scheduled sampling days and that cannot be used as make-up samples for missed or invalidated scheduled samples. Extra samples are used in mean calculations and are included in the series of all daily values subject to selection as a 98th percentile value, but are not used to determine which value in the sorted list represents the 98th percentile.
Make-up samples are samples collected to take the place of missed or invalidated required scheduled samples. Make-up samples can be made by either the primary or the collocated monitor. Make-up samples are either taken before the next required sampling day or exactly one week after the missed (or voided) sampling day.
The maximum quarterly value data substitution test substitutes actual “high” reported daily PM2.5 values from the same site (specifically, the highest reported non-excluded quarterly value(s) (year non-specific) contained in the combined site record for the evaluated 3-year period) for missing daily values.
The minimum quarterly value data substitution test substitutes actual “low” reported daily PM2.5 values from the same site (specifically, the lowest reported quarterly value(s) (year non-specific) contained in the combined site record for the evaluated 3-year period) for missing daily values.
98th percentile is the smallest daily value out of a year of PM2.5 mass monitoring data below which no more than 98 percent of all daily values fall using the ranking and selection method specified in section 4.5(a) of this appendix.
Primary monitors are suitable monitors designated by a state or local agency in their annual network plan (and in AQS) as the default data source for creating a combined site record for purposes of NAAQS comparisons. If there is only one suitable monitor at a particular site location, then it is presumed to be a primary monitor.
Quarter refers to a calendar quarter (e.g., January through March).
Quarterly data capture rate is the percentage of scheduled samples in a calendar quarter that have corresponding valid reported sample values. Quarterly data capture rates are specifically calculated as the number of creditable samples for the quarter divided by the number of scheduled samples for the quarter, the result then multiplied by 100 and rounded to the nearest integer.
Scheduled PM2.5samples refers to those reported daily values which are consistent with the required sampling frequency (per §58.12 of this chapter) for the primary monitor, or those that meet the special exception noted in section 3.0(e) of this appendix.
Seasonal sampling is the practice of collecting data at a reduced frequency during a season of expected low concentrations.
Suitable monitors are instruments that use sampling and analysis methods approved for NAAQS comparisons. For the annual and 24-hour PM2.5 NAAQS, suitable monitors include all FRMs, and all FEMs/ARMs except those specific continuous FEMs/ARMs disqualified by a particular monitoring agency network in accordance with §58.10(b)(13) and approved by the EPA Regional Administrator per §58.11(e) of this chapter.
Test design values (TDV) are numerical values that used in the data substitution tests described in sections 4.1(c)(i), 4.1(c)(ii) and 4.2(c)(i) of this appendix to determine if the PM2.5 NAAQS DV with incomplete data are judged to be valid for NAAQS comparisons. There are two TDVs: TDVmin to determine if the NAAQS is not met and is used in the “minimum quarterly value” data substitution test and TDVmax to determine if the NAAQS is met and is used in the “maximum quarterly value” data substitution test. These TDV's are derived by substituting historically low or historically high daily concentration values for missing data in an incomplete year(s).
Year refers to a calendar year.
2.0 Monitoring Considerations
(a) Section 58.30 of this chapter provides special considerations for data comparisons to the annual PM2.5 NAAQS.
(b) Monitors meeting the network technical requirements detailed in §58.11 of this chapter are suitable for comparison with the NAAQS for PM2.5.
(c) Section 58.12 of this chapter specifies the required minimum frequency of sampling for PM2.5. Exceptions to the specified sampling frequencies, such as seasonal sampling, are subject to the approval of the EPA Regional Administrator and must be documented in the state or local agency Annual Monitoring Network Plan as required in §58.10 of this chapter and also in AQS.
3.0 Requirements for Data Use and Data Reporting for Comparisons With the NAAQS for PM2.5
(a) Except as otherwise provided in this appendix, all valid FRM/FEM/ARM PM2.5 mass concentration data produced by suitable monitors that are required to be submitted to AQS, or otherwise available to EPA, meeting the requirements of §53.11 of this chapter including appendices A, C, and E shall be used in the DV calculations. Generally, EPA will only use such data if they have been certified by the reporting organization (as prescribed by §58.15 of this chapter); however, data not certified by the reporting organization can nevertheless be used, if the deadline for certification has passed and EPA judges the data to be complete and accurate.
(b) PM2.5 mass concentration data (typically collected hourly for continuous instruments and daily for filter-based instruments) shall be reported to AQS in micrograms per cubic meter (µg/m 3) to at least one decimal place. If concentrations are reported to one decimal place, additional digits to the right of the tenths decimal place shall be truncated. If concentrations are reported to AQS with more than one decimal place, AQS will truncate the value to one decimal place for NAAQS usage (i.e., for implementing the procedures in this appendix). In situations where suitable PM2.5 data are available to EPA but not reported to AQS, the same truncation protocol shall be applied to that data. In situations where PM2.5 mass data are submitted to AQS, or are otherwise available, with less precision than specified above, these data shall nevertheless still be deemed appropriate for NAAQS usage.
(c) Twenty-four-hour average concentrations will be computed in AQS from submitted hourly PM2.5 concentration data for each corresponding day of the year and the result will be stored in the first, or start, hour (i.e., midnight, hour ‘0’) of the 24-hour period. A 24-hour average concentration shall be considered valid if at least 75 percent of the hourly averages (i.e., 18 hourly values) for the 24-hour period are available. In the event that less than all 24 hourly average concentrations are available (i.e., less than 24, but at least 18), the 24-hour average concentration shall be computed on the basis of the hours available using the number of available hours within the 24-hour period as the divisor (e.g., 19, if 19 hourly values are available). Twenty-four-hour periods with seven or more missing hours shall also be considered valid if, after substituting zero for all missing hourly concentrations, the resulting 24-hour average daily value is greater than the level of the 24-hour PM2.5 NAAQS (i.e., greater than or equal to 35.5 µg/m 3). Twenty-four hour average PM2.5 mass concentrations that are averaged in AQS from hourly values will be truncated to one decimal place, consistent with the data handling procedure for the reported hourly (and also 24-hour filter-based) data.
(d) All calculations shown in this appendix shall be implemented on a site-level basis. Site level concentration data shall be processed as follows:
(1) The default dataset for PM2.5 mass concentrations for a site shall consist of the measured concentrations recorded from the designated primary monitor(s). All daily values produced by the primary monitor are considered part of the site record; this includes all creditable samples and all extra samples.
(2) Data for the primary monitors shall be augmented as much as possible with data from collocated monitors. If a valid daily value is not produced by the primary monitor for a particular day (scheduled or otherwise), but a value is available from a collocated monitor, then that collocated value shall be considered part of the combined site data record. If more than one collocated daily value is available, the average of those valid collocated values shall be used as the daily value. The data record resulting from this procedure is referred to as the “combined site data record.”
(3) In certain circumstances, including but not limited to site closures or relocations, data from two nearby sites may be combined into a single site data record for the purpose of calculating a valid design value. The appropriate Regional Administrator may approve such site combinations if the Regional Administrator determines that the measured concentrations do not differ substantially between the two sites, taking into consideration factors such as distance between sites, spatial and temporal patterns in air quality, local emissions and meteorology, jurisdictional boundaries, and terrain features.
(e) All daily values in a combined site data record are used in the calculations specified in this appendix; however, not all daily values are given credit towards data completeness requirements. Only creditable samples are given credit for data completeness. Creditable samples include daily values in the combined site record that are collected on scheduled sampling days and valid make-up samples taken for missed or invalidated samples on scheduled sampling days. Days are considered scheduled according to the required sampling frequency of the designated primary monitor with one exception. The exception is, if a collocated continuous FEM/ARM monitor has a more intensive sampling frequency than the primary FRM monitor, then samples contributed to the combined site record from that continuous FEM/ARM monitor are always considered scheduled and, hence, also creditable. Daily values in the combined site data record that are reported for nonscheduled days, but that are not valid make-up samples are referred to as extra samples.
4.0 Comparisons With the Annual and 24-Hour PM2.5 NAAQS
4.1 Annual PM2.5 NAAQS
(a) Levels of the primary and secondary annual PM 2.5 NAAQS are specified in §§50.7, 50.13, 50.18, and 50.20 as applicable.
(b) Three years of valid annual means are required to produce a valid annual PM2.5 NAAQS DV. A year meets data completeness requirements when quarterly data capture rates for all four quarters are at least 75 percent. However, years with at least 11 creditable samples in each quarter shall also be considered valid if the resulting annual mean or resulting annual PM2.5 NAAQS DV (rounded according to the conventions of section 4.3 of this appendix) is greater than the level of the applicable primary or secondary annual PM2.5 NAAQS. Furthermore, where the explicit 75 percent data capture and/or 11 sample minimum requirements are not met, the 3-year annual PM2.5 NAAQS DV shall still be considered valid if it passes at least one of the two data substitution tests stipulated below.
(c) In the case of one, two, or three years that do not meet the completeness requirements of section 4.1(b) of this appendix and thus would normally not be useable for the calculation of a valid annual PM2.5 NAAQS DV, the annual PM2.5 NAAQS DV shall nevertheless be considered valid if one of the test conditions specified in sections 4.1(c)(i) and 4.1(c)(ii) of this appendix is met.
(i) An annual PM2.5 NAAQS DV that is above the level of the NAAQS can be validated if it passes the minimum quarterly value data substitution test. This type of data substitution is permitted only if there are at least 30 days across the three quarters of the three years under consideration (e.g., collectively, quarter 1 of year 1, quarter 1 of year 2 and quarter 1 of year 3) from which to select the quarter-specific low value. Data substitution will be performed in all quarter periods that have less than 11 creditable samples.
Procedure: Identify for each deficient quarter (i.e., those with less than 11 creditable samples) the lowest reported daily value for that quarter, looking across those three months of all three years under consideration. If after substituting the lowest reported daily value for a quarter for (11− cn) daily values in the matching deficient quarter(s) (i.e., to bring the creditable number for those quarters up to 11), the procedure yields a recalculated annual PM2.5 NAAQS test DV (TDVmin) that is greater than the level of the standard, then the annual PM2.5 NAAQS DV is deemed to have passed the diagnostic test and is valid, and the annual PM2.5 NAAQS is deemed to have been violated in that 3-year period.
(ii) An annual PM2.5 NAAQS DV that is equal to or below the level of the NAAQS can be validated if it passes the maximum quarterly value data substitution test. This type of data substitution is permitted only if there is at least 50 percent data capture in each quarter that is deficient of 75 percent data capture in each of the three years under consideration. Data substitution will be performed in all quarter periods that have less than 75 percent data capture but at least 50 percent data capture. If any quarter has less than 50 percent data capture then this substitution test cannot be used.
Procedure: Identify for each deficient quarter (i.e., those with less than 75 percent but at least 50 percent data capture) the highest reported daily value for that quarter, excluding state-flagged data affected by exceptional events which have been approved for exclusion by the Administrator, looking across those three quarters of all three years under consideration. If after substituting the highest reported daily PM2.5 value for a quarter for all missing daily data in the matching deficient quarter(s) (i.e., to make those quarters 100 percent complete), the procedure yields a recalculated annual PM2.5 NAAQS test DV (TDVmax) that is less than or equal to the level of the standard, then the annual PM2.5 NAAQS DV is deemed to have passed the diagnostic test and is valid, and the annual PM2.5 NAAQS is deemed to have been met in that 3-year period.
(d) An annual PM2.5 NAAQS DV based on data that do not meet the completeness criteria stated in 4(b) and also do not satisfy the test conditions specified in section 4(c), may also be considered valid with the approval of, or at the initiative of, the EPA Administrator, who may consider factors such as monitoring site closures/moves, monitoring diligence, the consistency and levels of the daily values that are available, and nearby concentrations in determining whether to use such data.
(e) The equations for calculating the annual PM2.5 NAAQS DVs are given in section 4.4 of this appendix.
4.2 Twenty-four-hour PM2.5 NAAQS
(a) Levels of the primary and secondary 24-hour PM 2.5 NAAQS are specified in §§50.7, 50.13, 50.18, and 50.20 as applicable.
(b) Three years of valid annual PM2.5 98th percentile mass concentrations are required to produce a valid 24-hour PM2.5 NAAQS DV. A year meets data completeness requirements when quarterly data capture rates for all four quarters are at least 75 percent. However, years shall be considered valid, notwithstanding quarters with less than complete data (even quarters with less than 11 creditable samples, but at least one creditable sample must be present for the year), if the resulting annual 98th percentile value or resulting 24-hour NAAQS DV (rounded according to the conventions of section 4.3 of this appendix) is greater than the level of the standard. Furthermore, where the explicit 75 percent quarterly data capture requirement is not met, the 24-hour PM2.5 NAAQS DV shall still be considered valid if it passes the maximum quarterly value data substitution test.
(c) In the case of one, two, or three years that do not meet the completeness requirements of section 4.2(b) of this appendix and thus would normally not be useable for the calculation of a valid 24-hour PM2.5 NAAQS DV, the 24-hour PM2.5 NAAQS DV shall nevertheless be considered valid if the test conditions specified in section 4.2(c)(i) of this appendix are met.
(i) A PM2.5 24-hour mass NAAQS DV that is equal to or below the level of the NAAQS can be validated if it passes the maximum quarterly value data substitution test. This type of data substitution is permitted only if there is at least 50 percent data capture in each quarter that is deficient of 75 percent data capture in each of the three years under consideration. Data substitution will be performed in all quarters that have less than 75 percent data capture but at least 50 percent data capture. If any quarter has less than 50 percent data capture then this substitution test cannot be used.
Procedure: Identify for each deficient quarter (i.e., those with less than 75 percent but at least 50 percent data capture) the highest reported daily PM2.5 value for that quarter, excluding state-flagged data affected by exceptional events which have been approved for exclusion by the Regional Administrator, looking across those three quarters of all three years under consideration. If, after substituting the highest reported daily maximum PM2.5 value for a quarter for all missing daily data in the matching deficient quarter(s) (i.e., to make those quarters 100 percent complete), the procedure yields a recalculated 3-year 24-hour NAAQS test DV (TDVmax) less than or equal to the level of the standard, then the 24-hour PM2.5 NAAQS DV is deemed to have passed the diagnostic test and is valid, and the 24-hour PM2.5 NAAQS is deemed to have been met in that 3-year period.
(d) A 24-hour PM2.5 NAAQS DV based on data that do not meet the completeness criteria stated in section 4(b) of this appendix and also do not satisfy the test conditions specified in section 4(c) of this appendix, may also be considered valid with the approval of, or at the initiative of, the EPA Administrator, who may consider factors such as monitoring site closures/moves, monitoring diligence, the consistency and levels of the daily values that are available, and nearby concentrations in determining whether to use such data.
(e) The procedures and equations for calculating the 24-hour PM2.5 NAAQS DVs are given in section 4.5 of this appendix.
4.3 Rounding Conventions. For the purposes of comparing calculated PM2.5 NAAQS DVs to the applicable level of the standard, it is necessary to round the final results of the calculations described in sections 4.4 and 4.5 of this appendix. Results for all intermediate calculations shall not be rounded.
(a) Annual PM2.5 NAAQS DVs shall be rounded to the nearest tenth of a µg/m 3 (decimals x.x5 and greater are rounded up to the next tenth, and any decimal lower than x.x5 is rounded down to the nearest tenth).
(b) Twenty-four-hour PM2.5 NAAQS DVs shall be rounded to the nearest 1 µg/m 3 (decimals 0.5 and greater are rounded up to the nearest whole number, and any decimal lower than 0.5 is rounded down to the nearest whole number).
4.4 Equations for the Annual PM2.5 NAAQS.
(a) An annual mean value for PM2.5 is determined by first averaging the daily values of a calendar quarter using equation 1 of this appendix:
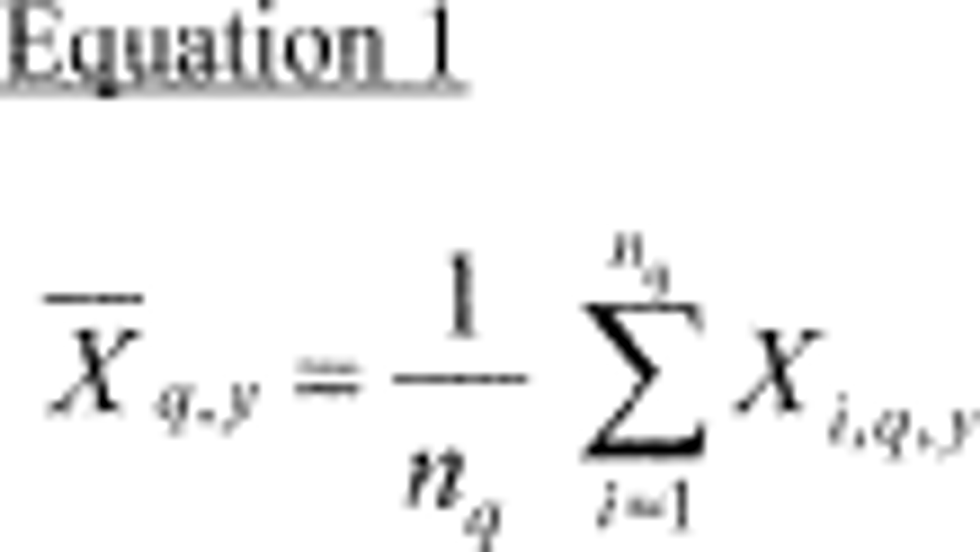
Where:
X͞ q,y = the mean for quarter q of the year y;
nq = the number of daily values in the quarter; and
xi q,y = the i th value in quarter q for year y.
(b) Equation 2 of this appendix is then used to calculate the site annual mean:
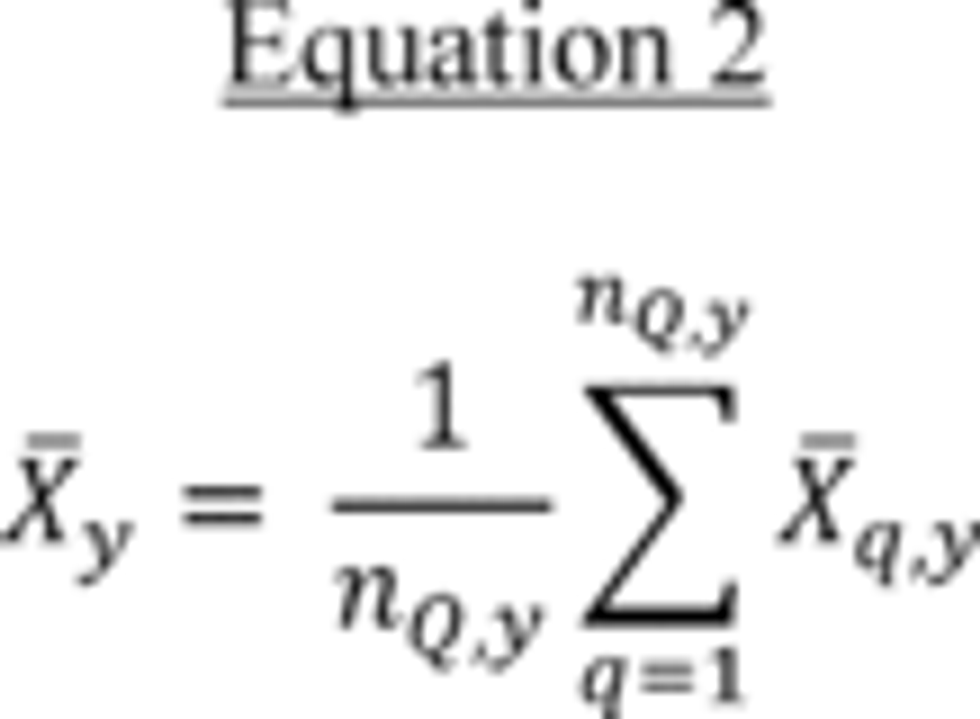
Where:
X͞ y = the annual mean concentration for year y (y = 1, 2, or 3);
nQ,y = the number of quarters Q in year y with at least one daily value; and
X͞ q,y = the mean for quarter q of year y (result of equation 1).
(c) The annual PM2.5 NAAQS DV is calculated using equation 3 of this appendix:
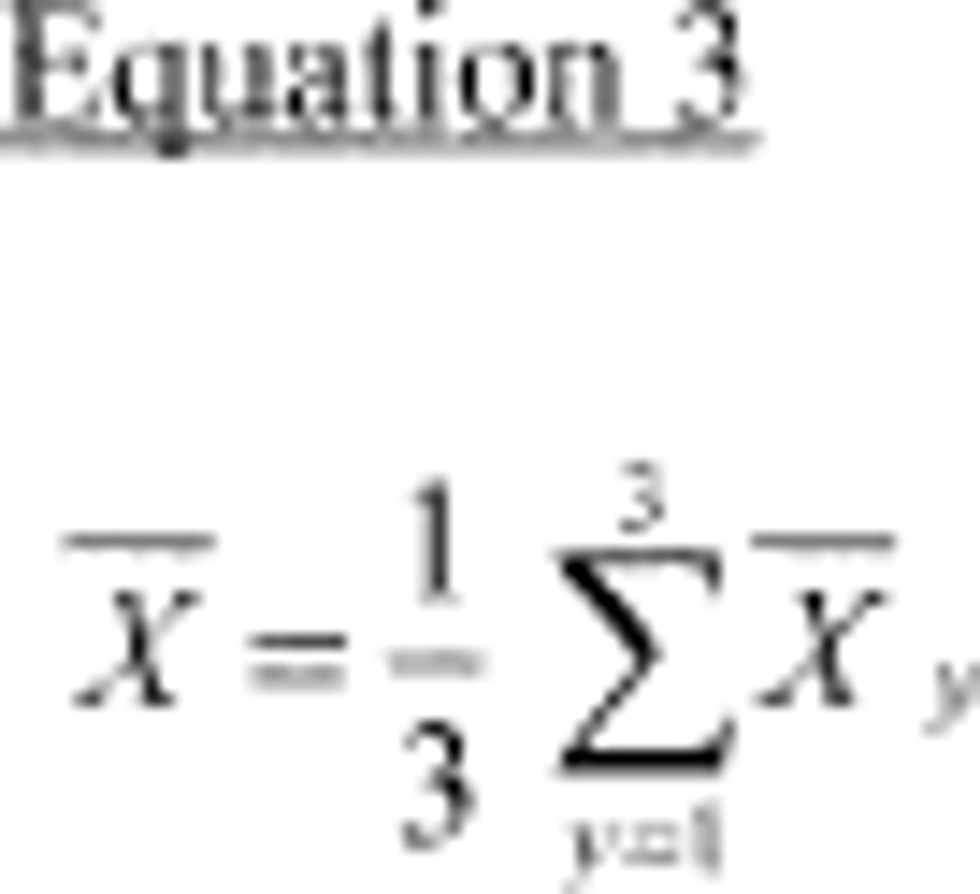
Where:
X͞ = the annual PM2.5 NAAQS DV; and
X͞ y = the annual mean for year y (result of equation 2)
(d) The annual PM2.5 NAAQS DV is rounded according to the conventions in section 4.3 of this appendix before comparisons with the levels of the primary and secondary annual PM2.5 NAAQS are made.
4.5 Procedures and Equations for the 24-Hour PM2.5 NAAQS
(a) When the data for a particular site and year meet the data completeness requirements in section 4.2 of this appendix, calculation of the 98th percentile is accomplished by the steps provided in this subsection. Table 1 of this appendix shall be used to identify annual 98th percentile values.
Identification of annual 98th percentile values using the Table 1 procedure will be based on the creditable number of samples (as described below), rather than on the actual number of samples. Credit will not be granted for extra (non-creditable) samples. Extra samples, however, are candidates for selection as the annual 98th percentile. [The creditable number of samples will determine how deep to go into the data distribution, but all samples (creditable and extra) will be considered when making the percentile assignment.] The annual creditable number of samples is the sum of the four quarterly creditable number of samples.
Procedure: Sort all the daily values from a particular site and year by descending value. (For example: (x[1], x[2], x[3], * * *, x[n]). In this case, x[1] is the largest number and x[n] is the smallest value.) The 98th percentile value is determined from this sorted series of daily values which is ordered from the highest to the lowest number. Using the left column of Table 1, determine the appropriate range for the annual creditable number of samples for year y (cny) (e.g., for 120 creditable samples per year, the appropriate range would be 101 to 150). The corresponding “n” value in the right column identifies the rank of the annual 98th percentile value in the descending sorted list of site specific daily values for year y (e.g., for the range of 101 to 150, n would be 3). Thus, P0.98, y = the n th largest value (e.g., for the range of 101 to 150, the 98th percentile value would be the third highest value in the sorted series of daily values.
| Annual number of creditable samples for year y (cny) | The 98th percentile for year y (P0.98,y), is the n th maximum 24-hour average value for the year where n is the listed number |
|---|---|
| 1 to 50 | 1 |
| 51 to 100 | 2 |
| 101 to 150 | 3 |
| 151 to 200 | 4 |
| 201 to 250 | 5 |
| 251 to 300 | 6 |
| 301 to 350 | 7 |
| 351 to 366 | 8 |
(b) The 24-hour PM2.5 NAAQS DV is then calculated by averaging the annual 98th percentiles using equation 4 of this appendix: P0.98,y
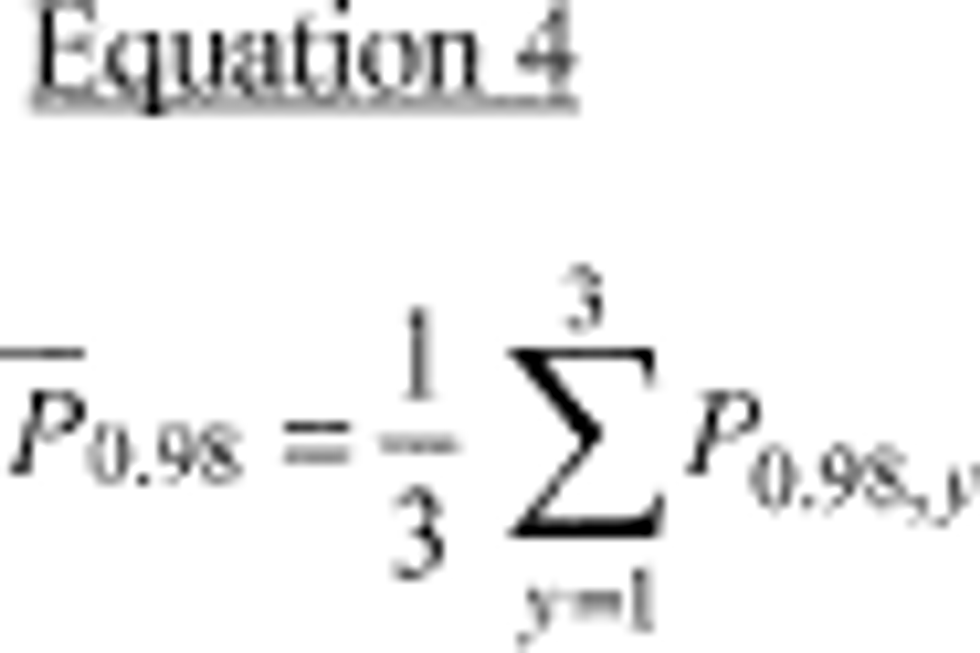
Where:
P͞ 0.98 = the 24-hour PM2.5 NAAQS DV; and
P0.98, y = the annual 98th percentile for year y
(c) The 24-hour PM2.5 NAAQS DV is rounded according to the conventions in section 4.3 of this appendix before a comparison with the level of the primary and secondary 24-hour NAAQS are made.
[78 FR 3277, Jan. 15, 2013, as amended at 82 FR 14327, Mar. 20, 2017; 89 FR 16381, March 6, 2024]
Appendix O to Part 50 - Reference Method for the Determination of Coarse Particulate Matter as PM10-2.5 in the Atmosphere
1.0 Applicability and Definition
1.1 This method provides for the measurement of the mass concentration of coarse particulate matter (PM10-2.5) in ambient air over a 24-hour period. In conjunction with additional analysis, this method may be used to develop speciated data.
1.2 For the purpose of this method, PM10-2.5 is defined as particulate matter having an aerodynamic diameter in the nominal range of 2.5 to 10 micrometers, inclusive.
1.3 For this reference method, PM10-2.5 concentrations shall be measured as the arithmetic difference between separate but concurrent, collocated measurements of PM10 and PM2.5, where the PM10 measurements are obtained with a specially approved sampler, identified as a “PM10c sampler,” that meets more demanding performance requirements than conventional PM10 samplers described in appendix J of this part. Measurements obtained with a PM10c sampler are identified as “PM10c measurements” to distinguish them from conventional PM10 measurements obtained with conventional PM10 samplers. Thus, PM10-2.5 = PM10c − PM2.5.
1.4 The PM10c and PM2.5 gravimetric measurement processes are considered to be nondestructive, and the PM10c and PM2.5 samples obtained in the PM10-2.5 measurement process can be subjected to subsequent physical or chemical analyses.
1.5 Quality assessment procedures are provided in part 58, appendix A of this chapter. The quality assurance procedures and guidance provided in reference 1 in section 13 of this appendix, although written specifically for PM2.5, are generally applicable for PM10c, and, hence, PM10-2.5 measurements under this method, as well.
1.6 A method based on specific model PM10c and PM2.5 samplers will be considered a reference method for purposes of §53.11 of this chapter only if:
(a) The PM10c and PM2.5 samplers and the associated operational procedures meet the requirements specified in this appendix and all applicable requirements in §53.11 of this chapter, and
(b) The method based on the specific samplers and associated operational procedures have been designated as a reference method in accordance with §53.11 of this chapter.
1.7 PM10-2.5 methods based on samplers that meet nearly all specifications set forth in this method but have one or more significant but minor deviations or modifications from those specifications may be designated as “Class I” equivalent methods for PM10-2.5 in accordance with §53.11 of this chapter.
1.8 PM2.5 measurements obtained incidental to the PM10-2.5 measurements by this method shall be considered to have been obtained with a reference method for PM2.5 in accordance with appendix L of this part.
1.9 PM10c measurements obtained incidental to the PM10-2.5 measurements by this method shall be considered to have been obtained with a reference method for PM10 in accordance with appendix J of this part, provided that:
(a) The PM10c measurements are adjusted to EPA reference conditions (25°C and 760 millimeters of mercury), and
(b) Such PM10c measurements are appropriately identified to differentiate them from PM10 measurements obtained with other (conventional) methods for PM10 designated in accordance with §53.11 of this chapter as reference or equivalent methods for PM10.
2.0 Principle
2.1 Separate, collocated, electrically powered air samplers for PM10c and PM2.5 concurrently draw ambient air at identical, constant volumetric flow rates into specially shaped inlets and through one or more inertial particle size separators where the suspended particulate matter in the PM10 or PM2.5 size range, as applicable, is separated for collection on a polytetrafluoroethylene (PTFE) filter over the specified sampling period. The air samplers and other aspects of this PM10-2.5 reference method are specified either explicitly in this appendix or by reference to other applicable regulations or quality assurance guidance.
2.2 Each PM10c and PM2.5 sample collection filter is weighed (after moisture and temperature conditioning) before and after sample collection to determine the net weight (mass) gain due to collected PM10c or PM2.5. The total volume of air sampled by each sampler is determined by the sampler from the measured flow rate at local ambient temperature and pressure and the sampling time. The mass concentrations of both PM10c and PM2.5 in the ambient air are computed as the total mass of collected particles in the PM10 or PM2.5 size range, as appropriate, divided by the total volume of air sampled by the respective samplers, and expressed in micrograms per cubic meter (µg/m 3)at local temperature and pressure conditions. The mass concentration of PM10-2.5 is determined as the PM10c concentration value less the corresponding, concurrently measured PM2.5 concentration value.
2.3 Most requirements for PM10-2.5 reference methods are similar or identical to the requirements for PM2.5 reference methods as set forth in appendix L to this part. To insure uniformity, applicable appendix L requirements are incorporated herein by reference in the sections where indicated rather than repeated in this appendix.
3.0 PM10-2.5Measurement Range
3.1 Lower concentration limit. The lower detection limit of the mass concentration measurement range is estimated to be approximately 3 µg/m 3, based on the observed precision of PM2.5 measurements in the national PM2.5 monitoring network, the probable similar level of precision for the matched PM10c measurements, and the additional variability arising from the differential nature of the measurement process. This value is provided merely as a guide to the significance of low PM10-2.5 concentration measurements.
3.2 Upper concentration limit. The upper limit of the mass concentration range is determined principally by the PM10c filter mass loading beyond which the sampler can no longer maintain the operating flow rate within specified limits due to increased pressure drop across the loaded filter. This upper limit cannot be specified precisely because it is a complex function of the ambient particle size distribution and type, humidity, the individual filter used, the capacity of the sampler flow rate control system, and perhaps other factors. All PM10c samplers are estimated to be capable of measuring 24-hour mass concentrations of at least 200 µg/m 3 while maintaining the operating flow rate within the specified limits. The upper limit for the PM10-2.5 measurement is likely to be somewhat lower because the PM10-2.5 concentration represents only a fraction of the PM10 concentration.
3.3 Sample period. The required sample period for PM10-2.5 concentration measurements by this method shall be at least 1,380 minutes but not more than 1,500 minutes (23 to 25 hours), and the start times of the PM2.5 and PM10c samples are within 10 minutes and the stop times of the samples are also within 10 minutes (see section 10.4 of this appendix).
4.0 Accuracy (bias)
4.1 Because the size, density, and volatility of the particles making up ambient particulate matter vary over wide ranges and the mass concentration of particles varies with particle size, it is difficult to define the accuracy of PM10-2.5 measurements in an absolute sense. Furthermore, generation of credible PM10-2.5 concentration standards at field monitoring sites and presenting or introducing such standards reliably to samplers or monitors to assess accuracy is still generally impractical. The accuracy of PM10-2.5 measurements is therefore defined in a relative sense as bias, referenced to measurements provided by other reference method samplers or based on flow rate verification audits or checks, or on other performance evaluation procedures.
4.2 Measurement system bias for monitoring data is assessed according to the procedures and schedule set forth in part 58, appendix A of this chapter. The goal for the measurement uncertainty (as bias) for monitoring data is defined in part 58, appendix A of this chapter as an upper 95 percent confidence limit for the absolute bias of 15 percent. Reference 1 in section 13 of this appendix provides additional information and guidance on flow rate accuracy audits and assessment of bias.
5.0 Precision
5.1 Tests to establish initial measurement precision for each sampler of the reference method sampler pair are specified as a part of the requirements for designation as a reference method under §53.11 of this chapter.
5.2 Measurement system precision is assessed according to the procedures and schedule set forth in appendix A to §53.11 of this chapter. The goal for acceptable measurement uncertainty, as precision, of monitoring data is defined in part 58, appendix A of this chapter as an upper 95 percent confidence limit for the coefficient of variation (CV) of 15 percent. Reference 1 in section 13 of this appendix provides additional information and guidance on this requirement.
6.0 Filters for PM10cand PM2.5Sample Collection. Sample collection filters for both PM10c and PM2.5 measurements shall be identical and as specified in section 6 of appendix L to this part.
7.0 Sampler. The PM10-2.5 sampler shall consist of a PM10c sampler and a PM2.5 sampler, as follows:
7.1 The PM2.5 sampler shall be as specified in section 7 of appendix L to this part.
7.2 The PM10c sampler shall be of like manufacturer, design, configuration, and fabrication to that of the PM2.5 sampler and as specified in section 7 of appendix L to this part, except as follows:
7.2.1 The particle size separator specified in section 7.3.4 of appendix L to this part shall be eliminated and replaced by a downtube extension fabricated as specified in Figure O-1 of this appendix.
7.2.2 The sampler shall be identified as a PM10c sampler on its identification label required under §53.9(d) of this chapter.
7.2.3 The average temperature and average barometric pressure measured by the sampler during the sample period, as described in Table L-1 of appendix L to this part, need not be reported to EPA's AQS data base, as required by section 7.4.19 and Table L-1 of appendix L to this part, provided such measurements for the sample period determined by the associated PM2.5 sampler are reported as required.
7.3 In addition to the operation/instruction manual required by section 7.4.18 of appendix L to this part for each sampler, supplemental operational instructions shall be provided for the simultaneous operation of the samplers as a pair to collect concurrent PM10c and PM2.5 samples. The supplemental instructions shall cover any special procedures or guidance for installation and setup of the samplers for PM10-2.5 measurements, such as synchronization of the samplers' clocks or timers, proper programming for collection of concurrent samples, and any other pertinent issues related to the simultaneous, coordinated operation of the two samplers.
7.4 Capability for electrical interconnection of the samplers to simplify sample period programming and further ensure simultaneous operation is encouraged but not required. Any such capability for interconnection shall not supplant each sampler's capability to operate independently, as required by section 7 of appendix L of this part.
8.0 Filter Weighing
8.1 Conditioning and weighing for both PM10c and PM2.5 sample filters shall be as specified in section 8 of appendix L to this part. See reference 1 of section 13 of this appendix for additional, more detailed guidance.
8.2 Handling, conditioning, and weighing for both PM10c and PM2.5 sample filters shall be matched such that the corresponding PM10c and PM2.5 filters of each filter pair receive uniform treatment. The PM10c and PM2.5 sample filters should be weighed on the same balance, preferably in the same weighing session and by the same analyst.
8.3 Due care shall be exercised to accurately maintain the paired relationship of each set of concurrently collected PM10c and PM2.5 sample filters and their net weight gain data and to avoid misidentification or reversal of the filter samples or weight data. See Reference 1 of section 13 of this appendix for additional guidance.
9.0 Calibration. Calibration of the flow rate, temperature measurement, and pressure measurement systems for both the PM10c and PM2.5 samplers shall be as specified in section 9 of appendix L to this part.
10.0 PM10-2.5 Measurement Procedure
10.1 The PM10c and PM2.5 samplers shall be installed at the monitoring site such that their ambient air inlets differ in vertical height by not more than 0.2 meter, if possible, but in any case not more than 1 meter, and the vertical axes of their inlets are separated by at least 1 meter but not more than 4 meters, horizontally.
10.2 The measurement procedure for PM10c shall be as specified in section 10 of appendix L to this part, with “PM10c” substituted for “PM2.5” wherever it occurs in that section.
10.3 The measurement procedure for PM2.5 shall be as specified in section 10 of appendix L to this part.
10.4 For the PM10-2.5 measurement, the PM10c and PM2.5 samplers shall be programmed to operate on the same schedule and such that the sample period start times are within 5 minutes and the sample duration times are within 5 minutes.
10.5 Retrieval, transport, and storage of each PM10c and PM2.5 sample pair following sample collection shall be matched to the extent practical such that both samples experience uniform conditions.
11.0 Sampler Maintenance. Both PM10c and PM2.5 samplers shall be maintained as described in section 11 of appendix L to this part.
12.0 Calculations
12.1 Both concurrent PM10c and PM2.5 measurements must be available, valid, and meet the conditions of section 10.4 of this appendix to determine the PM10-2.5 mass concentration.
12.2 The PM10c mass concentration is calculated using equation 1 of this section:
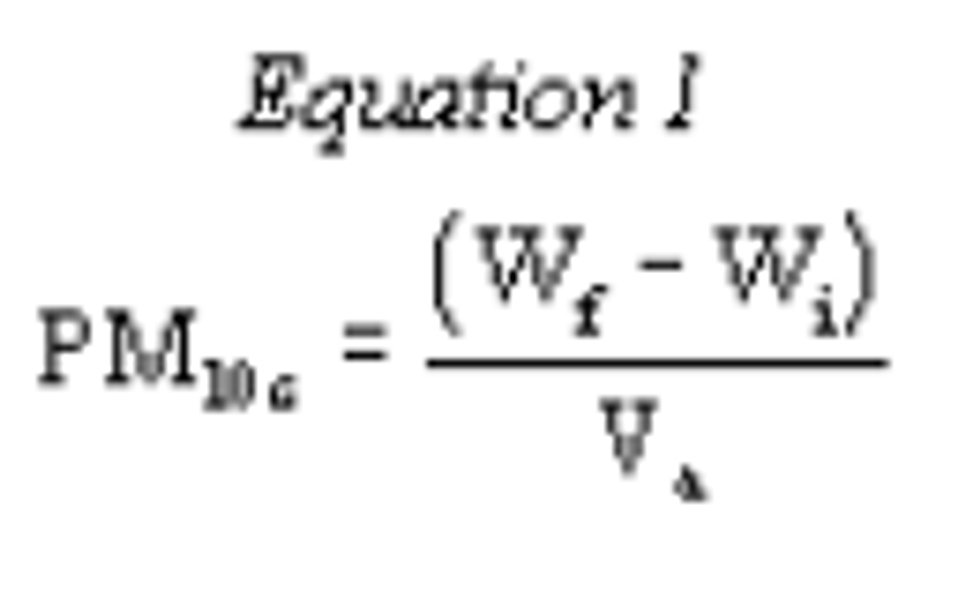
Where:
PM10c = mass concentration of PM10c, µg/m 3;
Wf, Wi = final and initial masses (weights), respectively, of the filter used to collect the PM10c particle sample, µg;
Va = total air volume sampled by the PM10c sampler in actual volume units measured at local conditions of temperature and pressure, as provided by the sampler, m 3.
Note:
Total sample time must be between 1,380 and 1,500 minutes (23 and 25 hrs) for a fully valid PM10c sample; however, see also section 3.3 of this appendix.
12.3 The PM2.5 mass concentration is calculated as specified in section 12 of appendix L to this part.
12.4 The PM10−2.5 mass concentration, in µg/m 3, is calculated using Equation 2 of this section:
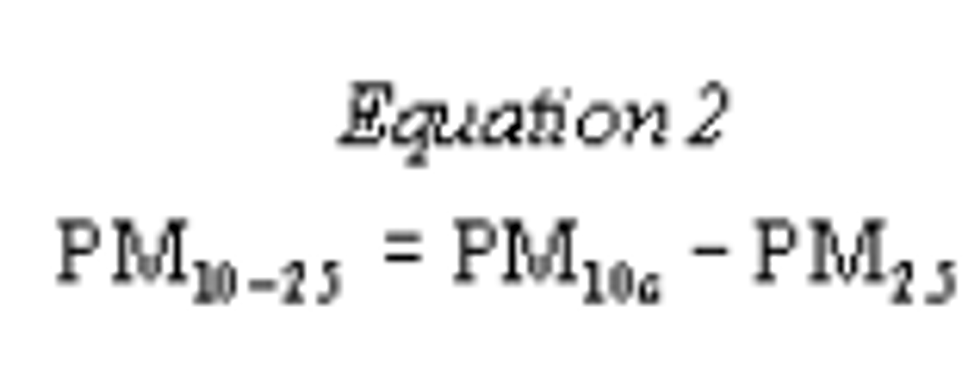
13.0 Reference
1. Quality Assurance Guidance Document 2.12. Monitoring PM2.5 in Ambient Air Using Designated Reference or Class I Equivalent Methods. Draft, November 1998 (or later version or supplement, if available). Available at: www.epa.gov/ttn/amtic/pgqa.html.
14.0 Figures
Figure O-1 is included as part of this appendix O.
[71 FR 61230, Oct. 17, 2006]
Appendix P to Part 50 - Interpretation of the Primary and Secondary National Ambient Air Quality Standards for Ozone
1. General
(a) This appendix explains the data handling conventions and computations necessary for determining whether the national 8-hour primary and secondary ambient air quality standards for ozone (O3) specified in §50.15 are met at an ambient O3 air quality monitoring site. Ozone is measured in the ambient air by a reference method based on appendix D of this part, as applicable, and designated in accordance with §53.11 of this chapter, or by an equivalent method designated in accordance with §53.11 of this chapter. Data reporting, data handling, and computation procedures to be used in making comparisons between reported O3 concentrations and the levels of the O3 standards are specified in the following sections. Whether to exclude, retain, or make adjustments to the data affected by exceptional events, including stratospheric O3 intrusion and other natural events, is determined by the requirements under §§50.1, 50.14 and 51.930.
(b) The terms used in this appendix are defined as follows:
8-hour average is the rolling average of eight hourly O3 concentrations as explained in section 2 of this appendix.
Annual fourth-highest daily maximum refers to the fourth highest value measured at a monitoring site during a particular year.
Daily maximum 8-hour average concentration refers to the maximum calculated 8-hour average for a particular day as explained in section 2 of this appendix.
Design values are the metrics (i.e., statistics) that are compared to the NAAQS levels to determine compliance, calculated as shown in section 3 of this appendix.
O3monitoring season refers to the span of time within a calendar year when individual States are required to measure ambient O3 concentrations as listed in §53.11 appendix D to this chapter.
Year refers to calendar year.
2. Primary and Secondary Ambient Air Quality Standards for Ozone
2.1 Data Reporting and Handling Conventions
Computing 8-hour averages. Hourly average concentrations shall be reported in parts per million (ppm) to the third decimal place, with additional digits to the right of the third decimal place truncated. Running 8-hour averages shall be computed from the hourly O3 concentration data for each hour of the year and shall be stored in the first, or start, hour of the 8-hour period. An 8-hour average shall be considered valid if at least 75% of the hourly averages for the 8-hour period are available. In the event that only 6 or 7 hourly averages are available, the 8-hour average shall be computed on the basis of the hours available using 6 or 7 as the divisor. 8-hour periods with three or more missing hours shall be considered valid also, if, after substituting one-half the minimum detectable limit for the missing hourly concentrations, the 8-hour average concentration is greater than the level of the standard. The computed 8-hour average O3 concentrations shall be reported to three decimal places (the digits to the right of the third decimal place are truncated, consistent with the data handling procedures for the reported data).
Daily maximum 8-hour average concentrations. (a) There are 24 possible running 8-hour average O3 concentrations for each calendar day during the O3 monitoring season. The daily maximum 8-hour concentration for a given calendar day is the highest of the 24 possible 8-hour average concentrations computed for that day. This process is repeated, yielding a daily maximum 8-hour average O3 concentration for each calendar day with ambient O3 monitoring data. Because the 8-hour averages are recorded in the start hour, the daily maximum 8-hour concentrations from two consecutive days may have some hourly concentrations in common. Generally, overlapping daily maximum 8-hour averages are not likely, except in those non-urban monitoring locations with less pronounced diurnal variation in hourly concentrations.
(b) An O3 monitoring day shall be counted as a valid day if valid 8-hour averages are available for at least 75% of possible hours in the day (i.e., at least 18 of the 24 averages). In the event that less than 75% of the 8-hour averages are available, a day shall also be counted as a valid day if the daily maximum 8-hour average concentration for that day is greater than the level of the standard.
2.2 Primary and Secondary Standard-related Summary Statistic
The standard-related summary statistic is the annual fourth-highest daily maximum 8-hour O3 concentration, expressed in parts per million, averaged over three years. The 3-year average shall be computed using the three most recent, consecutive calendar years of monitoring data meeting the data completeness requirements described in this appendix. The computed 3-year average of the annual fourth-highest daily maximum 8-hour average O3 concentrations shall be reported to three decimal places (the digits to the right of the third decimal place are truncated, consistent with the data handling procedures for the reported data).
2.3 Comparisons with the Primary and Secondary Ozone Standards
(a) The primary and secondary O3 ambient air quality standards are met at an ambient air quality monitoring site when the 3-year average of the annual fourth-highest daily maximum 8-hour average O3 concentration is less than or equal to 0.075 ppm.
(b) This comparison shall be based on three consecutive, complete calendar years of air quality monitoring data. This requirement is met for the 3-year period at a monitoring site if daily maximum 8-hour average concentrations are available for at least 90% of the days within the O3 monitoring season, on average, for the 3-year period, with a minimum data completeness requirement in any one year of at least 75% of the days within the O3 monitoring season. When computing whether the minimum data completeness requirements have been met, meteorological or ambient data may be sufficient to demonstrate that meteorological conditions on missing days were not conducive to concentrations above the level of the standard. Missing days assumed less then the level of the standard are counted for the purpose of meeting the data completeness requirement, subject to the approval of the appropriate Regional Administrator.
(c) Years with concentrations greater than the level of the standard shall be included even if they have less than complete data. Thus, in computing the 3-year average fourth maximum concentration, calendar years with less than 75% data completeness shall be included in the computation if the 3-year average fourth-highest 8-hour concentration is greater than the level of the standard.
(d) Comparisons with the primary and secondary O3 standards are demonstrated by examples 1 and 2 in paragraphs (d)(1) and (d)(2) respectively as follows:
| Year | Percent valid days (within the required monitoring season) | 1st Highest daily max 8-hour Conc. (ppm) | 2nd Highest daily max 8-hour Conc. (ppm) | 3rd Highest daily max 8-hour Conc. (ppm) | 4th Highest daily max 8-hour Conc. (ppm) | 5th Highest daily max 8-hour Conc. (ppm) |
|---|---|---|---|---|---|---|
| 2004 | 100 | 0.092 | 0.090 | 0.085 | 0.079 | 0.078 |
| 2005 | 96 | 0.084 | 0.083 | 0.075 | 0.072 | 0.070 |
| 2006 | 98 | 0.080 | 0.079 | 0.077 | 0.076 | 0.060 |
| Average | 98 | 0.075 |
(1) As shown in Example 1, this monitoring site meets the primary and secondary O3 standards because the 3-year average of the annual fourth-highest daily maximum 8-hour average O3 concentrations (i.e., 0.075666 * * * ppm, truncated to 0.075 ppm) is less than or equal to 0.075 ppm. The data completeness requirement is also met because the average percent of days within the required monitoring season with valid ambient monitoring data is greater than 90%, and no single year has less than 75% data completeness. In Example 1, the individual 8-hour averages used to determine the annual fourth maximum have also been truncated to the third decimal place.
| Year | Percent valid days (within the required monitoring season) | 1st Highest daily max 8-hour Conc. (ppm) | 2nd Highest daily max 8-hour Conc. (ppm) | 3rd Highest daily max 8-hour Conc. (ppm) | 4th Highest daily max 8-hour Conc. (ppm) | 5th Highest daily max 8-hour Conc. (ppm) |
|---|---|---|---|---|---|---|
| 2004 | 96 | 0.105 | 0.103 | 0.103 | 0.103 | 0.102 |
| 2005 | 74 | 0.104 | 0.103 | 0.092 | 0.091 | 0.088 |
| 2006 | 98 | 0.103 | 0.101 | 0.101 | 0.095 | 0.094 |
| Average | 89 | 0.096 |
As shown in Example 2, the primary and secondary O3 standards are not met for this monitoring site because the 3-year average of the fourth-highest daily maximum 8-hour average O3 concentrations (i.e., 0.096333 * * * ppm, truncated to 0.096 ppm) is greater than 0.075 ppm, even though the data capture is less than 75% and the average data capture for the 3 years is less than 90% within the required monitoring season. In Example 2, the individual 8-hour averages used to determine the annual fourth maximum have also been truncated to the third decimal place.
3. Design Values for Primary and Secondary Ambient Air Quality Standards for Ozone
The air quality design value at a monitoring site is defined as that concentration that when reduced to the level of the standard ensures that the site meets the standard. For a concentration-based standard, the air quality design value is simply the standard-related test statistic. Thus, for the primary and secondary standards, the 3-year average annual fourth-highest daily maximum 8-hour average O3 concentration is also the air quality design value for the site.
[73 FR 16511, Mar. 27, 2008]
Appendix Q to Part 50 - Reference Method for the Determination of Lead in Particulate Matter as PM10 Collected From Ambient Air
This Federal Reference Method (FRM) draws heavily from the specific analytical protocols used by the U.S. EPA.
1. Applicability and Principle
1.1 This method provides for the measurement of the lead (Pb) concentration in particulate matter that is 10 micrometers or less (PM10) in ambient air. PM10 is collected on an acceptable (see section 6.1.2) 46.2 mm diameter polytetrafluoroethylene (PTFE) filter for 24 hours using active sampling at local conditions with a low-volume air sampler. The low-volume sampler has an average flow rate of 16.7 liters per minute (Lpm) and total sampled volume of 24 cubic meters (m 3) of air. The analysis of Pb in PM10 is performed on each individual 24-hour sample. Gravimetric mass analysis of PM10c filters is not required for Pb analysis. For the purpose of this method, PM10 is defined as particulate matter having an aerodynamic diameter in the nominal range of 10 micrometers (10 µm) or less.
1.2 For this reference method, PM10 shall be collected with the PM10c federal reference method (FRM) sampler as described in appendix O to Part 50 using the same sample period, measurement procedures, and requirements specified in appendix L of Part 50. The PM10c sampler is also being used for measurement of PM10−2.5 mass by difference and as such, the PM10c sampler must also meet all of the performance requirements specified for PM2.5 in appendix L. The concentration of Pb in the atmosphere is determined in the total volume of air sampled and expressed in micrograms per cubic meter (µg/m 3) at local temperature and pressure conditions.
1.3 The FRM will serve as the basis for approving Federal Equivalent Methods (FEMs) as specified in 40 CFR Part 53 (Reference and Equivalent Methods). This FRM specifically applies to the analysis of Pb in PM10 filters collected with the PM10c sampler. If these filters are analyzed for elements other than Pb, then refer to the guidance provided in the EPA Inorganic Compendium Method IO-3.3 (Reference 1 of section 8) for multi-element analysis.
1.4 The PM10c air sampler draws ambient air at a constant volumetric flow rate into a specially shaped inlet and through an inertial particle size separator, where the suspended particulate matter in the PM10 size range is separated for collection on a PTFE filter over the specified sampling period. The Pb content of the PM10 sample is analyzed by energy-dispersive X-ray fluorescence spectrometry (EDXRF). Energy-dispersive X-ray fluorescence spectrometry provides a means for identification of an element by measurement of its characteristic X-ray emission energy. The method allows for quantification of the element by measuring the intensity of X-rays emitted at the characteristic photon energy and then relating this intensity to the elemental concentration. The number or intensity of X-rays produced at a given energy provides a measure of the amount of the element present by comparisons with calibration standards. The X-rays are detected and the spectral signals are acquired and processed with a personal computer. EDXRF is commonly used as a non-destructive method for quantifying trace elements in PM. A detailed explanation of quantitative X-ray spectrometry is described in references 2, 3 and 4.
1.5 Quality assurance (QA) procedures for the collection of monitoring data are contained in Part 58, appendix A.
2. PM10Pb Measurement Range and Detection Limit. The values given below in section 2.1 and 2.2 are typical of the method capabilities. Absolute values will vary for individual situations depending on the instrument, detector age, and operating conditions used. Data are typically reported in ng/m 3 for ambient air samples; however, for this reference method, data will be reported in µg/m 3 at local temperature and pressure conditions.
2.1 EDXRF Pb Measurement Range. The typical ambient air measurement range is 0.001 to 30 µg Pb/m 3, assuming an upper range calibration standard of about 60 µg Pb per square centimeter (cm 2), a filter deposit area of 11.86 cm 2, and an air volume of 24 m 3. The top range of the EDXRF instrument is much greater than what is stated here. The top measurement range of quantification is defined by the level of the high concentration calibration standard used and can be increased to expand the measurement range as needed.
2.2 Detection Limit (DL). A typical estimate of the one-sigma detection limit (DL) is about 2 ng Pb/cm 2 or 0.001 µg Pb/m 3, assuming a filter size of 46.2 mm (filter deposit area of 11.86 cm 2) and a sample air volume of 24 m 3. The DL is an estimate of the lowest amount of Pb that can be reliably distinguished from a blank filter. The one-sigma detection limit for Pb is calculated as the average overall uncertainty or propagated error for Pb, determined from measurements on a series of blank filters from the filter lot(s) in use. Detection limits must be determined for each filter lot in use. If a new filter lot is used, then a new DL must be determined. The sources of random error which are considered are calibration uncertainty; system stability; peak and background counting statistics; uncertainty in attenuation corrections; and uncertainty in peak overlap corrections, but the dominating source by far is peak and background counting statistics. At a minimum, laboratories are to determine annual estimates of the DL using the guidance provided in Reference 5.
3. Factors Affecting Bias and Precision of Lead Determination by EDXRF
3.1 Filter Deposit. X-ray spectra are subject to distortion if unusually heavy deposits are analyzed. This is the result of internal absorption of both primary and secondary X-rays within the sample; however, this is not an issue for Pb due to the energetic X-rays used to fluoresce Pb and the energetic characteristic X-rays emitted by Pb. The optimum mass filter loading for multi-elemental EDXRF analyis is about 100 µg/cm 2 or 1.2 mg/filter for a 46.2-mm filter. Too little deposit material can also be problematic due to low counting statistics and signal noise. The particle mass deposit should minimally be 15 µg/cm 2. The maximum PM10 filter loading or upper concentration limit of mass expected to be collected by the PM10c sampler is 200 µg/m 3 (Appendix O to Part 50, Section 3.2). This equates to a mass loading of about 400 µg/cm 2 and is the maximum expected loading for PM10c filters. This maximum loading is acceptable for the analysis of Pb and other high-Z elements with very energetic characteristic X-rays. A properly collected sample will have a uniform deposit over the entire collection area. Samples with physical deformities (including a visually non-uniform deposit area) should not be quantitatively analyzed. Tests on the uniformity of particle deposition on PM10C filters showed that the non-uniformity of the filter deposit represents a small fraction of the overall uncertainty in ambient Pb concentration measurement. The analysis beam of the XRF analyzer does not cover the entire filter collection area. The minimum allowable beam size is 10 mm.
3.2 Spectral Interferences and Spectral Overlap. Spectral interference occurs when the entirety of the analyte spectral lines of two species are nearly 100% overlapped. The presence of arsenic (As) is a problematic interference for EDXRF systems which use the Pb Lα line exclusively to quantify the Pb concentration. This is because the Pb Lα line and the As Kα lines severely overlap. The use of multiple Pb lines, including the Lβ and/or the Lγ lines for quantification must be used to reduce the uncertainty in the Pb determination in the presence of As. There can be instances when lines partially overlap the Pb spectral lines, but with the energy resolution of most detectors these overlaps are typically de-convoluted using standard spectral de-convolution software provided by the instrument vendor. An EDXRF protocol for Pb must define which Pb lines are used for quantification and where spectral overlaps occur. A de-convolution protocol must be used to separate all the lines which overlap with Pb.
3.3 Particle Size Effects and Attenuation Correction Factors. X-ray attenuation is dependent on the X-ray energy, mass sample loading, composition, and particle size. In some cases, the excitation and fluorescent X-rays are attenuated as they pass through the sample. In order to relate the measured intensity of the X-rays to the thin-film calibration standards used, the magnitude of any attenuation present must be corrected for. See references 6, 7, and 8 for more discussion on this issue. Essentially no attenuation corrections are necessary for Pb in PM10: Both the incoming excitation X-rays used for analyzing lead and the fluoresced Pb X-rays are sufficiently energetic that for particles in this size range and for normal filter loadings, the Pb X-ray yield is not significantly impacted by attenuation.
4. Precision
4.1 Measurement system precision is assessed according to the procedures set forth in appendix A to part 58. Measurement method precision is assessed from collocated sampling and analysis. The goal for acceptable measurement uncertainty, as precision, is defined as an upper 90 percent confidence limit for the coefficient of variation (CV) of 20 percent.
5. Bias
5.1 Measurement system bias for monitoring data is assessed according to the procedures set forth in appendix A of part 58. The bias is assessed through an audit using spiked filters. The goal for measurement bias is defined as an upper 95 percent confidence limit for the absolute bias of 15 percent.
6. Measurement of PTFE Filters by EDXRF
6.1 Sampling
6.1.1 Low-Volume PM10cSampler. The low-volume PM10c sampler shall be used for PM10 sample collection and operated in accordance with the performance specifications described in part 50, appendix L.
6.1.2 PTFE Filters and Filter Acceptance Testing. The PTFE filters used for PM10c sample collection shall meet the specifications provided in part 50, appendix L. The following requirements are similar to those currently specified for the acceptance of PM2.5 filters that are tested for trace elements by EDXRF. For large filter lots (greater than 500 filters) randomly select 20 filters from a given lot. For small lots (less than 500 filters) a lesser number of filters may be taken. Analyze each blank filter separately and calculate the average lead concentration in ng/cm 2. Ninety percent, or 18 of the 20 filters, must have an average lead concentration that is less than 4.8 ng Pb/cm 2.
6.1.2.1 Filter Blanks. Field blank filters shall be collected along with routine samples. Field blank filters will be collected that are transported to the sampling site and placed in the sampler for the duration of sampling without sampling. Laboratory blank filters from each filter lot used shall be analyzed with each batch of routine sample filters analyzed. Laboratory blank filters are used in background subtraction as discussed below in Section 6.2.4.
6.2 Analysis. The four main categories of random and systematic error encountered in X-ray fluorescence analysis include errors from sample collection, the X-ray source, the counting process, and inter-element effects. These errors are addressed through the calibration process and mathematical corrections in the instrument software. Spectral processing methods are well established and most commercial analyzers have software that can implement the most common approaches (references 9-11) to background subtraction, peak overlap correction, counting and deadtime corrections.
6.2.1 EDXRF Analysis Instrument. An energy-dispersive XRF system is used. Energy-dispersive XRF systems are available from a number of commercial vendors. Examples include Thermo (www.thermo.com), Spectro (http://www.spectro.com), Xenemetrix (http://www.xenemetrix.com) and PANalytical (http://www.panalytical.com). 1 The analysis is performed at room temperature in either vacuum or in a helium atmosphere. The specific details of the corrections and calibration algorithms are typically included in commercial analytical instrument software routines for automated spectral acquisition and processing and vary by manufacturer. It is important for the analyst to understand the correction procedures and algorithms of the particular system used, to ensure that the necessary corrections are applied.
1 These are examples of available systems and is not an all inclusive list. The mention of commercial products does not imply endorsement by the U.S. Environmental Protection Agency.
6.2.2 Thin film standards. Thin film standards are used for calibration because they most closely resemble the layer of particles on a filter. Thin films standards are typically deposited on Nuclepore substrates. The preparation of thin film standards is discussed in reference 8, and 10. The NIST SRM 2783 (Air Particulate on Filter Media) is currently available on polycarbonate filters and contains a certified concentration for Pb. Thin film standards at 15 and 50 µg/cm 2 are commercially available from MicroMatter Inc. (Arlington, WA).
6.2.3 Filter Preparation. Filters used for sample collection are 46.2-mm PTFE filters with a pore size of 2 microns and filter deposit area 11.86 cm 2. Cold storage is not a requirement for filters analyzed for Pb; however, if filters scheduled for XRF analysis were stored cold, they must be allowed to reach room temperature prior to analysis. All filter samples received for analysis are checked for any holes, tears, or a non-uniform deposit which would prevent quantitative analysis. Samples with physical deformities are not quantitatively analyzable. The filters are carefully removed with tweezers from the Petri dish and securely placed into the instrument-specific sampler holder for analysis. Care must be taken to protect filters from contamination prior to analysis. Filters must be kept covered when not being analyzed. No other preparation of filter samples is required.
6.2.4 Calibration. In general, calibration determines each element's sensitivity, i.e., its response in x-ray counts/sec to each µg/cm 2 of a standard and an interference coefficient for each element that causes interference with another one (See section 3.2 above). The sensitivity can be determined by a linear plot of count rate versus concentration (µg/cm 2) in which the slope is the instrument's sensitivity for that element. A more precise way, which requires fewer standards, is to fit sensitivity versus atomic number. Calibration is a complex task in the operation of an XRF system. Two major functions accomplished by calibration are the production of reference spectra which are used for fitting and the determination of the elemental sensitivities. Included in the reference spectra (referred to as “shapes”) are background-subtracted peak shapes of the elements to be analyzed (as well as interfering elements) and spectral backgrounds. Pure element thin film standards are used for the element peak shapes and clean filter blanks from the same lot as routine filter samples are used for the background. The analysis of Pb in PM filter deposits is based on the assumption that the thickness of the deposit is small with respect to the characteristic Pb X-ray transmission thickness. Therefore, the concentration of Pb in a sample is determined by first calibrating the spectrometer with thin film standards to determine the sensitivity factor for Pb and then analyzing the unknown samples under identical excitation conditions as used to determine the calibration. Calibration shall be performed annually or when significant repairs or changes occur (e.g., a change in fluorescers, X-ray tubes, or detector). Calibration establishes the elemental sensitivity factors and the magnitude of interference or overlap coefficients. See reference 7 for more detailed discussion of calibration and analysis of shapes standards for background correction, coarse particle absorption corrections, and spectral overlap.
6.2.4.1 Spectral Peak Fitting. The EPA uses a library of pure element peak shapes (shape standards) to extract the elemental background-free peak areas from an unknown spectrum. It is also possible to fit spectra using peak stripping or analytically defined functions such as modified Gaussian functions. The EPA shape standards are generated from pure, mono-elemental thin film standards. The shape standards are acquired for sufficiently long times to provide a large number of counts in the peaks of interest. It is not necessary for the concentration of the standard to be known. A slight contaminant in the region of interest in a shape standard can have a significant and serious effect on the ability of the least squares fitting algorithm to fit the shapes to the unknown spectrum. It is these elemental peak shapes that are fitted to the peaks in an unknown sample during spectral processing by the analyzer. In addition to this library of elemental shapes there is also a background shape spectrum for the filter type used as discussed below in section 6.2.4.2 of this section.
6.2.4.2 Background Measurement and Correction. A background spectrum generated by the filter itself must be subtracted from the X-ray spectrum prior to extracting peak areas. Background spectra must be obtained for each filter lot used for sample collection. The background shape standards which are used for background fitting are created at the time of calibration. If a new lot of filters is used, new background spectra must be obtained. A minimum of 20 clean blank filters from each filter lot are kept in a sealed container and are used exclusively for background measurement and correction. The spectra acquired on individual blank filters are added together to produce a single spectrum for each of the secondary targets or fluorescers used in the analysis of lead. Individual blank filter spectra which show atypical contamination are excluded from the summed spectra. The summed spectra are fitted to the appropriate background during spectral processing. Background correction is automatically included during spectral processing of each sample.
7. Calculation.
7.1 PM10Pb concentrations. The PM10 Pb concentration in the atmosphere (µg/m 3) is calculated using the following equation:
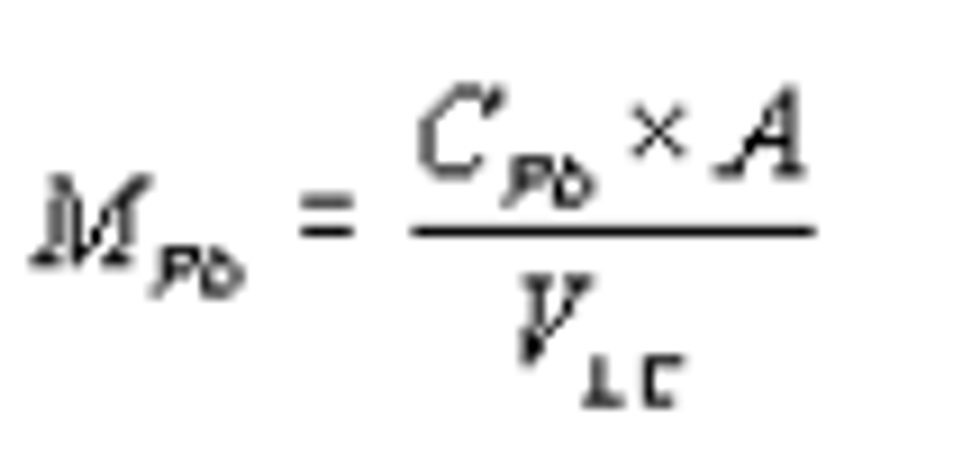
Where,
MPb is the mass per unit volume for lead in µg/m 3;
CPb is the mass per unit area for lead in µg/cm 2 as measured by XRF;
A is the filter deposit area in cm 2;
VLC is the total volume of air sampled by the PM10c sampler in actual volume units measured at local conditions of temperature and pressure, as provided by the sampler in m 3.
7.2 PM10Pb Uncertainty Calculations.
The principal contributors to total uncertainty of XRF values include: field sampling; filter deposit area; XRF calibration; attenuation or loss of the x-ray signals due to the other components of the particulate sample; and determination of the Pb X-ray emission peak area by curve fitting. See reference 12 for a detailed discussion of how uncertainties are similarly calculated for the PM2.5 Chemical Speciation program.
The model for calculating total uncertainty is:
δtot = (δf2 + δa2 + δc2 + δv2) 1/2
Where,
δf = fitting uncertainty (XRF-specific, from 2 to 100 + %)
δa = attenuation uncertainty (XRF-specific, insignificant for Pb)
δc = calibration uncertainty (combined lab uncertainty, assumed as 5%)
δv = volume/deposition size uncertainty (combined field uncertainty, assumed as 5%)
8. References
1. Inorganic Compendium Method IO-3.3; Determination of Metals in Ambient Particulate Matter Using X-Ray Fluorescence (XRF) Spectroscopy; U.S. Environmental Protection Agency, Cincinnati, OH 45268. EPA/625/R-96/010a. June 1999.
2. Jenkins, R., Gould, R.W., and Gedcke, D. Quantitative X-ray Spectrometry: Second Edition. Marcel Dekker, Inc., New York, NY. 1995.
3. Jenkins, R. X-Ray Fluorescence Spectrometry: Second Edition in Chemical Analysis, a Series of Monographs on Analytical Chemistry and Its Applications, Volume 152. Editor J.D.Winefordner; John Wiley & Sons, Inc., New York, NY. 1999.
4. Dzubay, T.G. X-ray Fluorescence Analysis of Environmental Samples, Ann Arbor Science Publishers Inc., 1977.
5. Code of Federal Regulations (CFR) 40, Part 136, Appendix B; Definition and Procedure for the Determination of the Method Detection Limit - Revision 1.1.
6. Drane, E.A, Rickel, D.G., and Courtney, W.J., “Computer Code for Analysis X-Ray Fluorescence Spectra of Airborne Particulate Matter,” in Advances in X-Ray Analysis, J.R. Rhodes, Ed., Plenum Publishing Corporation, New York, NY, p. 23 (1980).
7. Analysis of Energy-Dispersive X-ray Spectra of Ambient Aerosols with Shapes Optimization, Guidance Document; TR-WDE-06-02; prepared under contract EP-D-05-065 for the U.S. Environmental Protection Agency, National Exposure Research Laboratory. March 2006.
8. Billiet, J., Dams, R., and Hoste, J. (1980) Multielement Thin Film Standards for XRF Analysis, X-Ray Spectrometry, 9(4): 206-211.
9. Bonner, N.A.; Bazan, F.; and Camp, D.C. (1973). Elemental analysis of air filter samples using x-ray fluorescence. Report No. UCRL-51388. Prepared for U.S. Atomic Energy Commission, by Univ. of Calif., Lawrence Livermore Laboratory, Livermore, CA.
10. Dzubay, T.G.; Lamothe, P.J.; and Yoshuda, H. (1977). Polymer films as calibration standards for X-ray fluorescence analysis. Adv. X-Ray Anal., 20:411.
11. Giauque, R.D.; Garrett, R.B.; and Goda, L.Y. (1977). Calibration of energy-dispersive X-ray spectrometers for analysis of thin environmental samples. In X-Ray Fluorescence Analysis of Environmental Samples, T.G. Dzubay, Ed., Ann Arbor Science Publishers, Ann Arbor, MI, pp. 153-181.
12. Harmonization of Interlaboratory X-ray Fluorescence Measurement Uncertainties, Detailed Discussion Paper; August 4, 2006; prepared for the Office of Air Quality Planning and Standards under EPA contract 68-D-03-038. http://www.epa.gov/ttn/amtic/files/ambient/pm25/spec/xrfdet.pdf.
[73 FR 67052, Nov. 12, 2008]
Appendix R to Part 50 - Interpretation of the National Ambient Air Quality Standards for Lead
1. General.
(a) This appendix explains the data handling conventions and computations necessary for determining when the primary and secondary national ambient air quality standards (NAAQS) for lead (Pb) specified in §50.16 are met. The NAAQS indicator for Pb is defined as: lead and its compounds, measured as elemental lead in total suspended particulate (Pb-TSP), sampled and analyzed by a Federal reference method (FRM) based on appendix G to this part or by a Federal equivalent method (FEM) designated in accordance with §53.11 of this chapter. Although Pb-TSP is the lead NAAQS indicator, surrogate Pb-TSP concentrations shall also be used for NAAQS comparisons; specifically, valid surrogate Pb-TSP data are concentration data for lead and its compounds, measured as elemental lead, in particles with an aerodynamic size of 10 microns or less (Pb-PM10), sampled and analyzed by an FRM based on appendix Q to this part or by an FEM designated in accordance with §53.11 of this chapter. Surrogate Pb-TSP data (i.e., Pb-PM10 data), however, can only be used to show that the Pb NAAQS were violated (i.e., not met); they can not be used to demonstrate that the Pb NAAQS were met. Pb-PM10 data used as surrogate Pb-TSP data shall be processed at face value; that is, without any transformation or scaling. Data handling and computation procedures to be used in making comparisons between reported and/or surrogate Pb-TSP concentrations and the level of the Pb NAAQS are specified in the following sections.
(b) Whether to exclude, retain, or make adjustments to the data affected by exceptional events, including natural events, is determined by the requirements and process deadlines specified in §§50.1, 50.14, and 51.930 of this chapter.
(c) The terms used in this appendix are defined as follows:
Annual monitoring network plan refers to the plan required by section 58.10 of this chapter.
Creditable samples are samples that are given credit for data completeness. They include valid samples collected on required sampling days and valid “make-up” samples taken for missed or invalidated samples on required sampling days.
Daily values for Pb refer to the 24-hour mean concentrations of Pb (Pb-TSP or Pb-PM10), measured from midnight to midnight (local standard time), that are used in NAAQS computations.
Design value is the site-level metric (i.e., statistic) that is compared to the NAAQS level to determine compliance; the design value for the Pb NAAQS is selected according to the procedures in this appendix from among the valid three-month Pb-TSP and surrogate Pb-TSP (Pb-PM10) arithmetic mean concentration for the 38-month period consisting of the most recent 3-year calendar period plus two previous months (i.e., 36 3-month periods) using the last month of each 3-month period as the period of report.
Extra samples are non-creditable samples. They are daily values that do not occur on scheduled sampling days and that can not be used as “make-up samples” for missed or invalidated scheduled samples. Extra samples are used in mean calculations. For purposes of determining whether a sample must be treated as a make-up sample or an extra sample, Pb-TSP and Pb-PM10 data collected before January 1, 2009 will be treated with an assumed scheduled sampling frequency of every sixth day.
Make-up samples are samples taken to replace missed or invalidated required scheduled samples. Make-ups can be made by either the primary or collocated (same size fraction) instruments; to be considered a valid make-up, the sampling must be conducted with equipment and procedures that meet the requirements for scheduled sampling. Make-up samples are either taken before the next required sampling day or exactly one week after the missed (or voided) sampling day. Make-up samples can not span years; that is, if a scheduled sample for December is missed (or voided), it can not be made up in January. Make-up samples, however, may span months, for example a missed sample on January 31 may be made up on February 1, 2, 3, 4, 5, or 7 (with an assumed sampling frequency of every sixth day). Section 3(e) explains how such month-spanning make-up samples are to be treated for purposes of data completeness and mean calculations. Only two make-up samples are permitted each calendar month; these are counted according to the month in which the miss and not the makeup occurred. For purposes of determining whether a sample must be treated as a make-up sample or an extra sample, Pb-TSP and Pb-PM10 data collected before January 1, 2009 will be treated with an assumed scheduled sampling frequency of every sixth day.
Monthly mean refers to an arithmetic mean, calculated as specified in section 6(a) of this appendix. Monthly means are computed at each monitoring site separately for Pb-TSP and Pb-PM10 (i.e., by site-parameter-year-month).
Parameter refers either to Pb-TSP or to Pb-PM10.
Pollutant Occurrence Code (POC) refers to a numerical code (1, 2, 3, etc.) used to distinguish the data from two or more monitors for the same parameter at a single monitoring site.
Scheduled sampling day means a day on which sampling is scheduled based on the required sampling frequency for the monitoring site, as provided in section 58.12 of this chapter.
Three-month means are arithmetic averages of three consecutive monthly means. Three-month means are computed on a rolling, overlapping basis. Each distinct monthly mean will be included in three different 3-month means; for example, in a given year, a November mean would be included in: (1) The September-October-November 3-month mean, (2) the October-November-December 3-month mean, and (3) the November-December-January(of the following year) 3-month mean. Three-month means are computed separately for each parameter per section 6(a) (and are referred to as 3-month parameter means) and are validated according to the criteria specified in section 4(c). The parameter-specific 3-month means are then prioritized according to section 2(a) to determine a single 3-month site mean.
Year refers to a calendar year.
2. Use of Pb-PM10Data as Surrogate Pb-TSP Data.
(a) As stipulated in section 2.10 of Appendix C to 40 CFR part 58, at some mandatory Pb monitoring locations, monitoring agencies are required to sample for Pb as Pb-TSP, and at other mandatory Pb monitoring sites, monitoring agencies are permitted to monitor for Pb-PM10 in lieu of Pb-TSP. In either situation, valid collocated Pb data for the other parameter may be produced. Additionally, there may be non-required monitoring locations that also produce valid Pb-TSP and/or valid Pb-PM10 data. Pb-TSP data and Pb-PM10 data are always processed separately when computing monthly and 3-month parameter means; monthly and 3-month parameter means are validated according to the criteria stated in section 4 of this appendix. Three-month “site” means, which are the final valid 3-month mean from which a design value is identified, are determined from the one or two available valid 3-month parameter means according to the following prioritization which applies to all Pb monitoring locations.
(i) Whenever a valid 3-month Pb-PM10 mean shows a violation and either is greater than a corresponding (collocated) 3-month Pb-TSP mean or there is no corresponding valid 3-month Pb-TSP mean present, then that 3-month Pb-PM10 mean will be the site-level mean for that (site's) 3-month period.
(ii) Otherwise (i.e., there is no valid violating 3-month Pb-PM10 that exceeds a corresponding 3-month Pb-TSP mean),
(A) If a valid 3-month Pb-TSP mean exists, then it will be the site-level mean for that (site's) 3-month period, or
(B) If a valid 3-month Pb-TSP mean does not exist, then there is no valid 3-month site mean for that period (even if a valid non-violating 3-month Pb-PM10 mean exists).
(b) As noted in section 1(a) of this appendix, FRM/FEM Pb-PM10 data will be processed at face value (i.e., at reported concentrations) without adjustment when computing means and making NAAQS comparisons.
3. Requirements for Data Used for Comparisons With the Pb NAAQS and Data Reporting Considerations.
(a) All valid FRM/FEM Pb-TSP data and all valid FRM/FEM Pb-PM10 data submitted to EPA's Air Quality System (AQS), or otherwise available to EPA, meeting the requirements of §53.11 of this chapter including appendices A, C, and E shall be used in design value calculations. Pb-TSP and Pb-PM10 data representing sample collection periods prior to January 1, 2009 (i.e., “pre-rule” data) will also be considered valid for NAAQS comparisons and related attainment/nonattainment determinations if the sampling and analysis methods that were utilized to collect that data were consistent with previous or newly designated FRMs or FEMs and with either the provisions of §53.11 of this chapter including appendices A, C, and E that were in effect at the time of original sampling or that are in effect at the time of the attainment/nonattainment determination, and if such data are submitted to AQS prior to September 1, 2009.
(b) Pb-TSP and Pb-PM10 measurement data are reported to AQS in units of micrograms per cubic meter (µg/m 3) at local conditions (local temperature and pressure, LC) to three decimal places; any additional digits to the right of the third decimal place are truncated. Pre-rule Pb-TSP and Pb-PM10 concentration data that were reported in standard conditions (standard temperature and standard pressure, STP) will not require a conversion to local conditions but rather, after truncating to three decimal places and processing as stated in this appendix, shall be compared “as is” to the NAAQS (i.e., the LC to STP conversion factor will be assumed to be one). However, if the monitoring agency has retroactively resubmitted Pb-TSP or Pb-PM10 pre-rule data converted from STP to LC based on suitable meteorological data, only the LC data will be used.
(c) At each monitoring location (site), Pb-TSP and Pb-PM10 data are to be processed separately when selecting daily data by day (as specified in section 3(d) of this appendix), when aggregating daily data by month (per section 6(a)), and when forming 3-month means (per section 6(b)). However, when deriving (i.e., identifying) the design value for the 38-month period, 3-month means for the two data types may be considered together; see sections 2(a) and 4(e) of this appendix for details.
(d) Daily values for sites will be selected for a site on a size cut (Pb-TSP or Pb-PM10, i.e., “parameter”) basis; Pb-TSP concentrations and Pb-PM10 concentrations shall not be commingled in these determinations. Site level, parameter-specific daily values will be selected as follows:
(i) The starting dataset for a site-parameter shall consist of the measured daily concentrations recorded from the designated primary FRM/FEM monitor for that parameter. The primary monitor for each parameter shall be designated in the appropriate state or local agency annual Monitoring Network Plan. If no primary monitor is designated, the Administrator will select which monitor to treat as primary. All daily values produced by the primary sampler are considered part of the site-parameter data record (i.e., that site-parameter's set of daily values); this includes all creditable samples and all extra samples. For pre-rule Pb-TSP and Pb-PM10 data, valid data records present in AQS for the monitor with the lowest occurring Pollutant Occurrence Code (POC), as selected on a site-parameter-daily basis, will constitute the site-parameter data record. Where pre-rule Pb-TSP data (or subsequent non-required Pb-TSP or Pb-PM10 data) are reported in “composite” form (i.e., multiple filters for a month of sampling that are analyzed together), the composite concentration will be used as the site-parameter monthly mean concentration if there are no valid daily Pb-TSP data reported for that month with a lower POC.
(ii) Data for the primary monitor for each parameter shall be augmented as much as possible with data from collocated (same parameter) FRM/FEM monitors. If a valid 24-hour measurement is not produced from the primary monitor for a particular day (scheduled or otherwise), but a valid sample is generated by a collocated (same parameter) FRM/FEM instrument, then that collocated value shall be considered part of the site-parameter data record (i.e., that site-parameter's monthly set of daily values). If more than one valid collocated FRM/FEM value is available, the mean of those valid collocated values shall be used as the daily value. Note that this step will not be necessary for pre-rule data given the daily identification presumption for the primary monitor.
(e) All daily values in the composite site-parameter record are used in monthly mean calculations. However, not all daily values are given credit towards data completeness requirements. Only “creditable” samples are given credit for data completeness. Creditable samples include valid samples on scheduled sampling days and valid make-up samples. All other types of daily values are referred to as “extra” samples. Make-up samples taken in the (first week of the) month after the one in which the miss/void occurred will be credited for data capture in the month of the miss/void but will be included in the month actually taken when computing monthly means. For example, if a make-up sample was taken in February to replace a missed sample scheduled for January, the make-up concentration would be included in the February monthly mean but the sample credited in the January data capture rate.
4. Comparisons With the Pb NAAQS.
(a) The Pb NAAQS is met at a monitoring site when the identified design value is valid and less than or equal to 0.15 micrograms per cubic meter (µg/m 3). A Pb design value that meets the NAAQS (i.e., 0.15 µg/m 3 or less), is considered valid if it encompasses 36 consecutive valid 3-month site means (specifically for a 3-year calendar period and the two previous months). For sites that begin monitoring Pb after this rule is effective but before January 15, 2010 (or January 15, 2011), a 2010-2012 (or 2011-2013) Pb design value that meets the NAAQS will be considered valid if it encompasses at least 34 consecutive valid 3-month means (specifically encompassing only the 3-year calendar period). See 4(c) of this appendix for the description of a valid 3-month mean and section 6(d) for the definition of the design value.
(b) The Pb NAAQS is violated at a monitoring site when the identified design value is valid and is greater than 0.15 µg/m 3, no matter whether determined from Pb-TSP or Pb-PM10 data. A Pb design value greater than 0.15 µg/m 3 is valid no matter how many valid 3-month means in the 3-year period it encompasses; that is, a violating design value is valid even if it (i.e., the highest 3-month mean) is the only valid 3-month mean in the 3-year timeframe. Further, a site does not have to monitor for three full calendar years in order to have a valid violating design value; a site could monitor just three months and still produce a valid (violating) design value.
(c)(i) A 3-month parameter mean is considered valid (i.e., meets data completeness requirements) if the average of the data capture rate of the three constituent monthly means (i.e., the 3-month data capture rate) is greater than or equal to 75 percent. Monthly data capture rates (expressed as a percentage) are specifically calculated as the number of creditable samples for the month (including any make-up samples taken the subsequent month for missed samples in the month in question, and excluding any make-up samples taken in the month in question for missed samples in the previous month) divided by the number of scheduled samples for the month, the result then multiplied by 100 but not rounded. The 3-month data capture rate is the sum of the three corresponding unrounded monthly data capture rates divided by three and the result rounded to the nearest integer (zero decimal places). As noted in section 3(c), Pb-TSP and Pb-PM10 daily values are processed separately when calculating monthly means and data capture rates; a Pb-TSP value cannot be used as a make-up for a missing Pb-PM10 value or vice versa. For purposes of assessing data capture, Pb-TSP and Pb-PM10 data collected before January 1, 2009 will be treated with an assumed scheduled sampling frequency of every sixth day.
(ii) A 3-month parameter mean that does not have at least 75 percent data capture and thus is not considered valid under 4(c)(i) shall be considered valid (and complete) if it passes either of the two following “data substitution” tests, one such test for validating an above NAAQS-level (i.e., violating) 3-month Pb-TSP or Pb-PM10 mean (using actual “low” reported values from the same site at about the same time of the year (i.e., in the same month) looking across three or four years), and the second test for validating a below-NAAQS level 3-month Pb-TSP mean (using actual “high” values reported for the same site at about the same time of the year (i.e., in the same month) looking across three or four years). Note that both tests are merely diagnostic in nature intending to confirm that there is a very high likelihood if not certainty that the original mean (the one with less than 75% data capture) reflects the true over/under NAAQS-level status for that 3-month period; the result of one of these data substitution tests (i.e., a “test mean”, as defined in section 4(c)(ii)(A) or 4(c)(ii)(B)) is not considered the actual 3-month parameter mean and shall not be used in the determination of design values. For both types of data substitution, substitution is permitted only if there are available data points from which to identify the high or low 3-year month-specific values, specifically if there are at least 10 data points total from at least two of the three (or four for November and December) possible year-months. Data substitution may only use data of the same parameter type.
(A) The “above NAAQS level” test is as follows: Data substitution will be done in each month of the 3-month period that has less than 75 percent data capture; monthly capture rates are temporarily rounded to integers (zero decimals) for this evaluation. If by substituting the lowest reported daily value for that month (year non-specific; e.g., for January) over the 38-month design value period in question for missing scheduled data in the deficient months (substituting only enough to meet the 75 percent data capture minimum), the computation yields a recalculated test 3-month parameter mean concentration above the level of the standard, then the 3-month period is deemed to have passed the diagnostic test and the level of the standard is deemed to have been exceeded in that 3-month period. As noted in section 4(c)(ii), in such a case, the 3-month parameter mean of the data actually reported, not the recalculated (“test”) result including the low values, shall be used to determine the design value.
(B) The “below NAAQS level” test is as follows: Data substitution will be performed for each month of the 3-month period that has less than 75 percent but at least 50 percent data capture; if any month has less than 50% data capture then the 3-month mean can not utilize this substitution test. Also, incomplete 3-month Pb-PM10 means can not utilize this test. A 3-month Pb-TSP mean with less than 75% data capture shall still be considered valid (and complete) if, by substituting the highest reported daily value, month-specific, over the 3-year design value period in question, for all missing scheduled data in the deficient months (i.e., bringing the data capture rate up to 100%), the computation yields a recalculated 3-month parameter mean concentration equal or less than the level of the standard (0.15 µg/m 3), then the 3-month mean is deemed to have passed the diagnostic test and the level of the standard is deemed not to have been exceeded in that 3-month period (for that parameter). As noted in section 4(c)(ii), in such a case, the 3-month parameter mean of the data actually reported, not the recalculated (“test”) result including the high values, shall be used to determine the design value.
(d) Months that do not meet the completeness criteria stated in 4(c)(i) or 4(c)(ii), and design values that do not meet the completeness criteria stated in 4(a) or 4(b), may also be considered valid (and complete) with the approval of, or at the initiative of, the Administrator, who may consider factors such as monitoring site closures/moves, monitoring diligence, the consistency and levels of the valid concentration measurements that are available, and nearby concentrations in determining whether to use such data.
(e) The site-level design value for a 38-month period (three calendar years plus two previous months) is identified from the available (between one and 36) valid 3-month site means. In a situation where there are valid 3-month means for both parameters (Pb-TSP and Pb-PM10), the mean originating from the reported Pb-TSP data will be the one deemed the site-level monthly mean and used in design value identifications unless the Pb-PM10 mean shows a violation of the NAAQS and exceeds the Pb-TSP mean; see section 2(a) for details. A monitoring site will have only one site-level 3-month mean per 3-month period; however, the set of site-level 3-month means considered for design value identification (i.e., one to 36 site-level 3-month means) can be a combination of Pb-TSP and Pb-PM10 data.
(f) The procedures for calculating monthly means and 3-month means, and identifying Pb design values are given in section 6 of this appendix.
5. Rounding Conventions.
(a) Monthly means and monthly data capture rates are not rounded.
(b) Three-month means shall be rounded to the nearest hundredth µg/m 3 (0.xx). Decimals 0.xx5 and greater are rounded up, and any decimal lower than 0.xx5 is rounded down. E.g., a 3-month mean of 0.104925 rounds to 0.10 and a 3-month mean of .10500 rounds to 0.11. Three-month data capture rates, expressed as a percent, are round to zero decimal places.
(c) Because a Pb design value is simply a (highest) 3-month mean and because the NAAQS level is stated to two decimal places, no additional rounding beyond what is specified for 3-month means is required before a design value is compared to the NAAQS.
6. Procedures and Equations for the Pb NAAQS.
(a)(i) A monthly mean value for Pb-TSP (or Pb-PM10) is determined by averaging the daily values of a calendar month using equation 1 of this appendix, unless the Administrator chooses to exercise his discretion to use the alternate approach described in 6(a)(ii).
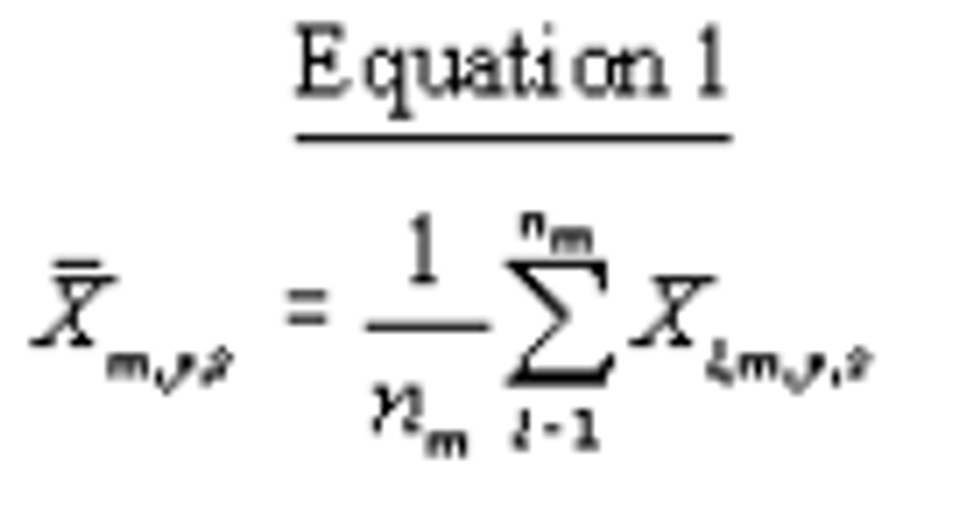
Where:
Xm,y,s = the mean for month m of the year y for sites; and
nm = the number of daily values in the month (creditable plus extra samples); and
Xi,m,y,s = the i th value in month m for year y for site s.
(a)(ii) The Administrator may at his discretion use the following alternate approach to calculating the monthly mean concentration if the number of extra sampling days during a month is greater than the number of successfully completed scheduled and make-up sample days in that month. In exercising his discretion, the Administrator will consider whether the approach specified in 6(a)(i) might in the Administrator's judgment result in an unrepresentative value for the monthly mean concentration. This provision is to protect the integrity of the monthly and 3-month mean concentration values in situations in which, by intention or otherwise, extra sampling days are concentrated in a period during which ambient concentrations are particularly high or low. The alternate approach is to average all extra and make-up samples (in the given month) taken after each scheduled sampling day (“Day X”) and before the next scheduled sampling day (e.g., “Day X + 6”, in the case of one-in-six sampling) with the sample taken on Day X (assuming valid data was obtained on the scheduled sampling day), and then averaging these averages to calculate the monthly mean. This approach has the effect of giving approximately equal weight to periods during a month that have equal number of days, regardless of how many samples were actually obtained during the periods, thus mitigating the potential for the monthly mean to be distorted. The first day of scheduled sampling typically will not fall on the first day of the calendar month, and there may be make-up and/or extra samples (in that same calendar month) preceding the first scheduled day of the month. These samples will not be shifted into the previous month's mean concentration, but rather will stay associated with their actual calendar month as follows. Any extra and make-up samples taken in a month before the first scheduled sampling day of the month will be associated with and averaged with the last scheduled sampling day of that same month.
(b) Three-month parameter means are determined by averaging three consecutive monthly means of the same parameter using Equation 2 of this appendix.
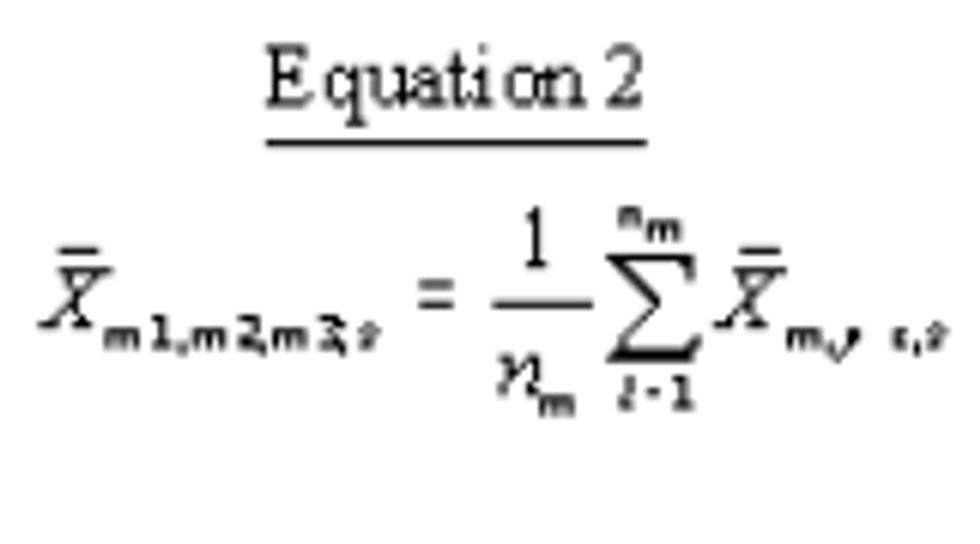
Where:
X͞ m1, m2, m3; s = the 3-month parameter mean for months m1, m2, and m3 for site s; and
nm = the number of monthly means available to be averaged (typically 3, sometimes 1 or 2 if one or two months have no valid daily values); and
Xm, y: z, s = The mean for month m of the year y (or z) for site s.
(c) Three-month site means are determined from available 3-month parameter means according to the hierarchy established in 2(a) of this appendix.
(d) The site-level Pb design value is the highest valid 3-month site-level mean over the most recent 38-month period (i.e., the most recent 3-year calendar period plus two previous months). Section 4(a) of this appendix explains when the identified design value is itself considered valid for purposes of determining that the NAAQS is met or violated at a site.
[73 FR 67054, Nov. 12, 2008]
Appendix S to Part 50 - Interpretation of the Primary National Ambient Air Quality Standards for Oxides of Nitrogen (Nitrogen Dioxide)
1. General
(a) This appendix explains the data handling conventions and computations necessary for determining when the primary national ambient air quality standards for oxides of nitrogen as measured by nitrogen dioxide (“NO2 NAAQS”) specified in 50.11 are met. Nitrogen dioxide (NO2) is measured in the ambient air by a Federal reference method (FRM) based on appendix F to this part or by a Federal equivalent method (FEM) designated in accordance with §53.11 of this chapter. Data handling and computation procedures to be used in making comparisons between reported NO2 concentrations and the levels of the NO2 NAAQS are specified in the following sections.
(b) Whether to exclude, retain, or make adjustments to the data affected by exceptional events, including natural events, is determined by the requirements and process deadlines specified in 50.1, 50.14 and 51.930 of this chapter.
(c) The terms used in this appendix are defined as follows:
Annual mean refers to the annual average of all of the 1-hour concentration values as defined in section 5.1 of this appendix.
Daily maximum 1-hour values for NO2 refers to the maximum 1-hour NO2 concentration values measured from midnight to midnight (local standard time) that are used in NAAQS computations.
Design values are the metrics (i.e., statistics) that are compared to the NAAQS levels to determine compliance, calculated as specified in section 5 of this appendix. The design values for the primary NAAQS are:
(1) The annual mean value for a monitoring site for one year (referred to as the “annual primary standard design value”).
(2) The 3-year average of annual 98th percentile daily maximum 1-hour values for a monitoring site (referred to as the “1-hour primary standard design value”).
98th percentile daily maximum 1-hour value is the value below which nominally 98 percent of all daily maximum 1-hour concentration values fall, using the ranking and selection method specified in section 5.2 of this appendix.
Quarter refers to a calendar quarter.
Year refers to a calendar year.
2. Requirements for Data Used for Comparisons With the NO2 NAAQS and Data Reporting Considerations
(a) All valid FRM/FEM NO2 hourly data required to be submitted to EPA's Air Quality System (AQS), or otherwise available to EPA, meeting the requirements of §53.11 of this chapter including appendices A, C, and E shall be used in design value calculations. Multi-hour average concentration values collected by wet chemistry methods shall not be used.
(b) When two or more NO2 monitors are operated at a site, the State may in advance designate one of them as the primary monitor. If the State has not made this designation, the Administrator will make the designation, either in advance or retrospectively. Design values will be developed using only the data from the primary monitor, if this results in a valid design value. If data from the primary monitor do not allow the development of a valid design value, data solely from the other monitor(s) will be used in turn to develop a valid design value, if this results in a valid design value. If there are three or more monitors, the order for such comparison of the other monitors will be determined by the Administrator. The Administrator may combine data from different monitors in different years for the purpose of developing a valid 1-hour primary standard design value, if a valid design value cannot be developed solely with the data from a single monitor. However, data from two or more monitors in the same year at the same site will not be combined in an attempt to meet data completeness requirements, except if one monitor has physically replaced another instrument permanently, in which case the two instruments will be considered to be the same monitor, or if the State has switched the designation of the primary monitor from one instrument to another during the year.
(c) Hourly NO2 measurement data shall be reported to AQS in units of parts per billion (ppb), to at most one place after the decimal, with additional digits to the right being truncated with no further rounding.
3. Comparisons With the NO2 NAAQS
3.1 The Annual Primary NO2 NAAQS
(a) The annual primary NO2 NAAQS is met at a site when the valid annual primary standard design value is less than or equal to 53 parts per billion (ppb).
(b) An annual primary standard design value is valid when at least 75 percent of the hours in the year are reported.
(c) An annual primary standard design value based on data that do not meet the completeness criteria stated in section 3.1(b) may also be considered valid with the approval of, or at the initiative of, the Administrator, who may consider factors such as monitoring site closures/moves, monitoring diligence, the consistency and levels of the valid concentration measurements that are available, and nearby concentrations in determining whether to use such data.
(d) The procedures for calculating the annual primary standard design values are given in section 5.1 of this appendix.
3.2 The 1-hour Primary NO2 NAAQS
(a) The 1-hour primary NO2 NAAQS is met at a site when the valid 1-hour primary standard design value is less than or equal to 100 parts per billion (ppb).
(b) An NO2 1-hour primary standard design value is valid if it encompasses three consecutive calendar years of complete data. A year meets data completeness requirements when all 4 quarters are complete. A quarter is complete when at least 75 percent of the sampling days for each quarter have complete data. A sampling day has complete data if 75 percent of the hourly concentration values, including State-flagged data affected by exceptional events which have been approved for exclusion by the Administrator, are reported.
(c) In the case of one, two, or three years that do not meet the completeness requirements of section 3.2(b) of this appendix and thus would normally not be useable for the calculation of a valid 3-year 1-hour primary standard design value, the 3-year 1-hour primary standard design value shall nevertheless be considered valid if one of the following conditions is true.
(i) At least 75 percent of the days in each quarter of each of three consecutive years have at least one reported hourly value, and the design value calculated according to the procedures specified in section 5.2 is above the level of the primary 1-hour standard.
(ii)(A) A 1-hour primary standard design value that is below the level of the NAAQS can be validated if the substitution test in section 3.2(c)(ii)(B) results in a “test design value” that is below the level of the NAAQS. The test substitutes actual “high” reported daily maximum 1-hour values from the same site at about the same time of the year (specifically, in the same calendar quarter) for unknown values that were not successfully measured. Note that the test is merely diagnostic in nature, intended to confirm that there is a very high likelihood that the original design value (the one with less than 75 percent data capture of hours by day and of days by quarter) reflects the true under-NAAQS-level status for that 3-year period; the result of this data substitution test (the “test design value”, as defined in section 3.2(c)(ii)(B)) is not considered the actual design value. For this test, substitution is permitted only if there are at least 200 days across the three matching quarters of the three years under consideration (which is about 75 percent of all possible daily values in those three quarters) for which 75 percent of the hours in the day, including State-flagged data affected by exceptional events which have been approved for exclusion by the Administrator, have reported concentrations. However, maximum 1-hour values from days with less than 75 percent of the hours reported shall also be considered in identifying the high value to be used for substitution.
(B) The substitution test is as follows: Data substitution will be performed in all quarter periods that have less than 75 percent data capture but at least 50 percent data capture, including State-flagged data affected by exceptional events which have been approved for exclusion by the Administrator; if any quarter has less than 50 percent data capture then this substitution test cannot be used. Identify for each quarter (e.g., January-March) the highest reported daily maximum 1-hour value for that quarter, excluding State-flagged data affected by exceptional events which have been approved for exclusion by the Administrator, looking across those three months of all three years under consideration. All daily maximum 1-hour values from all days in the quarter period shall be considered when identifying this highest value, including days with less than 75 percent data capture. If after substituting the highest non-excluded reported daily maximum 1-hour value for a quarter for as much of the missing daily data in the matching deficient quarter(s) as is needed to make them 100 percent complete, the procedure in section 5.2 yields a recalculated 3-year 1-hour standard “test design value” below the level of the standard, then the 1-hour primary standard design value is deemed to have passed the diagnostic test and is valid, and the level of the standard is deemed to have been met in that 3-year period. As noted in section 3.2(c)(i), in such a case, the 3-year design value based on the data actually reported, not the “test design value”, shall be used as the valid design value.
(iii)(A) A 1-hour primary standard design value that is above the level of the NAAQS can be validated if the substitution test in section 3.2(c)(iii)(B) results in a “test design value” that is above the level of the NAAQS. The test substitutes actual “low” reported daily maximum 1-hour values from the same site at about the same time of the year (specifically, in the same three months of the calendar) for unknown values that were not successfully measured. Note that the test is merely diagnostic in nature, intended to confirm that there is a very high likelihood that the original design value (the one with less than 75 percent data capture of hours by day and of days by quarter) reflects the true above-NAAQS-level status for that 3-year period; the result of this data substitution test (the “test design value”, as defined in section 3.2(c)(iii)(B)) is not considered the actual design value. For this test, substitution is permitted only if there are a minimum number of available daily data points from which to identify the low quarter-specific daily maximum 1-hour values, specifically if there are at least 200 days across the three matching quarters of the three years under consideration (which is about 75 percent of all possible daily values in those three quarters) for which 75 percent of the hours in the day have reported concentrations. Only days with at least 75 percent of the hours reported shall be considered in identifying the low value to be used for substitution.
(B) The substitution test is as follows: Data substitution will be performed in all quarter periods that have less than 75 percent data capture. Identify for each quarter (e.g., January-March) the lowest reported daily maximum 1-hour value for that quarter, looking across those three months of all three years under consideration. All daily maximum 1-hour values from all days with at least 75 percent capture in the quarter period shall be considered when identifying this lowest value. If after substituting the lowest reported daily maximum 1-hour value for a quarter for as much of the missing daily data in the matching deficient quarter(s) as is needed to make them 75 percent complete, the procedure in section 5.2 yields a recalculated 3-year 1-hour standard “test design value” above the level of the standard, then the 1-hour primary standard design value is deemed to have passed the diagnostic test and is valid, and the level of the standard is deemed to have been exceeded in that 3-year period. As noted in section 3.2(c)(i), in such a case, the 3-year design value based on the data actually reported, not the “test design value”, shall be used as the valid design value.
(d) A 1-hour primary standard design value based on data that do not meet the completeness criteria stated in 3.2(b) and also do not satisfy section 3.2(c), may also be considered valid with the approval of, or at the initiative of, the Administrator, who may consider factors such as monitoring site closures/moves, monitoring diligence, the consistency and levels of the valid concentration measurements that are available, and nearby concentrations in determining whether to use such data.
(e) The procedures for calculating the 1-hour primary standard design values are given in section 5.2 of this appendix.
4. Rounding Conventions
4.1 Rounding Conventions for the Annual Primary NO2 NAAQS
(a) Hourly NO2 measurement data shall be reported to AQS in units of parts per billion (ppb), to at most one place after the decimal, with additional digits to the right being truncated with no further rounding.
(b) The annual primary standard design value is calculated pursuant to section 5.1 and then rounded to the nearest whole number or 1 ppb (decimals 0.5 and greater are rounded up to the nearest whole number, and any decimal lower than 0.5 is rounded down to the nearest whole number).
4.2 Rounding Conventions for the 1-hour Primary NO2 NAAQS
(a) Hourly NO2 measurement data shall be reported to AQS in units of parts per billion (ppb), to at most one place after the decimal, with additional digits to the right being truncated with no further rounding.
(b) Daily maximum 1-hour values are not rounded.
(c) The 1-hour primary standard design value is calculated pursuant to section 5.2 and then rounded to the nearest whole number or 1 ppb (decimals 0.5 and greater are rounded up to the nearest whole number, and any decimal lower than 0.5 is rounded down to the nearest whole number).
5. Calculation Procedures for the Primary NO2 NAAQS
5.1 Procedures for the Annual Primary NO2 NAAQS
(a) When the data for a site and year meet the data completeness requirements in section 3.1(b) of this appendix, or if the Administrator exercises the discretionary authority in section 3.1(c), the annual mean is simply the arithmetic average of all of the reported 1-hour values.
(b) The annual primary standard design value for a site is the valid annual mean rounded according to the conventions in section 4.1.
5.2 Calculation Procedures for the 1-hour Primary NO2 NAAQS
(a) Procedure for identifying annual 98th percentile values. When the data for a particular site and year meet the data completeness requirements in section 3.2(b), or if one of the conditions of section 3.2(c) is met, or if the Administrator exercises the discretionary authority in section 3.2(d), identification of annual 98th percentile value is accomplished as follows.
(i) The annual 98th percentile value for a year is the higher of the two values resulting from the following two procedures.
(1) Procedure 1.
(A) For the year, determine the number of days with at least 75 percent of the hourly values reported including State-flagged data affected by exceptional events which have been approved for exclusion by the Administrator.
(B) For the year, from only the days with at least 75 percent of the hourly values reported, select from each day the maximum hourly value excluding State-flagged data affected by exceptional events which have been approved for exclusion by the Administrator.
(C) Sort all these daily maximum hourly values from a particular site and year by descending value. (For example: (x[1], x[2], x[3], * * *, x[n]). In this case, x[1] is the largest number and x[n] is the smallest value.) The 98th percentile is determined from this sorted series of daily values which is ordered from the highest to the lowest number. Using the left column of Table 1, determine the appropriate range (i.e., row) for the annual number of days with valid data for year y (cny) as determined from step (A). The corresponding “n” value in the right column identifies the rank of the annual 98th percentile value in the descending sorted list of daily site values for year y. Thus, P0.98, y = the nth largest value.
(2) Procedure 2.
(A) For the year, determine the number of days with at least one hourly value reported including State-flagged data affected by exceptional events which have been approved for exclusion by the Administrator.
(B) For the year, from all the days with at least one hourly value reported, select from each day the maximum hourly value excluding State-flagged data affected by exceptional events which have been approved for exclusion by the Administrator.
(C) Sort all these daily maximum values from a particular site and year by descending value. (For example: (x[1], x[2], x[3], * * *, x[n]). In this case, x[1] is the largest number and x[n] is the smallest value.) The 98th percentile is determined from this sorted series of daily values which is ordered from the highest to the lowest number. Using the left column of Table 1, determine the appropriate range (i.e., row) for the annual number of days with valid data for year y (cny) as determined from step (A). The corresponding “n” value in the right column identifies the rank of the annual 98th percentile value in the descending sorted list of daily site values for year y. Thus, P0.98, y = the nth largest value.
(b) The 1-hour primary standard design value for a site is mean of the three annual 98th percentile values, rounded according to the conventions in section 4.
| Annual number
of days with valid data for year “y” (cny) | P0.98, y is the
nth maximum value of the year, where n is the listed number |
|---|---|
| 1-50 | 1 |
| 51-100 | 2 |
| 101-150 | 3 |
| 151-200 | 4 |
| 201-250 | 5 |
| 251-300 | 6 |
| 301-350 | 7 |
| 351-366 | 8 |
[75 FR 6532, Feb. 9, 2010]
Appendix T to Part 50 - Interpretation of the Primary National Ambient Air Quality Standards for Oxides of Sulfur (Sulfur Dioxide)
1. General
(a) This appendix explains the data handling conventions and computations necessary for determining when the primary national ambient air quality standards for Oxides of Sulfur as measured by Sulfur Dioxide (“SO2 NAAQS”) specified in §50.17 are met at an ambient air quality monitoring site. Sulfur Dioxide (SO2) is measured in the ambient air by a Federal reference method (FRM) based on appendix A or A-1 to this part or by a Federal equivalent method (FEM) designated in accordance with §53.11 of this chapter. Data handling and computation procedures to be used in making comparisons between reported SO2 concentrations and the levels of the SO2 NAAQS are specified in the following sections.
(b) Decisions to exclude, retain, or make adjustments to the data affected by exceptional events, including natural events, are made according to the requirements and process deadlines specified in §§50.1, 50.14 and 51.930 of this chapter.
(c) The terms used in this appendix are defined as follows:
Daily maximum 1-hour values for SO2 refers to the maximum 1-hour SO2 concentration values measured from midnight to midnight (local standard time) that are used in NAAQS computations.
Design values are the metrics (i.e., statistics) that are compared to the NAAQS levels to determine compliance, calculated as specified in section 5 of this appendix. The design value for the primary 1-hour NAAQS is the 3-year average of annual 99th percentile daily maximum 1-hour values for a monitoring site (referred to as the “1-hour primary standard design value”).
99th percentile daily maximum 1-hour value is the value below which nominally 99 percent of all daily maximum 1-hour concentration values fall, using the ranking and selection method specified in section 5 of this appendix.
Pollutant Occurrence Code (POC) refers to a numerical code (1, 2, 3, etc.) used to distinguish the data from two or more monitors for the same parameter at a single monitoring site.
Quarter refers to a calendar quarter.
Year refers to a calendar year.
2. Requirements for Data Used for Comparisons With the SO2 NAAQS and Data Reporting Considerations
(a) All valid FRM/FEM SO2 hourly data required to be submitted to EPA's Air Quality System (AQS), or otherwise available to EPA, meeting the requirements of §53.11 of this chapter including appendices A, C, and E shall be used in design value calculations. Multi-hour average concentration values collected by wet chemistry methods shall not be used.
(b) Data from two or more monitors from the same year at the same site reported to EPA under distinct Pollutant Occurrence Codes shall not be combined in an attempt to meet data completeness requirements. The Administrator will combine annual 99th percentile daily maximum concentration values from different monitors in different years, selected as described here, for the purpose of developing a valid 1-hour primary standard design value. If more than one of the monitors meets the completeness requirement for all four quarters of a year, the steps specified in section 5(a) of this appendix shall be applied to the data from the monitor with the highest average of the four quarterly completeness values to derive a valid annual 99th percentile daily maximum concentration. If no monitor is complete for all four quarters in a year, the steps specified in section 3(c) and 5(a) of this appendix shall be applied to the data from the monitor with the highest average of the four quarterly completeness values in an attempt to derive a valid annual 99th percentile daily maximum concentration. This paragraph does not prohibit a monitoring agency from making a local designation of one physical monitor as the primary monitor for a Pollutant Occurrence Code and substituting the 1-hour data from a second physical monitor whenever a valid concentration value is not obtained from the primary monitor; if a monitoring agency substitutes data in this manner, each substituted value must be accompanied by an AQS qualifier code indicating that substitution with a value from a second physical monitor has taken place.
(c) Hourly SO2 measurement data shall be reported to AQS in units of parts per billion (ppb), to at most one place after the decimal, with additional digits to the right being truncated with no further rounding.
3. Comparisons With the 1-Hour Primary SO2 NAAQS
(a) The 1-hour primary SO2 NAAQS is met at an ambient air quality monitoring site when the valid 1-hour primary standard design value is less than or equal to 75 parts per billion (ppb).
(b) An SO2 1-hour primary standard design value is valid if it encompasses three consecutive calendar years of complete data. A year meets data completeness requirements when all 4 quarters are complete. A quarter is complete when at least 75 percent of the sampling days for each quarter have complete data. A sampling day has complete data if 75 percent of the hourly concentration values, including State-flagged data affected by exceptional events which have been approved for exclusion by the Administrator, are reported.
(c) In the case of one, two, or three years that do not meet the completeness requirements of section 3(b) of this appendix and thus would normally not be useable for the calculation of a valid 3-year 1-hour primary standard design value, the 3-year 1-hour primary standard design value shall nevertheless be considered valid if one of the following conditions is true.
(i) At least 75 percent of the days in each quarter of each of three consecutive years have at least one reported hourly value, and the design value calculated according to the procedures specified in section 5 is above the level of the primary 1-hour standard.
(ii)(A) A 1-hour primary standard design value that is equal to or below the level of the NAAQS can be validated if the substitution test in section 3(c)(ii)(B) results in a “test design value” that is below the level of the NAAQS. The test substitutes actual “high” reported daily maximum 1-hour values from the same site at about the same time of the year (specifically, in the same calendar quarter) for unknown values that were not successfully measured. Note that the test is merely diagnostic in nature, intended to confirm that there is a very high likelihood that the original design value (the one with less than 75 percent data capture of hours by day and of days by quarter) reflects the true under-NAAQS-level status for that 3-year period; the result of this data substitution test (the “test design value”, as defined in section 3(c)(ii)(B)) is not considered the actual design value. For this test, substitution is permitted only if there are at least 200 days across the three matching quarters of the three years under consideration (which is about 75 percent of all possible daily values in those three quarters) for which 75 percent of the hours in the day, including State-flagged data affected by exceptional events which have been approved for exclusion by the Administrator, have reported concentrations. However, maximum 1-hour values from days with less than 75 percent of the hours reported shall also be considered in identifying the high value to be used for substitution.
(B) The substitution test is as follows: Data substitution will be performed in all quarter periods that have less than 75 percent data capture but at least 50 percent data capture, including State-flagged data affected by exceptional events which have been approved for exclusion by the Administrator; if any quarter has less than 50 percent data capture then this substitution test cannot be used. Identify for each quarter (e.g., January-March) the highest reported daily maximum 1-hour value for that quarter, excluding State-flagged data affected by exceptional events which have been approved for exclusion by the Administrator, looking across those three months of all three years under consideration. All daily maximum 1-hour values from all days in the quarter period shall be considered when identifying this highest value, including days with less than 75 percent data capture. If after substituting the highest reported daily maximum 1-hour value for a quarter for as much of the missing daily data in the matching deficient quarter(s) as is needed to make them 100 percent complete, the procedure in section 5 yields a recalculated 3-year 1-hour standard “test design value” less than or equal to the level of the standard, then the 1-hour primary standard design value is deemed to have passed the diagnostic test and is valid, and the level of the standard is deemed to have been met in that 3-year period. As noted in section 3(c)(i), in such a case, the 3-year design value based on the data actually reported, not the “test design value”, shall be used as the valid design value.
(iii)(A) A 1-hour primary standard design value that is above the level of the NAAQS can be validated if the substitution test in section 3(c)(iii)(B) results in a “test design value” that is above the level of the NAAQS. The test substitutes actual “low” reported daily maximum 1-hour values from the same site at about the same time of the year (specifically, in the same three months of the calendar) for unknown hourly values that were not successfully measured. Note that the test is merely diagnostic in nature, intended to confirm that there is a very high likelihood that the original design value (the one with less than 75 percent data capture of hours by day and of days by quarter) reflects the true above-NAAQS-level status for that 3-year period; the result of this data substitution test (the “test design value”, as defined in section 3(c)(iii)(B)) is not considered the actual design value. For this test, substitution is permitted only if there are a minimum number of available daily data points from which to identify the low quarter-specific daily maximum 1-hour values, specifically if there are at least 200 days across the three matching quarters of the three years under consideration (which is about 75 percent of all possible daily values in those three quarters) for which 75 percent of the hours in the day have reported concentrations. Only days with at least 75 percent of the hours reported shall be considered in identifying the low value to be used for substitution.
(B) The substitution test is as follows: Data substitution will be performed in all quarter periods that have less than 75 percent data capture. Identify for each quarter (e.g., January-March) the lowest reported daily maximum 1-hour value for that quarter, looking across those three months of all three years under consideration. All daily maximum 1-hour values from all days with at least 75 percent capture in the quarter period shall be considered when identifying this lowest value. If after substituting the lowest reported daily maximum 1-hour value for a quarter for as much of the missing daily data in the matching deficient quarter(s) as is needed to make them 75 percent complete, the procedure in section 5 yields a recalculated 3-year 1-hour standard “test design value” above the level of the standard, then the 1-hour primary standard design value is deemed to have passed the diagnostic test and is valid, and the level of the standard is deemed to have been exceeded in that 3-year period. As noted in section 3(c)(i), in such a case, the 3-year design value based on the data actually reported, not the “test design value”, shall be used as the valid design value.
(d) A 1-hour primary standard design value based on data that do not meet the completeness criteria stated in 3(b) and also do not satisfy section 3(c), may also be considered valid with the approval of, or at the initiative of, the Administrator, who may consider factors such as monitoring site closures/moves, monitoring diligence, the consistency and levels of the valid concentration measurements that are available, and nearby concentrations in determining whether to use such data.
(e) The procedures for calculating the 1-hour primary standard design values are given in section 5 of this appendix.
4. Rounding Conventions for the 1-Hour Primary SO2 NAAQS
(a) Hourly SO2 measurement data shall be reported to AQS in units of parts per billion (ppb), to at most one place after the decimal, with additional digits to the right being truncated with no further rounding.
(b) Daily maximum 1-hour values and therefore the annual 99th percentile of those daily values are not rounded.
(c) The 1-hour primary standard design value is calculated pursuant to section 5 and then rounded to the nearest whole number or 1 ppb (decimals 0.5 and greater are rounded up to the nearest whole number, and any decimal lower than 0.5 is rounded down to the nearest whole number).
5. Calculation Procedures for the 1-Hour Primary SO2 NAAQS
(a) Procedure for identifying annual 99th percentile values. When the data for a particular ambient air quality monitoring site and year meet the data completeness requirements in section 3(b), or if one of the conditions of section 3(c) is met, or if the Administrator exercises the discretionary authority in section 3(d), identification of annual 99th percentile value is accomplished as follows.
(i) The annual 99th percentile value for a year is the higher of the two values resulting from the following two procedures.
(1) Procedure 1. For the year, determine the number of days with at least 75 percent of the hourly values reported.
(A) For the year, determine the number of days with at least 75 percent of the hourly values reported including State-flagged data affected by exceptional events which have been approved for exclusion by the Administrator.
(B) For the year, from only the days with at least 75 percent of the hourly values reported, select from each day the maximum hourly value excluding State-flagged data affected by exceptional events which have been approved for exclusion by the Administrator.
(C) Sort all these daily maximum hourly values from a particular site and year by descending value. (For example: (x[1], x[2], x[3], * * *, x[n]). In this case, x[1] is the largest number and x[n] is the smallest value.) The 99th percentile is determined from this sorted series of daily values which is ordered from the highest to the lowest number. Using the left column of Table 1, determine the appropriate range (i.e., row) for the annual number of days with valid data for year y (cny). The corresponding “n” value in the right column identifies the rank of the annual 99th percentile value in the descending sorted list of daily site values for year y. Thus, P0.99, y = the nth largest value.
(2) Procedure 2. For the year, determine the number of days with at least one hourly value reported.
(A) For the year, determine the number of days with at least one hourly value reported including State-flagged data affected by exceptional events which have been approved for exclusion by the Administrator.
(B) For the year, from all the days with at least one hourly value reported, select from each day the maximum hourly value excluding State-flagged data affected by exceptional events which have been approved for exclusion by the Administrator.
(C) Sort all these daily maximum values from a particular site and year by descending value. (For example: (x[1], x[2], x[3], * * *, x[n]). In this case, x[1] is the largest number and x[n] is the smallest value.) The 99th percentile is determined from this sorted series of daily values which is ordered from the highest to the lowest number. Using the left column of Table 1, determine the appropriate range (i.e., row) for the annual number of days with valid data for year y (cny). The corresponding “n” value in the right column identifies the rank of the annual 99th percentile value in the descending sorted list of daily site values for year y. Thus, P0.99,y = the nth largest value.
(b) The 1-hour primary standard design value for an ambient air quality monitoring site is mean of the three annual 99th percentile values, rounded according to the conventions in section 4.
| Annual number of days with valid data for year “y” (cny) | P0.99,y is the nth
maximum value of the year, where n is the listed number |
|---|---|
| 1-100 | 1 |
| 101-200 | 2 |
| 201-300 | 3 |
| 301-366 | 4 |
[75 FR 35595, June 23, 2010]
Appendix U to Part 50 - Interpretation of the Primary and Secondary National Ambient Air Quality Standards for Ozone
1. General
(a) This appendix explains the data handling conventions and computations necessary for determining whether the primary and secondary national ambient air quality standards (NAAQS) for ozone (O3) specified in §50.19 are met at an ambient O3 air quality monitoring site. Data reporting, data handling, and computation procedures to be used in making comparisons between reported O3 concentrations and the levels of the O3 NAAQS are specified in the following sections.
(b) Whether to exclude or retain the data affected by exceptional events is determined by the requirements under §§50.1, 50.14 and 51.930.
(c) The terms used in this appendix are defined as follows:
8-hour average refers to the moving average of eight consecutive hourly O3 concentrations measured at a site, as explained in section 3 of this appendix.
Annual fourth-highest daily maximum refers to the fourth highest value measured at a site during a year.
Collocated monitors refers to the instance of two or more O3 monitors operating at the same physical location.
Daily maximum 8-hour average O3concentration refers to the maximum calculated 8-hour average value measured at a site on a particular day, as explained in section 3 of this appendix.
Design value refers to the metric (i.e., statistic) that is used to compare ambient O3 concentration data measured at a site to the NAAQS in order to determine compliance, as explained in section 4 of this appendix.
Minimum data completeness requirements refer to the amount of data that a site is required to collect in order to make a valid determination that the site is meeting the NAAQS.
Monitor refers to a physical instrument used to measure ambient O3 concentrations.
O3monitoring season refers to the span of time within a year when individual states are required to measure ambient O3 concentrations, as listed in Appendix D to §53.11 of this chapter.
Site refers to an ambient O3 air quality monitoring site.
Site data record refers to the set of hourly O3 concentration data collected at a site for use in comparisons with the NAAQS.
Year refers to calendar year.
2. Selection of Data for use in Comparisons With the Primary and Secondary Ozone NAAQS
(a) All valid hourly O3 concentration data collected using a federal reference method specified in Appendix D to this part, or an equivalent method designated in accordance with §53.11 of this chapter, meeting all applicable requirements in §53.11 of this chapter, and submitted to EPA's Air Quality System (AQS) database or otherwise available to EPA, shall be used in design value calculations.
(b) All design value calculations shall be implemented on a site-level basis. If data are reported to EPA from collocated monitors, those data shall be combined into a single site data record as follows:
(i) The monitoring agency shall designate one monitor as the primary monitor for the site.
(ii) Hourly O3 concentration data from a secondary monitor shall be substituted into the site data record whenever a valid hourly O3 concentration is not obtained from the primary monitor. In the event that hourly O3 concentration data are available for more than one secondary monitor, the hourly concentration values from the secondary monitors shall be averaged and substituted into the site data record.
(c) In certain circumstances, including but not limited to site closures or relocations, data from two nearby sites may be combined into a single site data record for the purpose of calculating a valid design value. The appropriate Regional Administrator may approve such combinations after taking into consideration factors such as distance between sites, spatial and temporal patterns in air quality, local emissions and meteorology, jurisdictional boundaries, and terrain features.
3. Data Reporting and Data Handling Conventions
(a) Hourly average O3 concentrations shall be reported in parts per million (ppm) to the third decimal place, with additional digits to the right of the third decimal place truncated. Each hour shall be identified using local standard time (LST).
(b) Moving 8-hour averages shall be computed from the hourly O3 concentration data for each hour of the year and shall be stored in the first, or start, hour of the 8-hour period. An 8-hour average shall be considered valid if at least 6 of the hourly concentrations for the 8-hour period are available. In the event that only 6 or 7 hourly concentrations are available, the 8-hour average shall be computed on the basis of the hours available, using 6 or 7, respectively, as the divisor. In addition, in the event that 5 or fewer hourly concentrations are available, the 8-hour average shall be considered valid if, after substituting zero for the missing hourly concentrations, the resulting 8-hour average is greater than the level of the NAAQS, or equivalently, if the sum of the available hourly concentrations is greater than 0.567 ppm. The 8-hour averages shall be reported to three decimal places, with additional digits to the right of the third decimal place truncated. Hourly O3 concentrations that have been approved under §50.14 as having been affected by exceptional events shall be counted as missing or unavailable in the calculation of 8-hour averages.
(c) The daily maximum 8-hour average O3 concentration for a given day is the highest of the 17 consecutive 8-hour averages beginning with the 8-hour period from 7:00 a.m. to 3:00 p.m. and ending with the 8-hour period from 11:00 p.m. to 7:00 a.m. the following day (i.e., the 8-hour averages for 7:00 a.m. to 11:00 p.m.). Daily maximum 8-hour average O3 concentrations shall be determined for each day with ambient O3 monitoring data, including days outside the O3 monitoring season if those data are available.
(d) A daily maximum 8-hour average O3 concentration shall be considered valid if valid 8-hour averages are available for at least 13 of the 17 consecutive 8-hour periods starting from 7:00 a.m. to 11:00 p.m. In addition, in the event that fewer than 13 valid 8-hour averages are available, a daily maximum 8-hour average O3 concentration shall also be considered valid if it is greater than the level of the NAAQS. Hourly O3 concentrations that have been approved under §50.14 as having been affected by exceptional events shall be included when determining whether these criteria have been met.
(e) The primary and secondary O3 design value statistic is the annual fourth-highest daily maximum 8-hour O3 concentration, averaged over three years, expressed in ppm. The fourth-highest daily maximum 8-hour O3 concentration for each year shall be determined based only on days meeting the validity criteria in 3(d). The 3-year average shall be computed using the three most recent, consecutive years of ambient O3 monitoring data. Design values shall be reported in ppm to three decimal places, with additional digits to the right of the third decimal place truncated.
4. Comparisons With the Primary and Secondary Ozone NAAQS
(a) The primary and secondary national ambient air quality standards for O3 are met at an ambient air quality monitoring site when the 3-year average of the annual fourth-highest daily maximum 8-hour average O3 concentration (i.e., the design value) is less than or equal to 0.070 ppm.
(b) A design value greater than the level of the NAAQS is always considered to be valid. A design value less than or equal to the level of the NAAQS must meet minimum data completeness requirements in order to be considered valid. These requirements are met for a 3-year period at a site if valid daily maximum 8-hour average O3 concentrations are available for at least 90% of the days within the O3 monitoring season, on average, for the 3-year period, with a minimum of at least 75% of the days within the O3 monitoring season in any one year.
(c) When computing whether the minimum data completeness requirements have been met, meteorological or ambient data may be sufficient to demonstrate that meteorological conditions on missing days were not conducive to concentrations above the level of the NAAQS. Missing days assumed less than the level of the NAAQS are counted for the purpose of meeting the minimum data completeness requirements, subject to the approval of the appropriate Regional Administrator.
(d) Comparisons with the primary and secondary O3 NAAQS are demonstrated by examples 1 and 2 as follows:
| Year | Percent valid days within O3 monitoring season (Data
completeness) | 1st highest daily max
8-hour O3 (ppm) | 2nd highest daily max
8-hour O3 (ppm) | 3rd highest daily max
8-hour O3 (ppm) | 4th highest daily max
8-hour O3 (ppm) | 5th highest daily max
8-hour O3 (ppm) |
|---|---|---|---|---|---|---|
| 2014 | 100 | 0.082 | 0.080 | 0.075 | 0.069 | 0.068 |
| 2015 | 96 | 0.074 | 0.073 | 0.065 | 0.062 | 0.060 |
| 2016 | 98 | 0.070 | 0.069 | 0.067 | 0.066 | 0.060 |
| Average | 98 | 0.065 |
As shown in Example 1, this site meets the primary and secondary O3 NAAQS because the 3-year average of the annual fourth-highest daily maximum 8-hour average O3 concentrations (i.e., 0.065666 ppm, truncated to 0.065 ppm) is less than or equal to 0.070 ppm. The minimum data completeness requirements are also met (i.e., design value is considered valid) because the average percent of days within the O3 monitoring season with valid ambient monitoring data is greater than 90%, and no single year has less than 75% data completeness.
| Year | Percent valid days within O3 monitoring season (Data completeness) | 1st highest daily max
8-hour O3 (ppm) | 2nd highest daily max
8-hour O3 (ppm) | 3rd highest daily max
8-hour O3 (ppm) | 4th highest daily max
8-hour O3 (ppm) | 5th highest daily max
8-hour O3 (ppm) |
|---|---|---|---|---|---|---|
| 2014 | 96 | 0.085 | 0.080 | 0.079 | 0.074 | 0.072 |
| 2015 | 74 | 0.084 | 0.083 | 0.072 | 0.071 | 0.068 |
| 2016 | 98 | 0.083 | 0.081 | 0.081 | 0.075 | 0.074 |
| Average | 89 | 0.073 |
As shown in Example 2, this site fails to meet the primary and secondary O3 NAAQS because the 3-year average of the annual fourth-highest daily maximum 8-hour average O3 concentrations (i.e., 0.073333 ppm, truncated to 0.073 ppm) is greater than 0.070 ppm, even though the annual data completeness is less than 75% in one year and the 3-year average data completeness is less than 90% (i.e., design value would not otherwise be considered valid).
[80 FR 65458, Oct. 26, 2015]
Source: 36 FR 22384, Nov. 25, 1971, unless otherwise noted.
['Air Programs']
['Chlorofluorocarbons', 'Criteria Air Pollutants', 'Air Quality']
UPGRADE TO CONTINUE READING
ezExplanations
Results Aided by AI