Compliance Just Got Easier: Stay ahead of regulatory changes with instant notifications on updates that matter.
['Water Programs']
['Water Analysis', 'Water Quality']
05/11/2022

Copyright 2026 J. J. Keller & Associate, Inc. For re-use options please contact copyright@jjkeller.com or call 800-558-5011.
1.0 Scope and Application
1.1 Inductively coupled plasma-atomic emission spectrometry (ICP-AES) is used to determine metals and some nonmetals in solution. This method is a consolidation of existing methods for water, wastewater, and solid wastes. 1-4 (For analysis of petroleum products see References 5 and 6, Section 16.0). This method is applicable to the following analytes:
| Analyte | Chemical abstract services registry number (CASRN) |
| Aluminum (Al) | 7429-90-5 |
| Antimony (Sb) | 7440-36-0 |
| Arsenic (As) | 7440-38-2 |
| Barium (Ba) | 7440-39-3 |
| Beryllium (Be) | 7440-41-7 |
| Boron (B) | 7440-42-8 |
| Cadmium (Cd) | 7440-43-9 |
| Calcium (Ca) | 7440-70-2 |
| Ceriuma (Cr) | 7440-45-1 |
| Chromium (Cr) | 7440-47-3 |
| Cobalt (Co) | 7440-48-4 |
| Copper (Cu) | 7440-50-8 |
| Iron (Fe) | 7439-89-6 |
| Lead (Pb) | 7439-92-1 |
| Lithium (Li) | 7439-93-2 |
| Magnesium (Mg) | 7439-95-4 |
| Manganese (Mn) | 7439-96-5 |
| Mercury (Hg) | 7439-97-6 |
| Molybdenum (Mo) | 7439-98-7 |
| Nickel (Ni) | 7440-02-0 |
| Phosphorus (P) | 7723-14-0 |
| Potassium (K) | 7440-09-7 |
| Selenium (Se) | 7782-49-2 |
| Silicab(Si02) | 7631-86-9 |
| Silver (Ag) | 7440-22-4 |
| Sodium (Na) | 7440-23-5 |
| Strontium (Sr) | 7440-24-6 |
| Thallium (Tl) | 7440-28-0 |
| Tin (Sn) | 7440-31-5 |
| Titanium (Ti) | 7440-32-6 |
| Vanadium (V) | 7440-62-2 |
| Zinc (Zn) | 7440-66-6 |
aCerium has been included as method analyte for correction of potential interelement spectral interference.
bThis method is not suitable for the determination of silica in solids.
1.2 For reference where this method is approved for use in compliance monitoring programs [e.g., Clean Water Act (NPDES) or Safe Drinking Water Act (SDWA)] consult both the appropriate sections of the Code of Federal Regulation (40 CFR Part 136 Table 1B for NPDES, and Part 141 §141.23 for drinking water), and the latest Federal Register announcements.
1.3 ICP-AES can be used to determine dissolved analytes in aqueous samples after suitable filtration and acid preservation. To reduce potential interferences, dissolved solids should be <0.2% (w/v) (Section 4.2).
1.4 With the exception of silver, where this method is approved for the determination of certain metal and metalloid contaminants in drinking water, samples may be analyzed directly by pneumatic nebulization without acid digestion if the sample has been properly preserved with acid and has turbidity of <1 NTU at the time of analysis. This total recoverable determination procedure is referred to as “direct analysis”. However, in the determination of some primary drinking water metal contaminants, preconcentration of the sample may be required prior to analysis in order to meet drinking water acceptance performance criteria (Sections 11.2.2 through 11.2.7).
1.5 For the determination of total recoverable analytes in aqueous and solid samples a digestion/extraction is required prior to analysis when the elements are not in solution (e.g., soils, sludges, sediments and aqueous samples that may contain particulate and suspended solids). Aqueous samples containing suspended or particulate material 1% (w/v) should be extracted as a solid type sample.
1.6 When determining boron and silica in aqueous samples, only plastic, PTFE or quartz labware should be used from time of sample collection to completion of analysis. For accurate determination of boron in solid samples only quartz or PTFE beakers should be used during acid extraction with immediate transfer of an extract aliquot to a plastic centrifuge tube following dilution of the extract to volume. When possible, borosilicate glass should be avoided to prevent contamination of these analytes.
1.7 Silver is only slightly soluble in the presence of chloride unless there is a sufficient chloride concentration to form the soluble chloride complex. Therefore, low recoveries of silver may occur in samples, fortified sample matrices and even fortified blanks if determined as a dissolved analyte or by "direct analysis" where the sample has not been processed using the total recoverable mixed acid digestion. For this reason it is recommended that samples be digested prior to the determination of silver. The total recoverable sample digestion procedure given in this method is suitable for the determination of silver in aqueous samples containing concentrations up to 0.1 mg/L. For the analysis of wastewater samples containing higher concentrations of silver, succeeding smaller volume, well mixed aliquots should be prepared until the analysis solution contains <0.1 mg/L silver. The extraction of solid samples containing concentrations of silver >50 mg/kg should be treated in a similar manner. Also, the extraction of tin from solid samples should be prepared again using aliquots <1 g when determined sample concentrations exceed 1%.
1.8 The total recoverable sample digestion procedure given in this method will solubilize and hold in solution only minimal concentrations of barium in the presence of free sulfate. For the analysis of barium in samples having varying and unknown concentrations of sulfate, analysis should be completed as soon as possible after sample preparation.
1.9 The total recoverable sample digestion procedure given in this method is not suitable for the determination of volatile organo-mercury compounds. However, if digestion is not required (turbidity <1 NTU), the combined concentrations of inorganic and organo-mercury in solution can be determined by "direct analysis" pneumatic nebulization provided the sample solution is adjusted to contain the same mixed acid (HNO 3 HCl) matrix as the total recoverable calibration standards and blank solutions.
1.10 Detection limits and linear ranges for the elements will vary with the wavelength selected, the spectrometer, and the matrices. Table 1 provides estimated instrument detection limits for the listed wavelengths. 7 However, actual method detection limits and linear working ranges will be dependent on the sample matrix, instrumentation, and selected operating conditions.
1.11 Users of the method data should state the data-quality objectives prior to analysis. Users of the method must document and have on file the required initial demonstration performance data described in Section 9.2 prior to using the method for analysis.
2.0 Summary of Method
2.1 An aliquot of a well mixed, homogeneous aqueous or solid sample is accurately weighed or measured for sample processing. For total recoverable analysis of a solid or an aqueous sample containing undissolved material, analytes are first solubilized by gentle refluxing with nitric and hydrochloric acids. After cooling, the sample is made up to volume, is mixed and centrifuged or allowed to settle overnight prior to analysis. For the determination of dissolved analytes in a filtered aqueous sample aliquot, or for the “direct analysis” total recoverable determination of analytes in drinking water where sample turbidity is <1 NTU, the sample is made ready for analysis by the appropriate addition of nitric acid, and then diluted to a predetermined volume and mixed before analysis.
2.2 The analysis described in this method involves multielemental determinations by ICP-AES using sequential or simultaneous instruments. The instruments measure characteristic atomic-line emission spectra by optical spectrometry. Samples are nebulized and the resulting aerosol is transported to the plasma torch. Element specific emission spectra are produced by a radio-frequency inductively coupled plasma. The spectra are dispersed by a grating spectrometer, and the intensities of the line spectra are monitored at specific wavelengths by a photosensitive device. Photocurrents from the photosensitive device are processed and controlled by a computer system. A background correction technique is required to compensate for variable background contribution to the determination of the analytes. Background must be measured adjacent to the analyte wavelength during analysis. Various interferences must be considered and addressed appropriately as discussed in Sections 4.0, 7.0, 9.0, 10.0, and 11.0.
3.0 Definitions
3.1 Calibration Blank—A volume of reagent water acidified with the same acid matrix as in the calibration standards. The calibration blank is a zero standard and is used to calibrate the ICP instrument (Section 7.10.1).
3.2 Calibration Standard (CAL)—A solution prepared from the dilution of stock standard solutions. The CAL solutions are used to calibrate the instrument response with respect to analyte concentration (Section 7.9).
3.3 Dissolved Analyte—The concentration of analyte in an aqueous sample that will pass through a 0.45 μm membrane filter assembly prior to sample acidification (Section 11.1).
3.4 Field Reagent Blank (FRB)—An aliquot of reagent water or other blank matrix that is placed in a sample container in the laboratory and treated as a sample in all respects, including shipment to the sampling site, exposure to the sampling site conditions, storage, preservation, and all analytical procedures. The purpose of the FRB is to determine if method analytes or other interferences are present in the field environment (Section 8.5).
3.5 Instrument Detection Limit (IDL)—The concentration equivalent to the analyte signal which is equal to three times the standard deviation of a series of 10 replicate measurements of the calibration blank signal at the same wavelength (Table 1.).
3.6 Instrument Performance Check (IPC) Solution—A solution of method analytes, used to evaluate the performance of the instrument system with respect to a defined set of method criteria (Sections 7.11 and 9.3.4).
3.7 Internal Standard—Pure analyte(s) added to a sample, extract, or standard solution in known amount(s) and used to measure the relative responses of other method analytes that are components of the same sample or solution. The internal standard must be an analyte that is not a sample component (Section 11.5).
3.8 Laboratory Duplicates (LD1 and LD2)—Two aliquots of the same sample taken in the laboratory and analyzed separately with identical procedures. Analyses of LD1 and LD2 indicate precision associated with laboratory procedures, but not with sample collection, preservation, or storage procedures.
3.9 Laboratory Fortified Blank (LFB)—An aliquot of LRB to which known quantities of the method analytes are added in the laboratory. The LFB is analyzed exactly like a sample, and its purpose is to determine whether the methodology is in control and whether the laboratory is capable of making accurate and precise measurements (Sections 7.10.3 and 9.3.2).
3.10 Laboratory Fortified Sample Matrix (LFM)—An aliquot of an environmental sample to which known quantities of the method analytes are added in the laboratory. The LFM is analyzed exactly like a sample, and its purpose is to determine whether the sample matrix contributes bias to the analytical results. The background concentrations of the analytes in the sample matrix must be determined in a separate aliquot and the measured values in the LFM corrected for background concentrations (Section 9.4).
3.11 Laboratory Reagent Blank (LRB)—An aliquot of reagent water or other blank matrices that are treated exactly as a sample including exposure to all glassware, equipment, solvents, reagents, and internal standards that are used with other samples. The LRB is used to determine if method analytes or other interferences are present in the laboratory environment, reagents, or apparatus (Sections 7.10.2 and 9.3.1).
3.12 Linear Dynamic Range (LDR)—The concentration range over which the instrument response to an analyte is linear (Section 9.2.2).
3.13 Method Detection Limit (MDL)—The minimum concentration of an analyte that can be identified, measured, and reported with 99% confidence that the analyte concentration is greater than zero (Section 9.2.4 and Table 4.).
3.14 Plasma Solution—A solution that is used to determine the optimum height above the work coil for viewing the plasma (Sections 7.15 and 10.2.3).
3.15 Quality Control Sample (QCS)—A solution of method analytes of known concentrations which is used to fortify an aliquot of LRB or sample matrix. The QCS is obtained from a source external to the laboratory and different from the source of calibration standards. It is used to check either laboratory or instrument performance (Sections 7.12 and 9.2.3).
3.16 Solid Sample—For the purpose of this method, a sample taken from material classified as soil, sediment or sludge.
3.17 Spectral Interference Check (SIC) Solution—A solution of selected method analytes of higher concentrations which is used to evaluate the procedural routine for correcting known interelement spectral interferences with respect to a defined set of method criteria (Sections 7.13, 7.14 and 9.3.5).
3.18 Standard Addition—The addition of a known amount of analyte to the sample in order to determine the relative response of the detector to an analyte within the sample matrix. The relative response is then used to assess either an operative matrix effect or the sample analyte concentration (Sections 9.5.1 and 11.5).
3.19 Stock Standard Solution—A concentrated solution containing one or more method analytes prepared in the laboratory using assayed reference materials or purchased from a reputable commercial source (Section 7.8).
3.20 Total Recoverable Analyte—The concentration of analyte determined either by "direct analysis" of an unfiltered acid preserved drinking water sample with turbidity of <1 NTU (Section 11.2.1), or by analysis of the solution extract of a solid sample or an unfiltered aqueous sample following digestion by refluxing with hot dilute mineral acid(s) as specified in the method (Sections 11.2 and 11.3).
3.21 Water Sample—For the purpose of this method, a sample taken from one of the following sources: drinking, surface, ground, storm runoff, industrial or domestic wastewater.
4.0 Interferences
4.1 Spectral interferences are caused by background emission from continuous or recombination phenomena, stray light from the line emission of high concentration elements, overlap of a spectral line from another element, or unresolved overlap of molecular band spectra.
4.1.1 Background emission and stray light can usually be compensated for by subtracting the background emission determined by measurement(s) adjacent to the analyte wavelength peak. Spectral scans of samples or single element solutions in the analyte regions may indicate not only when alternate wavelengths are desirable because of severe spectral interference, but also will show whether the most appropriate estimate of the background emission is provided by an interpolation from measurements on both sides of the wavelength peak or by the measured emission on one side or the other. The location(s) selected for the measurement of background intensity will be determined by the complexity of the spectrum adjacent to the wavelength peak. The location(s) used for routine measurement must be free of off-line spectral interference (interelement or molecular) or adequately corrected to reflect the same change in background intensity as occurs at the wavelength peak.
4.1.2 Spectral overlaps may be avoided by using an alternate wavelength or can be compensated for by equations that correct for interelement contributions, which involves measuring the interfering elements. Some potential on-line spectral interferences observed for the recommended wavelengths are given in Table 2. When operative and uncorrected, these interferences will produce false-positive determinations and be reported as analyte concentrations. The interferences listed are only those that occur between method analytes. Only interferences of a direct overlap nature that were observed with a single instrument having a working resolution of 0.035 nm are listed. More extensive information on interferant effects at various wavelengths and resolutions is available in Boumans' Tables.8Users may apply interelement correction factors determined on their instruments within tested concentration ranges to compensate (off-line or on-line) for the effects of interfering elements.
4.1.3 When interelement corrections are applied, there is a need to verify their accuracy by analyzing spectral interference check solutions as described in Section 7.13. Interelement corrections will vary for the same emission line among instruments because of differences in resolution, as determined by the grating plus the entrance and exit slit widths, and by the order of dispersion. Interelement corrections will also vary depending upon the choice of background correction points. Selecting a background correction point where an interfering emission line may appear should be avoided when practical. Interelement corrections that constitute a major portion of an emission signal may not yield accurate data. Users should not forget that some samples may contain uncommon elements that could contribute spectral interferences. 7,8
4.1.4 The interference effects must be evaluated for each individual instrument whether configured as a sequential or simultaneous instrument. For each instrument, intensities will vary not only with optical resolution but also with operating conditions (such as power, viewing height and argon flow rate). When using the recommended wavelengths given in Table 1, the analyst is required to determine and document for each wavelength the effect from the known interferences given in Table 2, and to utilize a computer routine for their automatic correction on all analyses. To determine the appropriate location for off-line background correction, the user must scan the area on either side adjacent to the wavelength and record the apparent emission intensity from all other method analytes. This spectral information must be documented and kept on file. The location selected for background correction must be either free of off-line interelement spectral interference or a computer routine must be used for their automatic correction on all determinations. If a wavelength other than the recommended wavelength is used, the user must determine and document both the on-line and off-line spectral interference effect from all method analytes and provide for their automatic correction on all analyses. Tests to determine the spectral interference must be done using analyte concentrations that will adequately describe the interference. Normally, 100 mg/L single element solutions are sufficient, however, for analytes such as iron that may be found at high concentration a more appropriate test would be to use a concentration near the upper LDR limit. See Section 10.4 for required spectral interference test criteria.
4.1.5 When interelement corrections are not used, either on-going SIC solutions (Section 7.14) must be analyzed to verify the absence of interelement spectral interference or a computer software routine must be employed for comparing the determinative data to limits files for notifying the analyst when an interfering element is detected in the sample at a concentration that will produce either an apparent false positive concentration, greater than the analyte IDL, or false negative analyte concentration, less than the 99% lower control limit of the calibration blank. When the interference accounts for 10% or more of the analyte concentration, either an alternate wavelength free of interference or another approved test procedure must be used to complete the analysis. For example, the copper peak at 213.853 nm could be mistaken for the zinc peak at 213.856 nm in solutions with high copper and low zinc concentrations. For this example, a spectral scan in the 213.8 nm region would not reveal the misidentification because a single peak near the zinc location would be observed. The possibility of this misidentification of copper for the zinc peak at 213.856 nm can be identified by measuring the copper at another emission line, e.g ., 324.754 nm. Users should be aware that, depending upon the instrumental resolution, alternate wavelengths with adequate sensitivity and freedom from interference may not be available for all matrices. In these circumstances the analyte must be determined using another approved test procedure.
4.2 Physical interferences are effects associated with the sample nebulization and transport processes. Changes in viscosity and surface tension can cause significant inaccuracies, especially in samples containing high dissolved solids or high acid concentrations. If physical interferences are present, they must be reduced by such means as a high-solids nebulizer, diluting the sample, using a peristaltic pump, or using an appropriate internal standard element. Another problem that can occur with high dissolved solids is salt buildup at the tip of the nebulizer, which affects aerosol flow rate and causes instrumental drift. This problem can be controlled by a high-solids nebulizer, wetting the argon prior to nebulization, using a tip washer, or diluting the sample. Also, it has been reported that better control of the argon flow rates, especially for the nebulizer, improves instrument stability and precision; this is accomplished with the use of mass flow controllers.
4.3 Chemical interferences include molecular-compound formation, ionization effects, and solute-vaporization effects. Normally, these effects are not significant with the ICP-AES technique. If observed, they can be minimized by careful selection of operating conditions (such as incident power and observation height), by buffering of the sample, by matrix matching, and by standard-addition procedures. Chemical interferences are highly dependent on matrix type and the specific analyte element.
4.4 Memory interferences result when analytes in a previous sample contribute to the signals measured in a new sample. Memory effects can result from sample deposition on the uptake tubing to the nebulizer, and from the buildup of sample material in the plasma torch and spray chamber. The site where these effects occur is dependent on the element and can be minimized by flushing the system with a rinse blank between samples (Section 7.10.4). The possibility of memory interferences should be recognized within an analytical run and suitable rinse times should be used to reduce them. The rinse times necessary for a particular element must be estimated prior to analysis. This may be achieved by aspirating a standard containing elements corresponding to either their LDR or a concentration ten times those usually encountered. The aspiration time should be the same as a normal sample analysis period, followed by analysis of the rinse blank at designated intervals. The length of time required to reduce analyte signals to within a factor of two of the method detection limit, should be noted. Until the required rinse time is established, this method requires a rinse period of at least 60 seconds between samples and standards. If a memory interference is suspected, the sample must be re-analyzed after a long rinse period.
5.0 Safety
5.1 The toxicity or carcinogenicity of each reagent used in this method have not been fully established. Each chemical should be regarded as a potential health hazard and exposure to these compounds should be as low as reasonably achievable. Each laboratory is responsible for maintaining a current awareness file of OSHA regulations regarding the safe handling of the chemicals specified in this method. 9-12 A reference file of material data handling sheets should also be made available to all personnel involved in the chemical analysis. Specifically, concentrated nitric and hydrochloric acids present various hazards and are moderately toxic and extremely irritating to skin and mucus membranes. Use these reagents in a fume hood whenever possible and if eye or skin contact occurs, flush with large volumes of water. Always wear safety glasses or a shield for eye protection, protective clothing and observe proper mixing when working with these reagents.
5.2 The acidification of samples containing reactive materials may result in the release of toxic gases, such as cyanides or sulfides. Acidification of samples should be done in a fume hood.
5.3 All personnel handling environmental samples known to contain or to have been in contact with human waste should be immunized against known disease causative agents.
5.4 The inductively coupled plasma should only be viewed with proper eye protection from the ultraviolet emissions.
5.5 It is the responsibility of the user of this method to comply with relevant disposal and waste regulations. For guidance see Sections 14.0 and 15.0.
6.0 Equipment and Supplies
6.1 Inductively coupled plasma emission spectrometer:
6.1.1 Computer-controlled emission spectrometer with background-correction capability.
6.1.2 Radio-frequency generator compliant with FCC regulations.
6.1.3 Argon gas supply—High purity grade (99.99%). When analyses are conducted frequently, liquid argon is more economical and requires less frequent replacement of tanks than compressed argon in conventional cylinders.
6.1.4 A variable speed peristaltic pump is required to deliver both standard and sample solutions to the nebulizer.
6.1.5 (Optional) Mass flow controllers to regulate the argon flow rates, especially the aerosol transport gas, are highly recommended. Their use will provide more exacting control of reproducible plasma conditions.
6.2 Analytical balance, with capability to measure to 0.1 mg, for use in weighing solids, for preparing standards, and for determining dissolved solids in digests or extracts.
6.3 A temperature adjustable hot plate capable of maintaining a temperature of 95°C.
6.4 (Optional) A temperature adjustable block digester capable of maintaining a temperature of 95°C and equipped with 250 mL constricted digestion tubes.
6.5 (Optional) A steel cabinet centrifuge with guard bowl, electric timer and brake.
6.6 A gravity convection drying oven with thermostatic control capable of maintaining 180°C ± 5°C.
6.7 (Optional) An air displacement pipetter capable of delivering volumes ranging from 0.1-2500 μL with an assortment of high quality disposable pipet tips.
6.8 Mortar and pestle, ceramic or nonmetallic material.
6.9 Polypropylene sieve, 5-mesh (4 mm opening).
6.10 Labware—For determination of trace levels of elements, contamination and loss are of prime consideration. Potential contamination sources include improperly cleaned laboratory apparatus and general contamination within the laboratory environment from dust, etc. A clean laboratory work area designated for trace element sample handling must be used. Sample containers can introduce positive and negative errors in the determination of trace elements by contributing contaminants through surface desorption or leaching, or depleting element concentrations through adsorption processes. All reusable labware (glass, quartz, polyethylene, PTFE, FEP, etc.) should be sufficiently clean for the task objectives. Several procedures found to provide clean labware include washing with a detergent solution, rinsing with tap water, soaking for four hours or more in 20% (v/v) nitric acid or a mixture of HNO 3 and HCl (1 2 9), rinsing with reagent water and storing clean. 2 3 Chromic acid cleaning solutions must be avoided because chromium is an analyte.
6.10.1 Glassware—Volumetric flasks, graduated cylinders, funnels and centrifuge tubes (glass and/or metal-free plastic).
6.10.2 Assorted calibrated pipettes.
6.10.3 Conical Phillips beakers (Corning 1080-250 or equivalent), 250 mL with 50 mm watch glasses.
6.10.4 Griffin beakers, 250 mL with 75 mm watch glasses and (optional) 75 mm ribbed watch glasses.
6.10.5 (Optional) PTFE and/or quartz Griffin beakers, 250 mL with PTFE covers.
6.10.6 Evaporating dishes or high-form crucibles, porcelain, 100 mL capacity.
6.10.7 Narrow-mouth storage bottles, FEP (fluorinated ethylene propylene) with screw closure, 125 mL to 1 L capacities.
6.10.8 One-piece stem FEP wash bottle with screw closure, 125 mL capacity.
7.0 Reagents and Standards
7.1 Reagents may contain elemental impurities which might affect analytical data. Only high-purity reagents that conform to the American Chemical Society specifications 13 should be used whenever possible. If the purity of a reagent is in question, analyze for contamination. All acids used for this method must be of ultra high-purity grade or equivalent. Suitable acids are available from a number of manufacturers. Redistilled acids prepared by sub-boiling distillation are acceptable.
7.2 Hydrochloric acid, concentrated (sp.gr. 1.19)—HCl.
7.2.1 Hydrochloric acid (1 1)—Add 500 mL concentrated HCl to 400 mL reagent water and dilute to 1 L.
7.2.2 Hydrochloric acid (1 4)—Add 200 mL concentrated HCl to 400 mL reagent water and dilute to 1 L.
7.2.3 Hydrochloric acid (1 20)—Add 10 mL concentrated HCl to 200 mL reagent water.
7.3 Nitric acid, concentrated (sp.gr. 1.41)—HNO 3 .
7.3.1 Nitric acid (1 1)—Add 500 mL concentrated HNO 3 to 400 mL reagent water and dilute to 1 L.
7.3.2 Nitric acid (1 2)—Add 100 mL concentrated HNO 3 to 200 mL reagent water.
7.3.3 Nitric acid (1 5)—Add 50 mL concentrated HNO 3 to 250 mL reagent water.
7.3.4 Nitric acid (1 9)—Add 10 mL concentrated HNO 3 to 90 mL reagent water.
7.4 Reagent water. All references to water in this method refer to ASTM Type I grade water. 14
7.5 Ammonium hydroxide, concentrated (sp.gr. 0.902).
7.6 Tartaric acid, ACS reagent grade.
7.7 Hydrogen peroxide, 50%, stabilized certified reagent grade.
7.8 Standard Stock Solutions—Stock standards may be purchased or prepared from ultra-high purity grade chemicals (99.99–99.999% pure). All compounds must be dried for one hour at 105 °C, unless otherwise specified. It is recommended that stock solutions be stored in FEP bottles. Replace stock standards when succeeding dilutions for preparation of calibration standards cannot be verified.
CAUTION: Many of these chemicals are extremely toxic if inhaled or swallowed (Section 5.1). Wash hands thoroughly after handling.
Typical stock solution preparation procedures follow for 1 L quantities, but for the purpose of pollution prevention, the analyst is encouraged to prepare smaller quantities when possible. Concentrations are calculated based upon the weight of the pure element or upon the weight of the compound multiplied by the fraction of the analyte in the compound
From pure element,

where: gravimetric factor = the weight fraction of the analyte in the compound
7.8.1 Aluminum solution, stock, 1 mL = 1000 μg Al: Dissolve 1.000 g of aluminum metal, weighed accurately to at least four significant figures, in an acid mixture of 4.0 mL of (1 1) HCl and 1 mL of concentrated HNO3 in a beaker. Warm beaker slowly to effect solution. When dissolution is complete, transfer solution quantitatively to a 1 L flask, add an additional 10.0 mL of (1 1) HCl and dilute to volume with reagent water.
7.8.2 Antimony solution, stock, 1 mL = 1000 μg Sb: Dissolve 1.000 g of antimony powder, weighed accurately to at least four significant figures, in 20.0 mL (1 1) HNO 3 and 10.0 mL concentrated HCl. Add 100 mL reagent water and 1.50 g tartaric acid. Warm solution slightly to effect complete dissolution. Cool solution and add reagent water to volume in a 1 L volumetric flask.
7.8.3 Arsenic solution, stock, 1 mL = 1000 μg As: Dissolve 1.320 g of As 2 O 3 (As fraction = 0.7574), weighed accurately to at least four significant figures, in 100 mL of reagent water containing 10.0 mL concentrated NH 4 OH. Warm the solution gently to effect dissolution. Acidify the solution with 20.0 mL concentrated HNO 3 and dilute to volume in a 1 L volumetric flask with reagent water.
7.8.4 Barium solution, stock, 1 mL = 1000 μg Ba: Dissolve 1.437 g BaCO 3 (Ba fraction = 0.6960), weighed accurately to at least four significant figures, in 150 mL (1 2) HNO 3 with heating and stirring to degas and dissolve compound. Let solution cool and dilute with reagent water in 1 L volumetric flask.
7.8.5 Beryllium solution, stock, 1 mL = 1000 μg Be: DO NOT DRY . Dissolve 19.66 g BeSO 4 •4H 2 O (Be fraction = 0.0509), weighed accurately to at least four significant figures, in reagent water, add 10.0 mL concentrated HNO 3 , and dilute to volume in a 1 L volumetric flask with reagent water.
7.8.6 Boron solution, stock, 1 mL = 1000 μg B: DO NOT DRY. Dissolve 5.716 g anhydrous H3BO3 (B fraction = 0.1749), weighed accurately to at least four significant figures, in reagent water and dilute in a 1 L volumetric flask with reagent water. Transfer immediately after mixing to a clean FEP bottle to minimize any leaching of boron from the glass volumetric container. Use of a nonglass volumetric flask is recommended to avoid boron contamination from glassware.
7.8.7 Cadmium solution, stock, 1 mL = 1000 μg Cd: Dissolve 1.000 g Cd metal, acid cleaned with (1 9) HNO 3 , weighed accurately to at least four significant figures, in 50 mL (1 1) HNO 3 with heating to effect dissolution. Let solution cool and dilute with reagent water in a 1 L volumetric flask.
7.8.8 Calcium solution, stock, 1 mL = 1000 μg Ca: Suspend 2.498 g CaCO 3 (Ca fraction = 0.4005), dried at 180 °C for one hour before weighing, weighed accurately to at least four significant figures, in reagent water and dissolve cautiously with a minimum amount of (1 1) HNO 3 . Add 10.0 mL concentrated HNO 3 and dilute to volume in a 1 L volumetric flask with reagent water.
7.8.9 Cerium solution, stock, 1 mL = 1000 μg Ce: Slurry 1.228 g CeO 2 (Ce fraction = 0.8141), weighed accurately to at least four significant figures, in 100 mL concentrated HNO 3 and evaporate to dryness. Slurry the residue in 20 mL H 2 O, add 50 mL concentrated HNO 3 , with heat and stirring add 60 mL 50% H 2 O 2 dropwise in 1 mL increments allowing periods of stirring between the 1 mL additions. Boil off excess H 2 O 2 before diluting to volume in a 1 L volumetric flask with reagent water.
7.8.10 Chromium solution, stock, 1 mL = 1000 μg Cr: Dissolve 1.923 g CrO 3 (Cr fraction = 0.5200), weighed accurately to at least four significant figures, in 120 mL (1 5) HNO 3 . When solution is complete, dilute to volume in a 1 L volumetric flask with reagent water.
7.8.11 Cobalt solution, stock, 1 mL = 1000 μg Co: Dissolve 1.000 g Co metal, acid cleaned with (1 9) HNO 3 , weighed accurately to at least four significant figures, in 50.0 mL (1 1) HNO 3 . Let solution cool and dilute to volume in a 1 L volumetric flask with reagent water.
7.8.12 Copper solution, stock, 1 mL = 1000 μg Cu: Dissolve 1.000 g Cu metal, acid cleaned with (1 9) HNO 3 , weighed accurately to at least four significant figures, in 50.0 mL (1 1) HNO 3 with heating to effect dissolution. Let solution cool and dilute in a 1 L volumetric flask with reagent water.
7.8.13 Iron solution, stock, 1 mL = 1000 μg Fe: Dissolve 1.000 g Fe metal, acid cleaned with (1 1) HCl, weighed accurately to four significant figures, in 100 mL (1 1) HCl with heating to effect dissolution. Let solution cool and dilute with reagent water in a 1 L volumetric flask.
7.8.14 Lead solution, stock, 1 mL = 1000 μg Pb: Dissolve 1.599 g Pb(NO 3 ) 2 (Pb fraction = 0.6256), weighed accurately to at least four significant figures, in a minimum amount of (1 1) HNO 3 . Add 20.0 mL (1 1) HNO 3 and dilute to volume in a 1 L volumetric flask with reagent water.
7.8.15 Lithium solution, stock, 1 mL = 1000 μg Li: Dissolve 5.324 g Li 2 CO 3 (Li fraction = 0.1878), weighed accurately to at least four significant figures, in a minimum amount of (1 1) HCl and dilute to volume in a 1 L volumetric flask with reagent water.
7.8.16 Magnesium solution, stock, 1 mL = 1000 μg Mg: Dissolve 1.000 g cleanly polished Mg ribbon, accurately weighed to at least four significant figures, in slowly added 5.0 mL (1 1) HCl (CAUTION: reaction is vigorous). Add 20.0 mL (1 1) HNO 3 and dilute to volume in a 1 L volumetric flask with reagent water.
7.8.17 Manganese solution, stock, 1 mL = 1000 μg Mn: Dissolve 1.000 g of manganese metal, weighed accurately to at least four significant figures, in 50 mL (1 1) HNO 3 and dilute to volume in a 1 L volumetric flask with reagent water.
7.8.18 Mercury solution, stock, 1 mL = 1000 μg Hg: DO NOT DRY . CAUTION: highly toxic element. Dissolve 1.354 g HgCl 2 (Hg fraction = 0.7388) in reagent water. Add 50.0 mL concentrated HNO 3 and dilute to volume in 1 L volumetric flask with reagent water.
7.8.19 Molybdenum solution, stock, 1 mL = 1000 μg Mo: Dissolve 1.500 g MoO 3 (Mo fraction = 0.6666), weighed accurately to at least four significant figures, in a mixture of 100 mL reagent water and 10.0 mL concentrated NH 4 OH, heating to effect dissolution. Let solution cool and dilute with reagent water in a 1 L volumetric flask.
7.8.20 Nickel solution, stock, 1 mL = 1000 μg Ni: Dissolve 1.000 g of nickel metal, weighed accurately to at least four significant figures, in 20.0 mL hot concentrated HNO 3 , cool, and dilute to volume in a 1 L volumetric flask with reagent water.
7.8.21 Phosphorus solution, stock, 1 mL = 1000 μg P: Dissolve 3.745 g NH 4 H 2 PO 4 (P fraction = 0.2696), weighed accurately to at least four significant figures, in 200 mL reagent water and dilute to volume in a 1 L volumetric flask with reagent water.
7.8.22 Potassium solution, stock, 1 mL = 1000 μg K: Dissolve 1.907 g KCl (K fraction = 0.5244) dried at 110 °C, weighed accurately to at least four significant figures, in reagent water, add 20 mL (1 1) HCl and dilute to volume in a 1 L volumetric flask with reagent water.
7.8.23 Selenium solution, stock, 1 mL = 1000 μg Se: Dissolve 1.405 g SeO 2 (Se fraction = 0.7116), weighed accurately to at least four significant figures, in 200 mL reagent water and dilute to volume in a 1 L volumetric flask with reagent water.
7.8.24 Silica solution, stock, 1 mL = 1000 μg SiO 2 : DO NOT DRY . Dissolve 2.964 g (NH 4 ) 2 SiF 6 , weighed accurately to at least four significant figures, in 200 mL (1 20) HCl with heating at 85 °C to effect dissolution. Let solution cool and dilute to volume in a 1 L volumetric flask with reagent water.
7.8.25 Silver solution, stock, 1 mL = 1000 μg Ag: Dissolve 1.000 g Ag metal, weighed accurately to at least four significant figures, in 80 mL (1 1) HNO 3 with heating to effect dissolution. Let solution cool and dilute with reagent water in a 1 L volumetric flask. Store solution in amber bottle or wrap bottle completely with aluminum foil to protect solution from light.
7.8.26 Sodium solution, stock, 1 mL = 1000 μg Na: Dissolve 2.542 g NaCl (Na fraction = 0.3934), weighed accurately to at least four significant figures, in reagent water. Add 10.0 mL concentrated HNO3 and dilute to volume in a 1 L volumetric flask with reagent water.
7.8.27 Strontium solution, stock, 1 mL = 1000 μg Sr: Dissolve 1.685 g SrCO 3 (Sr fraction = 0.5935), weighed accurately to at least four significant figures, in 200 mL reagent water with dropwise addition of 100 mL (1 1) HCl. Dilute to volume in a 1 L volumetric flask with reagent water.
7.8.28 Thallium solution, stock, 1 mL = 1000 μg Tl: Dissolve 1.303 g TlNO 3 (Tl fraction = 0.7672), weighed accurately to at least four significant figures, in reagent water. Add 10.0 mL concentrated HNO 3 and dilute to volume in a 1 L volumetric flask with reagent water.
7.8.29 Tin solution, stock, 1 mL = 1000 μg Sn: Dissolve 1.000 g Sn shot, weighed accurately to at least four significant figures, in an acid mixture of 10.0 mL concentrated HCl and 2.0 mL (1 1) HNO 3 with heating to effect dissolution. Let solution cool, add 200 mL concentrated HCl, and dilute to volume in a 1 L volumetric flask with reagent water.
7.8.30 Titanium solution, stock, 1 mL = 1000 μg Ti: DO NOT DRY . Dissolve 6.138 g (NH 4 ) 2 TiO(C 2 O 4 ) 2 •H 2 O (Ti fraction = 0.1629), weighed accurately to at least four significant figures, in 100 mL reagent water. Dilute to volume in a 1 L volumetric flask with reagent water.
7.8.31 Vanadium solution, stock, 1 mL = 1000 μg V: Dissolve 1.000 g V metal, acid cleaned with (1 9) HNO 3 , weighed accurately to at least four significant figures, in 50 mL (1 1) HNO 3 with heating to effect dissolution. Let solution cool and dilute with reagent water to volume in a 1 L volumetric flask.
7.8.32 Yttrium solution, stock 1 mL = 1000 μg Y: Dissolve 1.270 g Y 2 O 3 (Y fraction = 0.7875), weighed accurately to at least four significant figures, in 50 mL (1 1) HNO 3 , heating to effect dissolution. Cool and dilute to volume in a 1 L volumetric flask with reagent water.
7.8.33 Zinc solution, stock, 1 mL = 1000 μg Zn: Dissolve 1.000 g Zn metal, acid cleaned with (1 9) HNO 3 , weighed accurately to at least four significant figures, in 50 mL (1 1) HNO 3 with heating to effect dissolution. Let solution cool and dilute with reagent water to volume in a 1 L volumetric flask.
7.9 Mixed Calibration Standard Solutions—For the analysis of total recoverable digested samples prepare mixed calibration standard solutions (see Table 3) by combining appropriate volumes of the stock solutions in 500 mL volumetric flasks containing 20 mL (1 1) HNO 3 and 20 mL (1 1) HCl and dilute to volume with reagent water. Prior to preparing the mixed standards, each stock solution should be analyzed separately to determine possible spectral interferences or the presence of impurities. Care should be taken when preparing the mixed standards to ensure that the elements are compatible and stable together. To minimize the opportunity for contamination by the containers, it is recommended to transfer the mixed-standard solutions to acid-cleaned, never-used FEP fluorocarbon (FEP) bottles for storage. Fresh mixed standards should be prepared, as needed, with the realization that concentrations can change on aging. Calibration standards not prepared from primary standards must be initially verified using a certified reference solution. For the recommended wavelengths listed in Table 1 some typical calibration standard combinations are given in Table 3.
Note: If the addition of silver to the recommended mixed-acid calibration standard results in an initial precipitation, add 15 mL of reagent water and warm the flask until the solution clears. For this acid combination, the silver concentration should be limited to 0.5 mg/L.
7.10 Blanks—Four types of blanks are required for the analysis. The calibration blank is used in establishing the analytical curve, the laboratory reagent blank is used to assess possible contamination from the sample preparation procedure, the laboratory fortified blank is used to assess routine laboratory performance and a rinse blank is used to flush the instrument uptake system and nebulizer between standards, check solutions, and samples to reduce memory interferences.
7.10.1 The calibration blank for aqueous samples and extracts is prepared by acidifying reagent water to the same concentrations of the acids as used for the standards. The calibration blank should be stored in a FEP bottle.
7.10.2 The laboratory reagent blank (LRB) must contain all the reagents in the same volumes as used in the processing of the samples. The LRB must be carried through the same entire preparation scheme as the samples including sample digestion, when applicable.
7.10.3 The laboratory fortified blank (LFB) is prepared by fortifying an aliquot of the laboratory reagent blank with all analytes to a suitable concentration using the following recommended criteria: Ag 0.1 mg/L, K 5.0 mg/L and all other analytes 0.2 mg/L or a concentration approximately 100 times their respective MDL, whichever is greater. The LFB must be carried through the same entire preparation scheme as the samples including sample digestion, when applicable.
7.10.4 The rinse blank is prepared by acidifying reagent water to the same concentrations of acids as used in the calibration blank and stored in a convenient manner.
7.11 Instrument Performance Check (IPC) Solution—The IPC solution is used to periodically verify instrument performance during analysis. It should be prepared in the same acid mixture as the calibration standards by combining method analytes at appropriate concentrations. Silver must be limited to <0.5 mg/L; while potassium and phosphorus because of higher MDLs and silica because of potential contamination should be at concentrations of 10 mg/L. For other analytes a concentration of 2 mg/L is recommended. The IPC solution should be prepared from the same standard stock solutions used to prepare the calibration standards and stored in an FEP bottle. Agency programs may specify or request that additional instrument performance check solutions be prepared at specified concentrations in order to meet particular program needs.
7.12 Quality Control Sample (QCS)—Analysis of a QCS is required for initial and periodic verification of calibration standards or stock standard solutions in order to verify instrument performance. The QCS must be obtained from an outside source different from the standard stock solutions and prepared in the same acid mixture as the calibration standards. The concentration of the analytes in the QCS solution should be 1 mg/L, except silver, which must be limited to a concentration of 0.5 mg/L for solution stability. The QCS solution should be stored in a FEP bottle and analyzed as needed to meet data-quality needs. A fresh solution should be prepared quarterly or more frequently as needed.
7.13 Spectral Interference Check (SIC) Solutions—When interelement corrections are applied, SIC solutions are needed containing concentrations of the interfering elements at levels that will provide an adequate test of the correction factors.
7.13.1 SIC solutions containing (a) 300 mg/L Fe; (b) 200 mg/L AL; (c) 50 mg/L Ba; (d) 50 mg/L Be; (e) 50 mg/L Cd; (f) 50 mg/L Ce; (g) 50 mg/L Co; (h) 50 mg/L Cr; (i) 50 mg/L Cu; (j) 50 mg/L Mn; (k) 50 mg/L Mo; (l) 50 mg/L Ni; (m) 50 mg/L Sn; (n) 50 mg/L SiO2; (o) 50 mg/L Ti; (p) 50 mg/L Tl and (q) 50 mg/L V should be prepared in the same acid mixture as the calibration standards and stored in FEP bottles. These solutions can be used to periodically verify a partial list of the on-line (and possible off-line) interelement spectral correction factors for the recommended wavelengths given in Table 1. Other solutions could achieve the same objective as well. (Multielement SIC solutions3may be prepared and substituted for the single element solutions provided an analyte is not subject to interference from more than one interferant in the solution.)
Note: If wavelengths other than those recommended in Table 1 are used, other solutions different from those above (a through q) may be required.
7.13.2 For interferences from iron and aluminum, only those correction factors (positive or negative) when multiplied by 100 to calculate apparent analyte concentrations that exceed the determined analyte IDL or fall below the lower 3-sigma control limit of the calibration blank need be tested on a daily basis.
7.13.3 For the other interfering elements, only those correction factors (positive or negative) when multiplied by 10 to calculate apparent analyte concentrations that exceed the determined analyte IDL or fall below the lower 3-sigma control limit of the calibration blank need be tested on a daily basis.
7.13.4 If the correction routine is operating properly, the determined apparent analyte(s) concentration from analysis of each interference solution (a through q) should fall within a specific concentration range bracketing the calibration blank. This concentration range is calculated by multiplying the concentration of the interfering element by the value of the correction factor being tested and dividing by 10. If after subtraction of the calibration blank the apparent analyte concentration is outside (above or below) this range, a change in the correction factor of more than 10% should be suspected. The cause of the change should be determined and corrected and the correction factor should be updated.
Note: The SIC solution should be analyzed more than once to confirm a change has occurred with adequate rinse time between solutions and before subsequent analysis of the calibration blank.
7.13.5 If the correction factors tested on a daily basis are found to be within the 10% criteria for five consecutive days, the required verification frequency of those factors in compliance may be extended to a weekly basis. Also, if the nature of the samples analyzed is such (e.g., finished drinking water) that they do not contain concentrations of the interfering elements at the 10 mg/L level, daily verification is not required; however, all interelement spectral correction factors must be verified annually and updated, if necessary.
7.13.6 If the instrument does not display negative concentration values, fortify the SIC solutions with the elements of interest at 1 mg/L and test for analyte recoveries that are below 95%. In the absence of measurable analyte, over-correction could go undetected because a negative value could be reported as zero.
7.14 For instruments without interelement correction capability or when interelement corrections are not used, SIC solutions (containing similar concentrations of the major components in the samples,e.g.,10 mg/L) can serve to verify the absence of effects at the wavelengths selected. These data must be kept on file with the sample analysis data. If the SIC solution confirms an operative interference that is 10% of the analyte concentration, the analyte must be determined using a wavelength and background correction location free of the interference or by another approved test procedure. Users are advised that high salt concentrations can cause analyte signal suppressions and confuse interference tests.
7.15 Plasma Solution—The plasma solution is used for determining the optimum viewing height of the plasma above the work coil prior to using the method (Section 10.2). The solution is prepared by adding a 5 mL aliquot from each of the stock standard solutions of arsenic, lead, selenium, and thallium to a mixture of 20 mL (1 1) nitric acid and 20 mL (1 1) hydrochloric acid and diluting to 500 mL with reagent water. Store in a FEP bottle.
8.0 Sample Collection, Preservation, and Storage
8.1 Prior to the collection of an aqueous sample, consideration should be given to the type of data required, (i.e.,dissolved or total recoverable), so that appropriate preservation and pretreatment steps can be taken. The pH of all aqueous samples must be tested immediately prior to aliquoting for processing or "direct analysis" to ensure the sample has been properly preserved. If properly acid preserved, the sample can be held up to six months before analysis.
8.2 For the determination of the dissolved elements, the sample must be filtered through a 0.45 μm pore diameter membrane filter at the time of collection or as soon thereafter as practically possible. (Glass or plastic filtering apparatus are recommended to avoid possible contamination. Only plastic apparatus should be used when the determinations of boron and silica are critical.) Use a portion of the filtered sample to rinse the filter flask, discard this portion and collect the required volume of filtrate. Acidify the filtrate with (1 1) nitric acid immediately following filtration to pH <2.
8.3 For the determination of total recoverable elements in aqueous samples, samples are not filtered, but acidified with (1 1) nitric acid to pH <2 (normally, 3 mL of (1 1) acid per liter of sample is sufficient for most ambient and drinking water samples). Preservation may be done at the time of collection, however, to avoid the hazards of strong acids in the field, transport restrictions, and possible contamination it is recommended that the samples be returned to the laboratory within two weeks of collection and acid preserved upon receipt in the laboratory. Following acidification, the sample should be mixed, held for 16 hours, and then verified to be pH <2 just prior withdrawing an aliquot for processing or "direct analysis". If for some reason such as high alkalinity the sample pH is verified to be >2, more acid must be added and the sample held for 16 hours until verified to be pH <2. See Section 8.1.
Note: When the nature of the sample is either unknown or is known to be hazardous, acidification should be done in a fume hood. See Section 5.2.
8.4 Solid samples require no preservation prior to analysis other than storage at 4 °C. There is no established holding time limitation for solid samples.
8.5 For aqueous samples, a field blank should be prepared and analyzed as required by the data user. Use the same container and acid as used in sample collection.
9.0 Quality Control
9.1 Each laboratory using this method is required to operate a formal quality control (QC) program. The minimum requirements of this program consist of an initial demonstration of laboratory capability, and the periodic analysis of laboratory reagent blanks, fortified blanks and other laboratory solutions as a continuing check on performance. The laboratory is required to maintain performance records that define the quality of the data thus generated.
9.2 Initial Demonstration of Performance (mandatory).
9.2.1 The initial demonstration of performance is used to characterize instrument performance (determination of linear dynamic ranges and analysis of quality control samples) and laboratory performance (determination of method detection limits) prior to analyses conducted by this method.
9.2.2 Linear dynamic range (LDR)—The upper limit of the LDR must be established for each wavelength utilized. It must be determined from a linear calibration prepared in the normal manner using the established analytical operating procedure for the instrument. The LDR should be determined by analyzing succeedingly higher standard concentrations of the analyte until the observed analyte concentration is no more than 10% below the stated concentration of the standard. Determined LDRs must be documented and kept on file. The LDR which may be used for the analysis of samples should be judged by the analyst from the resulting data. Determined sample analyte concentrations that are greater than 90% of the determined upper LDR limit must be diluted and reanalyzed. The LDRs should be verified annually or whenever, in the judgment of the analyst, a change in analytical performance caused by either a change in instrument hardware or operating conditions would dictate they be redetermined.
9.2.3 Quality control sample (QCS)—When beginning the use of this method, on a quarterly basis, after the preparation of stock or calibration standard solutions or as required to meet data-quality needs, verify the calibration standards and acceptable instrument performance with the preparation and analyses of a QCS (Section 7.12). To verify the calibration standards the determined mean concentrations from three analyses of the QCS must be within 5% of the stated values. If the calibration standard cannot be verified, performance of the determinative step of the method is unacceptable. The source of the problem must be identified and corrected before either proceeding on with the initial determination of method detection limits or continuing with on-going analyses.
9.2.4 Method detection limit (MDL)—MDLs must be established for all wavelengths utilized, using reagent water (blank) fortified at a concentration of two to three times the estimated instrument detection limit.15To determine MDL values, take seven replicate aliquots of the fortified reagent water and process through the entire analytical method. Perform all calculations defined in the method and report the concentration values in the appropriate units. Calculate the MDL as follows:
MDL = (t) x (S)
Where:
t = students' t value for a 99% confidence level and a standard deviation estimate with n-1 degrees of freedom [t = 3.14 for seven replicates]
S= standard deviation of the replicate analysis
Note: If additional confirmation is desired, reanalyze the seven replicate aliquots on two more nonconsecutive days and again calculate the MDL values for each day. An average of the three MDL values for each analyte may provide for a more appropriate MDL estimate. If the relative standard deviation (RSD) from the analyses of the seven aliquots is <10%, the concentration used to determine the analyte MDL may have been inappropriately high for the determination. If so, this could result in the calculation of an unrealistically low MDL. Concurrently, determination of MDL in reagent water represents a best case situation and does not reflect possible matrix effects of real world samples. However, successful analyses of LFMs (Section 9.4) and the analyte addition test described in Section 9.5.1 can give confidence to the MDL value determined in reagent water. Typical single laboratory MDL values using this method are given in Table 4.
The MDLs must be sufficient to detect analytes at the required levels according to compliance monitoring regulation (Section 1.2). MDLs should be determined annually, when a new operator begins work or whenever, in the judgment of the analyst, a change in analytical performance caused by either a change in instrument hardware or operating conditions would dictate they be redetermined.
9.3 Assessing Laboratory Performance (mandatory)
9.3.1 Laboratory reagent blank (LRB)—The laboratory must analyze at least one LRB (Section 7.10.2) with each batch of 20 or fewer samples of the same matrix. LRB data are used to assess contamination from the laboratory environment. LRB values that exceed the MDL indicate laboratory or reagent contamination should be suspected. When LRB values constitute 10% or more of the analyte level determined for a sample or is 2.2 times the analyte MDL whichever is greater, fresh aliquots of the samples must be prepared and analyzed again for the affected analytes after the source of contamination has been corrected and acceptable LRB values have been obtained.
9.3.2 Laboratory fortified blank (LFB)—The laboratory must analyze at least one LFB (Section 7.10.3) with each batch of samples. Calculate accuracy as percent recovery using the following equation:
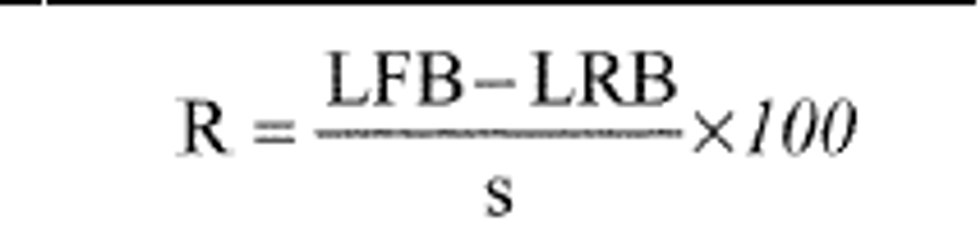
Where:
R = percent recovery
LFB = laboratory fortified blank
LRB = laboratory reagent blank
s = concentration equivalent of analyte added to fortify the LBR solution
If the recovery of any analyte falls outside the required control limits of 85-115%, that analyte is judged out of control, and the source of the problem should be identified and resolved before continuing analyses.
9.3.3 The laboratory must use LFB analyses data to assess laboratory performance against the required control limits of 85-115% (Section 9.3.2). When sufficient internal performance data become available (usually a minimum of 20-30 analyses), optional control limits can be developed from the mean percent recovery (x) and the standard deviation (S) of the mean percent recovery. These data can be used to establish the upper and lower control limits as follows:
UPPER CONTROL LIMIT = x 3S
LOWER CONTROL LIMIT = x - 3S
The optional control limits must be equal to or better than the required control limits of 85-115%. After each five to 10 new recovery measurements, new control limits can be calculated using only the most recent 20-30 data points. Also, the standard deviation (S) data should be used to establish an on-going precision statement for the level of concentrations included in the LFB. These data must be kept on file and be available for review.
9.3.4 Instrument performance check (IPC) solution—For all determinations the laboratory must analyze the IPC solution (Section 7.11) and a calibration blank immediately following daily calibration, after every 10th sample (or more frequently, if required) and at the end of the sample run. Analysis of the calibration blank should always be < the analyte IDL, but greater than the lower 3-sigma control limit of the calibration blank. Analysis of the IPC solution immediately following calibration must verify that the instrument is within 5% of calibration with a relative standard deviation <3% from replicate integrations 4. Subsequent analyses of the IPC solution must be within 10% of calibration. If the calibration cannot be verified within the specified limits, reanalyze either or both the IPC solution and the calibration blank. If the second analysis of the IPC solution or the calibration blank confirm calibration to be outside the limits, sample analysis must be discontinued, the cause determined, corrected and/or the instrument recalibrated. All samples following the last acceptable IPC solution must be reanalyzed. The analysis data of the calibration blank and IPC solution must be kept on file with the sample analyses data.
9.3.5 Spectral interference check (SIC) solution—For all determinations the laboratory must periodically verify the interelement spectral interference correction routine by analyzing SIC solutions. The preparation and required periodic analysis of SIC solutions and test criteria for verifying the interelement interference correction routine are given in Section 7.13. Special cases where on-going verification is required are described in Section 7.14.
9.4 Assessing Analyte Recovery and Data Quality.
9.4.1 Sample homogeneity and the chemical nature of the sample matrix can affect analyte recovery and the quality of the data. Taking separate aliquots from the sample for replicate and fortified analyses can in some cases assess the effect. Unless otherwise specified by the data user, laboratory or program, the following laboratory fortified matrix (LFM) procedure (Section 9.4.2) is required. Also, other tests such as the analyte addition test (Section 9.5.1) and sample dilution test (Section 9.5.2) can indicate if matrix effects are operative.
9.4.2 The laboratory must add a known amount of each analyte to a minimum of 10% of the routine samples. In each case the LFM aliquot must be a duplicate of the aliquot used for sample analysis and for total recoverable determinations added prior to sample preparation. For water samples, the added analyte concentration must be the same as that used in the laboratory fortified blank (Section 7.10.3). For solid samples, however, the concentration added should be expressed as mg/kg and is calculated for a one gram aliquot by multiplying the added analyte concentration (mg/L) in solution by the conversion factor 100 (mg/L × 0.1L/0.001kg = 100, Section 12.5). (For notes on Ag, Ba, and Sn see Sections 1.7 and 1.8.) Over time, samples from all routine sample sources should be fortified.
Note: The concentration of calcium, magnesium, sodium and strontium in environmental waters, along with iron and aluminum in solids can vary greatly and are not necessarily predictable. Fortifying these analytes in routine samples at the same concentration used for the LFB may prove to be of little use in assessing data quality for these analytes. For these analytes sample dilution and reanalysis using the criteria given in Section 9.5.2 is recommended. Also, if specified by the data user, laboratory or program, samples can be fortified at higher concentrations, but even major constituents should be limited to <25 mg/L so as not to alter the sample matrix and affect the analysis.
9.4.3 Calculate the percent recovery for each analyte, corrected for background concentrations measured in the unfortified sample, and compare these values to the designated LFM recovery range of 70-130% or a 3-sigma recovery range calculated from the regression equations given in Table 9.16 Recovery calculations are not required if the concentration added is less than 30% of the sample background concentration. Percent recovery may be calculated in units appropriate to the matrix, using the following equation:
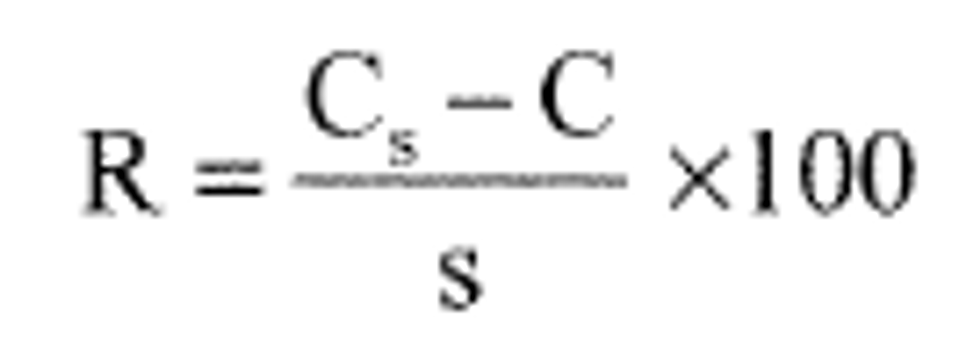
Where:
R = percent recovery
Cs = fortified sample concentration
C = sample background concentration
s = concentration equivalent of analyte added to fortify the sample
9.4.4 If the recovery of any analyte falls outside the designated LFM recovery range, and the laboratory performance for that analyte is shown to be in control (Section 9.3), the recovery problem encountered with the fortified sample is judged to be matrix related, not system related. The data user should be informed that the result for that analyte in the unfortified sample is suspect due to either the heterogeneous nature of the sample or matrix effects and analysis by method of standard addition or the use of an internal standard(s) (Section 11.5) should be considered.
9.4.5 Where reference materials are available, they should be analyzed to provide additional performance data. The analysis of reference samples is a valuable tool for demonstrating the ability to perform the method acceptably. Reference materials containing high concentrations of analytes can provide additional information on the performance of the spectral interference correction routine.
9.5 Assess the possible need for the method of standard additions (MSA) or internal standard elements by the following tests. Directions for using MSA or internal standard(s) are given in Section 11.5.
9.5.1 Analyte addition test: An analyte(s) standard added to a portion of a prepared sample, or its dilution, should be recovered to within 85% to 115% of the known value. The analyte(s) addition should produce a minimum level of 20 times and a maximum of 100 times the method detection limit. If the analyte addition is <20% of the sample analyte concentration, the following dilution test should be used. If recovery of the analyte(s) is not within the specified limits, a matrix effect should be suspected, and the associated data flagged accordingly. The method of additions or the use of an appropriate internal standard element may provide more accurate data.
9.5.2 Dilution test: If the analyte concentration is sufficiently high (minimally, a factor of 50 above the instrument detection limit in the original solution but <90% of the linear limit), an analysis of a 1 4 dilution should agree (after correction for the fivefold dilution) within 10% of the original determination. If not, a chemical or physical interference effect should be suspected and the associated data flagged accordingly. The method of standard additions or the use of an internal-standard element may provide more accurate data for samples failing this test.
10.0 Calibration and Standardization
10.1 Specific wavelengths are listed in Table 1. Other wavelengths may be substituted if they can provide the needed sensitivity and are corrected for spectral interference. However, because of the difference among various makes and models of spectrometers, specific instrument operating conditions cannot be given. The instrument and operating conditions utilized for determination must be capable of providing data of acceptable quality to the program and data user. The analyst should follow the instructions provided by the instrument manufacturer unless other conditions provide similar or better performance for a task. Operating conditions for aqueous solutions usually vary from 1100-1200 watts forward power, 15-16 mm viewing height, 15-19 L/min. argon coolant flow, 0.6-1 L/min. argon aerosol flow, 1-1.8 mL/min. sample pumping rate with a one minute preflush time and measurement time near 1 s per wavelength peak (for sequential instruments) and near 10 s per sample (for simultaneous instruments). Use of the Cu/Mn intensity ratio at 324.754 nm and 257.610 nm (by adjusting the argon aerosol flow) has been recommended as a way to achieve repeatable interference correction factors.17
10.2 Prior to using this method optimize the plasma operating conditions. The following procedure is recommended for vertically configured plasmas. The purpose of plasma optimization is to provide a maximum signal-to-background ratio for the least sensitive element in the analytical array. The use of a mass flow controller to regulate the nebulizer gas flow rate greatly facilitates the procedure.
10.2.1 Ignite the plasma and select an appropriate incident rf power with minimum reflected power. Allow the instrument to become thermally stable before beginning. This usually requires at least 30 to 60 minutes of operation. While aspirating the 1000 μg/mL solution of yttrium (Section 7.8.32), follow the instrument manufacturer's instructions and adjust the aerosol carrier gas flow rate through the nebulizer so a definitive blue emission region of the plasma extends approximately from 5-20 mm above the top of the work coil.18Record the nebulizer gas flow rate or pressure setting for future reference.
10.2.2 After establishing the nebulizer gas flow rate, determine the solution uptake rate of the nebulizer in mL/min. by aspirating a known volume calibration blank for a period of at least three minutes. Divide the spent volume by the aspiration time (in minutes) and record the uptake rate. Set the peristaltic pump to deliver the uptake rate in a steady even flow.
10.2.3 After horizontally aligning the plasma and/or optically profiling the spectrometer, use the selected instrument conditions from Sections 10.2.1 and 10.2.2, and aspirate the plasma solution (Section 7.15), containing 10 μg/mL each of As, Pb, Se and Tl. Collect intensity data at the wavelength peak for each analyte at 1 mm intervals from 14-18 mm above the top of the work coil. (This region of the plasma is commonly referred to as the analytical zone.)19Repeat the process using the calibration blank. Determine the net signal to blank intensity ratio for each analyte for each viewing height setting. Choose the height for viewing the plasma that provides the largest intensity ratio for the least sensitive element of the four analytes. If more than one position provides the same ratio, select the position that provides the highest net intensity counts for the least sensitive element or accept a compromise position of the intensity ratios of all four analytes.
10.2.4 The instrument operating condition finally selected as being optimum should provide the lowest reliable instrument detection limits and method detection limits. Refer to Tables 1 and 4 for comparison of IDLs and MDLs, respectively.
10.2.5 If either the instrument operating conditions, such as incident power and/or nebulizer gas flow rate are changed, or a new torch injector tube having a different orifice i.d. is installed, the plasma and plasma viewing height should be reoptimized.
10.2.6 Before daily calibration and after the instrument warmup period, the nebulizer gas flow must be reset to the determined optimized flow. If a mass flow controller is being used, it should be reset to the recorded optimized flow rate. In order to maintain valid spectral interelement correction routines the nebulizer gas flow rate should be the same from day-to-day (<2% change). The change in signal intensity with a change in nebulizer gas flow rate for both "hard" (Pb 220.353 nm) and "soft" (Cu 324.754) lines is illustrated in Figure 1.
10.3 Before using the procedure (Section 11.0) to analyze samples, there must be data available documenting initial demonstration of performance. The required data and procedure is described in Section 9.2. This data must be generated using the same instrument operating conditions and calibration routine (Section 11.4) to be used for sample analysis. These documented data must be kept on file and be available for review by the data user.
10.4 After completing the initial demonstration of performance, but before analyzing samples, the laboratory must establish and initially verify an interelement spectral interference correction routine to be used during sample analysis. A general description concerning spectral interference and the analytical requirements for background correction and for correction of interelement spectral interference in particular are given in Section 4.1. To determine the appropriate location for background correction and to establish the interelement interference correction routine, repeated spectral scan about the analyte wavelength and repeated analyses of the single element solutions may be required. Criteria for determining an interelement spectral interference is an apparent positive or negative concentration on the analyte that is outside the 3-sigma control limits of the calibration blank for the analyte. (The upper-control limit is the analyte IDL.) Once established, the entire routine must be initially and periodically verified annually, or whenever there is a change in instrument operating conditions (Section 10.2.5). Only a portion of the correction routine must be verified more frequently or on a daily basis. Test criteria and required solutions are described in Section 7.13. Initial and periodic verification data of the routine should be kept on file. Special cases where on-going verification are required is described in Section 7.14.
11.0 Procedure
11.1 Aqueous Sample Preparation—Dissolved Analytes
11.1.1 For the determination of dissolved analytes in ground and surface waters, pipet an aliquot (20 mL) of the filtered, acid preserved sample into a 50 mL polypropylene centrifuge tube. Add an appropriate volume of (1 1) nitric acid to adjust the acid concentration of the aliquot to approximate a 1% (v/v) nitric acid solution (e.g., add 0.4 mL (1 1) HNO3 to a 20 mL aliquot of sample). Cap the tube and mix. The sample is now ready for analysis (Section 1.3). Allowance for sample dilution should be made in the calculations. (If mercury is to be determined, a separate aliquot must be additionally acidified to contain 1% (v/v) HCl to match the signal response of mercury in the calibration standard and reduce memory interference effects. Section 1.9).
Note: If a precipitate is formed during acidification, transport, or storage, the sample aliquot must be treated using the procedure described in Sections 11.2.2 through 11.2.7 prior to analysis.
11.2 Aqueous Sample Preparation—Total Recoverable Analytes
11.2.1 For the "direct analysis" of total recoverable analytes in drinking water samples containing turbidity <1 NTU, treat an unfiltered acid preserved sample aliquot using the sample preparation procedure described in Section 11.1.1 while making allowance for sample dilution in the data calculation (Section 1.2). For the determination of total recoverable analytes in all other aqueous samples or for preconcentrating drinking water samples prior to analysis follow the procedure given in Sections 11.2.2 through 11.2.7.
11.2.2 For the determination of total recoverable analytes in aqueous samples (other than drinking water with <1 NTU turbidity), transfer a 100 mL (1 mL) aliquot from a well mixed, acid preserved sample to a 250 mL Griffin beaker (Sections 1.2, 1.3, 1.6, 1.7, 1.8, and 1.9). (When necessary, smaller sample aliquot volumes may be used.)
Note: If the sample contains undissolved solids >1%, a well mixed, acid preserved aliquot containing no more than 1 g particulate material should be cautiously evaporated to near 10 mL and extracted using the acid-mixture procedure described in Sections 11.3.3 through 11.3.6.
11.2.3 Add 2 mL (1 1) nitric acid and 1.0 mL of (1 1) hydrochloric acid to the beaker containing the measured volume of sample. Place the beaker on the hot plate for solution evaporation. The hot plate should be located in a fume hood and previously adjusted to provide evaporation at a temperature of approximately but no higher than 85 °C. (See the following note.) The beaker should be covered with an elevated watch glass or other necessary steps should be taken to prevent sample contamination from the fume hood environment.
Note: For proper heating adjust the temperature control of the hot plate such that an uncovered Griffin beaker containing 50 mL of water placed in the center of the hot plate can be maintained at a temperature approximately but no higher than 85 °C. (Once the beaker is covered with a watch glass the temperature of the water will rise to approximately 95 °C.)
11.2.4 Reduce the volume of the sample aliquot to about 20 mL by gentle heating at 85°C. DO NOT BOIL. This step takes about two hours for a 100 mL aliquot with the rate of evaporation rapidly increasing as the sample volume approaches 20 mL. (A spare beaker containing 20 mL of water can be used as a gauge.)
11.2.5 Cover the lip of the beaker with a watch glass to reduce additional evaporation and gently reflux the sample for 30 minutes. (Slight boiling may occur, but vigorous boiling must be avoided to prevent loss of the HCl-H2O azeotrope.)
11.2.6 Allow the beaker to cool. Quantitatively transfer the sample solution to a 50 mL volumetric flask, make to volume with reagent water, stopper and mix.
11.2.7 Allow any undissolved material to settle overnight, or centrifuge a portion of the prepared sample until clear. (If after centrifuging or standing overnight the sample contains suspended solids that would clog the nebulizer, a portion of the sample may be filtered for their removal prior to analysis. However, care should be exercised to avoid potential contamination from filtration.) The sample is now ready for analysis. Because the effects of various matrices on the stability of diluted samples cannot be characterized, all analyses should be performed as soon as possible after the completed preparation.
11.3 Solid Sample Preparation—Total Recoverable Analytes
11.3.1 For the determination of total recoverable analytes in solid samples, mix the sample thoroughly and transfer a portion (>20 g) to tared weighing dish, weigh the sample and record the wet weight (WW). (For samples with <35% moisture a 20 g portion is sufficient. For samples with moisture >35% a larger aliquot 50-100 g is required.) Dry the sample to a constant weight at 60 °C and record the dry weight (DW) for calculation of percent solids (Section 12.6). (The sample is dried at 60 °C to prevent the loss of mercury and other possible volatile metallic compounds, to facilitate sieving, and to ready the sample for grinding.)
11.3.2 To achieve homogeneity, sieve the dried sample using a 5-mesh polypropylene sieve and grind in a mortar and pestle. (The sieve, mortar and pestle should be cleaned between samples.) From the dried, ground material weigh accurately a representative 1.0 ± 0.01 g aliquot (W) of the sample and transfer to a 250 mL Phillips beaker for acid extraction (Sections 1.6, 1.7, 1.8, and 1.9).
11.3.3 To the beaker add 4 mL of (1 1) HNO3 and 10 mL of (1 4) HCl. Cover the lip of the beaker with a watch glass. Place the beaker on a hot plate for reflux extraction of the analytes. The hot plate should be located in a fume hood and previously adjusted to provide a reflux temperature of approximately 95 °C. (See the following note.)
Note: For proper heating adjust the temperature control of the hot plate such that an uncovered Griffin beaker containing 50 mL of water placed in the center of the hot plate can be maintained at a temperature approximately but no higher than 85 °C. (Once the beaker is covered with a watch glass the temperature of the water will rise to approximately 95 °C.) Also, a block digester capable of maintaining a temperature of 95 °C and equipped with 250 mL constricted volumetric digestion tubes may be substituted for the hot plate and conical beakers in the extraction step.
11.3.4 Heat the sample and gently reflux for 30 minutes. Very slight boiling may occur, however vigorous boiling must be avoided to prevent loss of the HCl-H2O azeotrope. Some solution evaporation will occur (3-4 mL).
11.3.5 Allow the sample to cool and quantitatively transfer the extract to a 100 mL volumetric flask. Dilute to volume with reagent water, stopper and mix.
11.3.6 Allow the sample extract solution to stand overnight to separate insoluble material or centrifuge a portion of the sample solution until clear. (If after centrifuging or standing overnight the extract solution contains suspended solids that would clog the nebulizer, a portion of the extract solution may be filtered for their removal prior to analysis. However, care should be exercised to avoid potential contamination from filtration.) The sample extract is now ready for analysis. Because the effects of various matrices on the stability of diluted samples cannot be characterized, all analyses should be performed as soon as possible after the completed preparation.
11.4 Sample Analysis
11.4.1 Prior to daily calibration of the instrument inspect the sample introduction system including the nebulizer, torch, injector tube and uptake tubing for salt deposits, dirt and debris that would restrict solution flow and affect instrument performance. Clean the system when needed or on a daily basis.
11.4.2 Configure the instrument system to the selected power and operating conditions as determined in Sections 10.1 and 10.2.
11.4.3 The instrument must be allowed to become thermally stable before calibration and analyses. This usually requires at least 30 to 60 minutes of operation. After instrument warmup, complete any required optical profiling or alignment particular to the instrument.
11.4.4 For initial and daily operation calibrate the instrument according to the instrument manufacturer's recommended procedures, using mixed calibration standard solutions (Section 7.9) and the calibration blank (Section 7.10.1). A peristaltic pump must be used to introduce all solutions to the nebulizer. To allow equilibrium to be reached in the plasma, aspirate all solutions for 30 seconds after reaching the plasma before beginning integration of the background corrected signal to accumulate data. When possible, use the average value of replicate integration periods of the signal to be correlated to the analyte concentration. Flush the system with the rinse blank (Section 7.10.4) for a minimum of 60 seconds (Section 4.4) between each standard. The calibration line should consist of a minimum of a calibration blank and a high standard. Replicates of the blank and highest standard provide an optimal distribution of calibration standards to minimize the confidence band for a straight-line calibration in a response region with uniform variance.20
11.4.5 After completion of the initial requirements of this method (Sections 10.3 and 10.4), samples should be analyzed in the same operational manner used in the calibration routine with the rinse blank also being used between all sample solutions, LFBs, LFMs, and check solutions (Section 7.10.4).
11.4.6 During the analysis of samples, the laboratory must comply with the required quality control described in Sections 9.3 and 9.4. Only for the determination of dissolved analytes or the "direct analysis" of drinking water with turbidity of <1 NTU is the sample digestion step of the LRB, LFB, and LFM not required.
11.4.7 Determined sample analyte concentrations that are 90% or more of the upper limit of the analyte LDR must be diluted with reagent water that has been acidified in the same manner as calibration blank and reanalyzed (see Section 11.4.8). Also, for the interelement spectral interference correction routines to remain valid during sample analysis, the interferant concentration must not exceed its LDR. If the interferant LDR is exceeded, sample dilution with acidified reagent water and reanalysis is required. In these circumstances analyte detection limits are raised and determination by another approved test procedure that is either more sensitive and/or interference free is recommended.
11.4.8 When it is necessary to assess an operative matrix interference (e.g.,signal reduction due to high dissolved solids), the tests described in Section 9.5 are recommended.
11.4.9 Report data as directed in Section 12.0.
11.5 If the method of standard additions (MSA) is used, standards are added at one or more levels to portions of a prepared sample. This technique21 compensates for enhancement or depression of an analyte signal by a matrix. It will not correct for additive interferences such as contamination, interelement interferences, or baseline shifts. This technique is valid in the linear range when the interference effect is constant over the range, the added analyte responds the same as the endogenous analyte, and the signal is corrected for additive interferences. The simplest version of this technique is the single-addition method. This procedure calls for two identical aliquots of the sample solution to be taken. To the first aliquot, a small volume of standard is added; while to the second aliquot, a volume of acid blank is added equal to the standard addition. The sample concentration is calculated by the following:
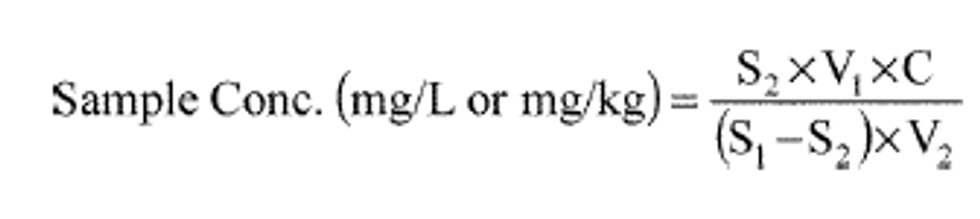
Where:
C = Concentration of the standard solution (mg/L)
S1 = Signal for fortified aliquot
S2 = Signal for unfortified aliquot
V1 = Volume of the standard addition (L)
V2 = Volume of the sample aliquot (L) used for MSA
For more than one fortified portion of the prepared sample, linear regression analysis can be applied using a computer or calculator program to obtain the concentration of the sample solution. An alternative to using the method of standard additions is use of the internal standard technique by adding one or more elements (not in the samples and verified not to cause an uncorrected interelement spectral interference) at the same concentration (which is sufficient for optimum precision) to the prepared samples (blanks and standards) that are affected the same as the analytes by the sample matrix. Use the ratio of analyte signal to the internal standard signal for calibration and quantitation.
12.0 Data Analysis and Calculations
12.1 Sample data should be reported in units of mg/L for aqueous samples and mg/kg dry weight for solid samples.
12.2 For dissolved aqueous analytes (Section 11.1) report the data generated directly from the instrument with allowance for sample dilution. Do not report analyte concentrations below the IDL.
12.3 For total recoverable aqueous analytes (Section 11.2), multiply solution analyte concentrations by the dilution factor 0.5, when 100 mL aliquot is used to produce the 50 mL final solution, and report data as instructed in Section 12.4. If a different aliquot volume other than 100 mL is used for sample preparation, adjust the dilution factor accordingly. Also, account for any additional dilution of the prepared sample solution needed to complete the determination of analytes exceeding 90% or more of the LDR upper limit. Do not report data below the determined analyte MDL concentration or below an adjusted detection limit reflecting smaller sample aliquots used in processing or additional dilutions required to complete the analysis.
12.4 For analytes with MDLs <0.01 mg/L, round the data values to the thousandth place and report analyte concentrations up to three significant figures. For analytes with MDLs <0.01 mg/L round the data values to the 100th place and report analyte concentrations up to three significant figures. Extract concentrations for solids data should be rounded in a similar manner before calculations in Section 12.5 are performed.
12.5 For total recoverable analytes in solid samples (Section 11.3), round the solution analyte concentrations (mg/L) as instructed in Section 12.4. Report the data up to three significant figures as mg/kg dry-weight basis unless specified otherwise by the program or data user. Calculate the concentration using the equation below:
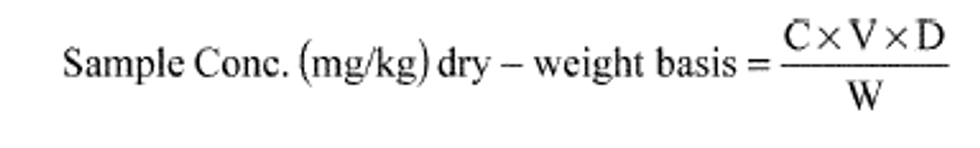
Where:
C = Concentration in extract (mg/L)
V = Volume of extract (L, 100 mL = 0.1L)
D = Dilution factor (undiluted = 1)
W = Weight of sample aliquot extracted (g x 0.001 = kg)
Do not report analyte data below the estimated solids MDL or an adjusted MDL because of additional dilutions required to complete the analysis.
12.6 To report percent solids in solid samples (Section 11.3) calculate as follows:
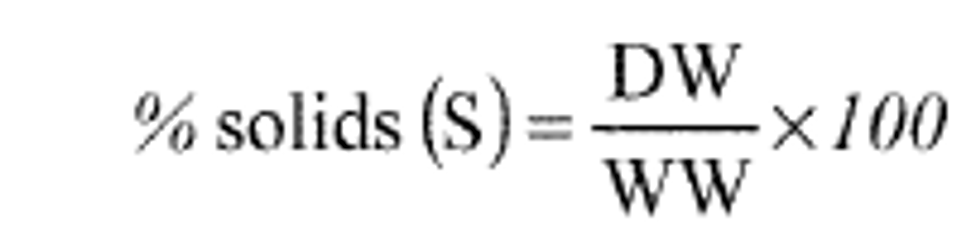
Where:
DW = Sample weight (g) dried at 60°C
WW = Sample weight (g) before drying
Note: If the data user, program or laboratory requires that the reported percent solids be determined by drying at 105 °C, repeat the procedure given in Section 11.3 using a separate portion (>20 g) of the sample and dry to constant weight at 103-105°C.
12.7 The QC data obtained during the analyses provide an indication of the quality of the sample data and should be provided with the sample results.
13.0 Method Performance
13.1 Listed in Table 4 are typical single laboratory total recoverable MDLs determined for the recommended wavelengths using simultaneous ICP-AES and the operating conditions given in Table 5. The MDLs were determined in reagent blank matrix (best case situation). PTFE beakers were used to avoid boron and silica contamination from glassware with the final dilution to 50 mL completed in polypropylene centrifuged tubes. The listed MDLs for solids are estimates and were calculated from the aqueous MDL determinations.
13.2 Data obtained from single laboratory method testing are summarized in Table 6 for five types of water samples consisting of drinking water, surface water, ground water, and two wastewater effluents. The data presented cover all analytes except cerium and titanium. Samples were prepared using the procedure described in Section 11.2. For each matrix, five replicate aliquots were prepared, analyzed and the average of the five determinations used to define the sample background concentration of each analyte. In addition, two pairs of duplicates were fortified at different concentration levels. For each method analyte, the sample background concentration, mean percent recovery, standard deviation of the percent recovery, and relative percent difference between the duplicate fortified samples are listed in Table 6. The variance of the five replicate sample background determinations is included in the calculated standard deviation of the percent recovery when the analyte concentration in the sample was greater than the MDL. The tap and well waters were processed in Teflon and quartz beakers and diluted in polypropylene centrifuged tubes. The nonuse of borosilicate glassware is reflected in the precision and recovery data for boron and silica in those two sample types.
13.3 Data obtained from single laboratory method testing are summarized in Table 7 for three solid samples consisting of EPA 884 Hazardous Soil, SRM 1645 River Sediment, and EPA 286 Electroplating Sludge. Samples were prepared using the procedure described in Section 11.3. For each method analyte, the sample background concentration, mean percent recovery of the fortified additions, the standard deviation of the percent recovery, and relative percent difference between duplicate additions were determined as described in Section 13.2. Data presented are for all analytes except cerium, silica, and titanium. Limited comparative data to other methods and SRM materials are presented in Reference 23 of Section 16.0.
13.4 Performance data for aqueous solutions independent of sample preparation from a multilaboratory study are provided in Table 8.22
13.5 Listed in Table 9 are regression equations for precision and bias for 25 analytes abstracted from EPA Method Study 27, a multilaboratory validation study of Method 200.7.1These equations were developed from data received from 12 laboratories using the total recoverable sample preparation procedure on reagent water, drinking water, surface water and three industrial effluents. For a complete review and description of the study, see Reference 16 of Section 16.0.
14.0 Pollution Prevention
14.1 Pollution prevention encompasses any technique that reduces or eliminates the quantity or toxicity of waste at the point of generation. Numerous opportunities for pollution prevention exist in laboratory operation. The EPA has established a preferred hierarchy of environmental management techniques that places pollution prevention as the management option of first choice. Whenever feasible, laboratory personnel should use pollution prevention techniques to address their waste generation (e.g., Section 7.8). When wastes cannot be feasibly reduced at the source, the Agency recommends recycling as the next best option.
14.2 For information about pollution prevention that may be applicable to laboratories and research institutions, consult "Less is Better: Laboratory Chemical Management for Waste Reduction", available from the American Chemical Society's Department of Government Relations and Science Policy, 1155 16th Street NW., Washington, DC 20036, (202) 872-4477.
15.0 Waste Management
15.1 The Environmental Protection Agency requires that laboratory waste management practices be conducted consistent with all applicable rules and regulations. The Agency urges laboratories to protect the air, water, and land by minimizing and controlling all releases from hoods and bench operations, complying with the letter and spirit of any sewer discharge permits and regulations, and by complying with all solid and hazardous waste regulations, particularly the hazardous waste identification rules and land disposal restrictions. For further information on waste management consult "The Waste Management Manual for Laboratory Personnel", available from the American Chemical Society at the address listed in the Section 14.2.
16.0 References
- U.S. Environmental Protection Agency. Inductively Coupled Plasma—Atomic Emission Spectrometric Method for Trace Element Analysis of Water and Wastes—Method 200.7, Dec. 1982. EPA-600/4-79-020, revised March 1983.
- U.S. Environmental Protection Agency. Inductively Coupled Plasma Atomic Emission Spectroscopy Method 6010, SW- 846 Test Methods for Evaluating Solid Waste, 3rd Edition, 1986.
- U.S. Environmental Protection Agency. Method 200.7: Determination of Metals and Trace Elements in Water and Wastes by Inductively Coupled Plasma- Atomic Emission Spectrometry, revision 3.3, EPA 600 4-91/010, June 1991.
- U.S. Environmental Protection Agency. Inductively Coupled Plasma—Atomic Emission Spectrometry Method for the Analysis of Waters and Solids, EMMC, July 1992.
- Fassel, V.A. et al. Simultaneous Determination of Wear Metals in Lubricating Oils by Inductively-Coupled Plasma Atomic Emission Spectrometry. Anal. Chem. 48:516-519, 1976.
- Merryfield, R.N. and R.C. Loyd. Simultaneous Determination of Metals in Oil by Inductively Coupled Plasma Emission Spectrometry. Anal. Chem. 51:1965-1968, 1979.
- Winge, R.K. et al. Inductively Coupled Plasma—Atomic Emission Spectroscopy: An Atlas of Spectral Information, Physical Science Data 20. Elsevier Science Publishing, New York, New York, 1985.
- Boumans, P.W.J.M. Line Coincidence Tables for Inductively Coupled Plasma Atomic Emission Spectrometry, 2nd edition. Pergamon Press, Oxford, United Kingdom, 1984.
- Carcinogens—Working With Carcinogens, Department of Health, Education, and Welfare, Public Health Service, Center for Disease Control, National Institute for Occupational Safety and Health, Publication No. 77-206, Aug. 1977. Available from the National Technical Information Service (NTIS) as PB-277256.
- OSHA Safety and Health Standards, General Industry, (29 CFR 1910), Occupational Safety and Health Administration, OSHA 2206, (Revised, January 1976).
- Safety in Academic Chemistry Laboratories, American Chemical Society Publication, Committee on Chemical Safety, 3rd Edition, 1979.
- Proposed OSHA Safety and Health Standards, Laboratories, Occupational Safety and Health Administration, Federal Register, July 24, 1986.
- Rohrbough, W.G. et al. Reagent Chemicals, American Chemical Society Specifications, 7th edition. American Chemical Society, Washington, DC, 1986.
- American Society for Testing and Materials. Standard Specification for Reagent Water, D1193-77. Annual Book of ASTM Standards, Vol. 11.01. Philadelphia, PA, 1991.
- Code of Federal Regulations 40, Ch. 1, Pt. 136 Appendix B.
- Maxfield, R. and B. Mindak. EPA Method Study 27, Method 200.7 Trace Metals by ICP, Nov. 1983. Available from National Technical Information Service (NTIS) as PB 85-248-656.
- Botto, R.I. Quality Assurance in Operating a Multielement ICP Emission Spectrometer. Spectrochim. Acta, 39B(1):95-113, 1984.
- Wallace, G.F., Some Factors Affecting the Performance of an ICP Sample Introduction System. Atomic Spectroscopy, Vol. 4, p. 188-192, 1983.
- Koirtyohann, S.R. et al. Nomenclature System for the Low-Power Argon Inductively Coupled Plasma, Anal. Chem. 52:1965, 1980.
- Deming, S.N. and S.L. Morgan. Experimental Design for Quality and Productivity in Research, Development, and Manufacturing, Part III, pp. 119-123. Short course publication by Statistical Designs, 9941 Rowlett, Suite 6, Houston, TX 77075, 1989.
- Winefordner, J.D., Trace Analysis: Spectroscopic Methods for Elements, Chemical Analysis, Vol. 46, pp. 41-42.
- Jones, C.L. et al. An Interlaboratory Study of Inductively Coupled Plasma Atomic Emission Spectroscopy Method 6010 and Digestion Method 3050. EPA-600/ 4-87-032, U.S. Environmental Protection Agency, Las Vegas, Nevada, 1987.
- Martin, T.D., E.R. Martin and SE. Long. Method 200.2: Sample Preparation Procedure for Spectrochemical Analyses of Total Recoverable Elements, EMSL ORD, USEPA, 1989.
17.0 Tables, Diagrams, Flowcharts, and Validation Data
| Analyte | Wavelengtha (nm) | Estimated detection limitb (μg/L) | Calibratec to (mg/L) |
| Aluminum | 308.215 | 45 | 10 |
| Antimony | 206.833 | 32 | 5 |
| Arsenic | 193.759 | 53 | 10 |
| Barium | 493.409 | 2.3 | 1 |
| Beryllium | 313.042 | 0.27 | 1 |
| Boron | 249.678 | 5.7 | 1 |
| Cadmium | 226.502 | 3.4 | 2 |
| Calcium | 315.887 | 30 | 10 |
| Cerium | 413.765 | 48 | 2 |
| Chromium | 205.552 | 6.1 | 5 |
| Cobalt | 228.616 | 7.0 | 2 |
| Copper | 324.754 | 5.4 | 2 |
| Iron | 259.940 | 6.2 | 10 |
| Lead | 220.353 | 42 | 10 |
| Lithium | 670.784 | d3.7 | 5 |
| Magnesium | 279.079 | 30 | 10 |
| Manganese | 257.610 | 1.4 | 2 |
| Mercury | 194.227 | 2.5 | 2 |
| Molybdenum | 203.844 | 12 | 10 |
| Nickel | 231.604 | 15 | 2 |
| Phosphorus | 214.914 | 76 | 10 |
| Potassium | 766.491 | e700 | 20 |
| Selenium | 196.090 | 75 | 5 |
| Silica (SiO2) | 251.611 | d26 (SiO2) | 10 |
| Silver | 328.068 | 7.0 | 0.5 |
| Sodium | 588.995 | 29 | 10 |
| Strontium | 421.552 | 0.77 | 1 |
| Thallium | 190.864 | 40 | 5 |
| Tin | 189.980 | 25 | 4 |
| Titanium | 334.941 | 3.8 | 10 |
| Vanadium | 292.402 | 7.5 | 2 |
| Zinc | 213.856 | 1.8 | 5 |
aThe wavelengths listed are recommended because of their sensitivity and overall acceptability. Other wavelengths may be substituted if they can provide the needed sensitivity and are treated with the same corrective techniques for spectral interference (see Section 4.1).
bThese estimated 3-sigma instrumental detection limits16 are provided only as a guide to instrumental limits. The method detection limits are sample dependent and may vary as the sample matrix varies. Detection limits for solids can be estimated by dividing these values by the grams extracted per liter, which depends upon the extraction procedure. Divide solution detection limits by 10 for 1 g extracted to 100 mL for solid detection limits.
cSuggested concentration for instrument calibration.2 Other calibration limits in the linear ranges may be used.
dCalculated from 2-sigma data.5
eHighly dependent on operating conditions and plasma position.
| Analyte | Wavelength (nm) | Interferant* |
| Ag | 328.068 | Ce, Ti, Mn |
| Al | 308.215 | V, Mo, Ce, Mn |
| B | 193.759 | V, Al, Co, Fe, Ni |
| Ba | 249.678 | None |
| Be | 493.409 | None |
| Ca | 313.042 | V, Ce |
| Cd | 315.887 | Co, Mo, Ce |
| Ce | 226.502 | Ni, Ti, Fe, Ce |
| Co | 228.616 | None |
| Cr | 205.552 | Ti, Ba, Cd, Ni, Cr, Mo, Ce |
| Cu | 324.754 | Be, Mo, Ni |
| Fe | 259.940 | Mo, Ti |
| Hg | 194.227 | None |
| K | 766.941 | V, Mo |
| Li | 670.784 | None |
| Mg | 279.079 | None |
| Mn | 257.610 | Ce |
| Mo | 203.844 | Ce |
| Na | 588.995 | Ce |
| Ni | 231.604 | None |
| P | 214.914 | Co, Tl |
| Pb | 220.353 | Cu, Mo |
| Sb | 206.833 | Co, Al, Ce, Cu, Ni, Ti, Fe |
| Se | 196.099 | Cr, Mo, Sn, Ti, Ce, Fe |
| SiO2 | 251.611 | Fe |
| Sn | 189.980 | None |
| Sr | 421.552 | Mo, Ti, Fe, Mn, Si |
| Tl | 190.864 | Ti, Mo, Co, Ce, Al, V, Mn |
| Ti | 334.941 | None |
| V | 292.402 | Ti, Mo, Co, Ce Al, V, Mn |
| Zn | 213.856 | Ni, Cu, Fe |
* These on-line interferences from method analytes and titanium only were observed using an instrument with 0.035 nm resolution (see Section 4.1.2). Interferant ranked by magnitude of intensity with the most severe interferant listed first in the row.
| Solution | Analytes |
| I | Ag, As, B, Ba, Ca, Cd, Cu, Mn, Sb, and Se |
| II | K, Li, Mo, Na, Sr, and Ti |
| III | Co, P, V, and Ce |
| IV | Al, Cr, Hg, SiO2, Sn, Zn |
| V | Be, Fe, Mg, Ni, Pb, and Tl |
| Analyte | MDLs Aqueous, mg/L(1) | Solids, mg/kg(2) |
| Ag | 0.002 | 0.3 |
| Al | 0.02 | 3 |
| As | 0.008 | 2 |
| B | 0.003 | - |
| Ba | 0.001 | 0.2 |
| Be | 0.0003 | 0.1 |
| Ca | 0.01 | 2 |
| Cd | 0.001 | 0.2 |
| Ce | 0.02 | 3 |
| Co | 0.002 | 0.4 |
| Cr | 0.004 | 0.8 |
| Cu | 0.003 | 0.5 |
| Fe | *0.03 | 6 |
| Hg | 0.007 | 2 |
| K | 0.3 | 6 |
| Li | 0.001 | 0.2 |
| Mg | 0.02 | 3 |
| Mn | 0.001 | 0.2 |
| Mo | 0.004 | 1 |
| Na | 0.03 | 6 |
| Ni | 0.005 | 1 |
| P | 0.06 | 12 |
| Pb | 0.01 | 2 |
| Sb | 0.008 | 2 |
| Se | 0.02 | 5 |
| SiO2 | 0.02 | - |
| Sn | 0.007 | 2 |
| Sr | 0.0003 | 0.1 |
| Tl | 0.001 | 0.2 |
| Ti | 0.02 | 3 |
| V | 0.003 | 1 |
| Zn | 0.002 | 0.3 |
| (1)MDL concentrations are computed for original matrix with allowance for 2x sample preconcentration during preparation. Samples were processed in PTFE and diluted in 50-mL plastic centrifuge tubes. | ||
| (2)Estimated, calculated from aqueous MDL determinations. | ||
| —Boron not reported because of glassware contamination. Silica not determined in solid samples. | ||
| * Elevated value due to fume-hood contamination. | ||
| Incident rf power | 1100 watts |
| Reflected rf power | <5 watts |
| Viewing height above work coil | 15 mm |
| Injector tube orifice i.d. | 1 mm |
| Argon supply | liquid argon |
| Argon pressure | 40 psi |
| Coolant argon flow rate | 19 L/min. |
| Aerosol carrier argon flow rate | 620 mL/min. |
| Auxiliary (plasma) argon flow rate | 300 mL/min. |
| Sample uptake rate controlled to | 1.2 mL/min. |
| Analyte | Sample conc. mg/L | Low spike mg/L | Average recovery R (%) | S (R) | RPD | High spike mg/L | Average recovery R (%) | S (R) | RPD |
| Tap Water | |||||||||
| Ag | <0.002 | 0.05 | 95 | 0.7 | 2.1 | 0.2 | 96 | 0.0 | 0.0 |
| Al | 0.185 | 0.05 | 98 | 8.8 | 1.7 | 0.2 | 105 | 3.0 | 3.1 |
| As | <0.008 | 0.05 | 108 | 1.4 | 3.7 | 0.2 | 101 | 0.7 | 2.0 |
| B | 0.023 | 0.1 | 98 | 0.2 | 0.0 | 0.4 | 98 | 0.2 | 0.5 |
| Ba | 0.042 | 0.05 | 102 | 1.6 | 2.2 | 0.2 | 98 | 0.4 | 0.8 |
| Be | <0.0003 | 0.01 | 100 | 0.0 | 0.0 | 0.1 | 99 | 0.0 | 0.0 |
| Ca | 35.2 | 5.0 | 101 | 8.8 | 1.7 | 20.0 | 103 | 2.0 | 0.9 |
| Cd | <0.001 | 0.01 | 105 | 3.5 | 9.5 | 0.1 | 98 | 0.0 | 0.0 |
| Co | <0.002 | 0.02 | 100 | 0.0 | 0.0 | 0.2 | 99 | 0.5 | 1.5 |
| Cr | <0.004 | 0.01 | 110 | 0.0 | 0.0 | 0.1 | 102 | 0.0 | 0.0 |
| Cu | <0.003 | 0.02 | 103 | 1.8 | 4.9 | 0.2 | 101 | 1.2 | 3.5 |
| Fe | 0.008 | 0.1 | 106 | 1.0 | 1.8 | 0.4 | 105 | 0.3 | 0.5 |
| Hg | <0.007 | 0.05 | 103 | 0.7 | 1.9 | 0.2 | 100 | 0.4 | 1.0 |
| K | 1.98 | 5.0 | 109 | 1.4 | 2.3 | 20. | 107 | 0.7 | 1.7 |
| Li | 0.006 | 0.02 | 103 | 6.9 | 3.8 | 0.2 | 110 | 1.9 | 4.4 |
| Mg | 8.08 | 5.0 | 104 | 2.2 | 1.5 | 20.0 | 100 | 0.7 | 1.1 |
| Mn | <0.001 | 0.01 | 100 | 0.0 | 0.0 | 0.1 | 99 | 0.0 | 0.0 |
| Mo | <0.004 | 0.02 | 95 | 3.5 | 10.5 | 0.2 | 108 | 0.5 | 1.4 |
| Na | 10.3 | 5.0 | 99 | 3.0 | 2.0 | 20.0 | 106 | 1.0 | 1.6 |
| Ni | <0.005 | 0.02 | 108 | 1.8 | 4.7 | 0.2 | 104 | 1.1 | 2.9 |
| P | 0.045 | 0.1 | 102 | 13.1 | 9.4 | 0.4 | 104 | 3.2 | 1.3 |
| Pb | <0.01 | 0.05 | 95 | 0.7 | 2.1 | 0.2 | 100 | 0.2 | 0.5 |
| Sb | <0.008 | 0.05 | 99 | 0.7 | 2.0 | 0.2 | 102 | 0.7 | 2.0 |
| Se | <0.02 | 0.1 | 87 | 1.1 | 3.5 | 0.4 | 99 | 0.8 | 2.3 |
| SiO2 | 6.5 | 5.0 | 104 | 3.3 | 3.4 | 20.0 | 96 | 1.1 | 2.3 |
| Sn | <0.007 | 0.05 | 103 | 2.1 | 5.8 | 0.2 | 101 | 1.8 | 5.0 |
| Sr | 0.181 | 0.1 | 102 | 3.3 | 2.1 | 0.4 | 105 | 0.8 | 1.0 |
| Tl | <0.02 | 0.1 | 101 | 3.9 | 10.9 | 0.4 | 101 | 0.1 | 0.3 |
| V | <0.003 | 0.05 | 101 | 0.7 | 2.0 | 0.2 | 99 | 0.2 | 0.5 |
| Zn | 0.005 | 0.05 | 101 | 3.7 | 9.0 | 0.2 | 98 | 0.9 | 2.5 |
| Pond Water | |||||||||
| Ag | <0.002 | 0.05 | 92 | 0.0 | 0.0 | 0.2 | 94 | 0.0 | 0.0 |
| Al | 0.819 | 0.2 | 88 | 10.0 | 5.0 | 0.8 | 100 | 2.9 | 3.7 |
| As | <0.008 | 0.05 | 102 | 0.0 | 0.0 | 0.2 | 98 | 1.4 | 4.1 |
| B | 0.034 | 0.1 | 111 | 8.9 | 6.9 | 0.4 | 103 | 2.0 | 0.0 |
| Ba | 0.029 | 0.05 | 96 | 0.9 | 0.0 | 0.2 | 97 | 0.3 | 0.5 |
| Be | <0.0003 | 0.01 | 95 | 0.4 | 1.1 | 0.2 | 95 | 0.0 | 0.0 |
| Ca | 53.9 | 5.0 | * | * | 0.7 | 20.0 | 100 | 2.0 | 1.5 |
| Cd | <0.001 | 0.01 | 107 | 0.0 | 0.0 | 0.1 | 97 | 0.0 | 0.0 |
| Co | <0.002 | 0.02 | 100 | 2.7 | 7.5 | 0.2 | 97 | 0.7 | 2.1 |
| Cr | <0.004 | 0.01 | 105 | 3.5 | 9.5 | 0.1 | 103 | 1.1 | 2.9 |
| Cu | <0.003 | 0.02 | 98 | 2.1 | 4.4 | 0.2 | 100 | 0.5 | 1.5 |
| Fe | 0.875 | 0.2 | 95 | 8.9 | 2.8 | 0.8 | 97 | 3.2 | 3.6 |
| Hg | <0.007 | 0.05 | 97 | 3.5 | 10.3 | 0.2 | 98 | 0.0 | 0.0 |
| K | 2.48 | 5.0 | 106 | 0.3 | 0.1 | 20.0 | 103 | 0.2 | 0.4 |
| Li | <0.001 | 0.02 | 110 | 0.0 | 0.0 | 0.2 | 106 | 0.2 | 0.5 |
| Mg | 10.8 | 5.0 | 102 | 0.5 | 0.0 | 20.0 | 96 | 0.7 | 1.3 |
| Mn | 0.632 | 0.01 | * | * | 0.2 | 0.1 | 97 | 2.3 | 0.3 |
| Mo | <0.004 | 0.02 | 105 | 3.5 | 9.5 | 0.2 | 103 | 0.4 | 1.0 |
| Na | 17.8 | 5.0 | 103 | 1.3 | 0.4 | 20.0 | 94 | 0.3 | 0.0 |
| Ni | <0.005 | 0.02 | 96 | 5.6 | 9.1 | 0.2 | 100 | 0.7 | 1.5 |
| P | 0.196 | 0.1 | 91 | 14.7 | 0.3 | 0.4 | 108 | 3.9 | 1.3 |
| Pb | <0.01 | 0.05 | 96 | 2.6 | 7.8 | 0.2 | 100 | 0.7 | 2.0 |
| Sb | <0.008 | 0.05 | 102 | 2.8 | 7.8 | 0.2 | 104 | 0.4 | 1.0 |
| Se | <0.02 | 0.1 | 104 | 2.1 | 5.8 | 0.4 | 103 | 1.6 | 4.4 |
| SiO2 | 7.83 | 5.0 | 151 | 1.6 | 1.3 | 20.0 | 117 | 0.4 | 0.6 |
| Sn | <0.007 | 0.05 | 98 | 0.0 | 0.0 | 0.2 | 99 | 1.1 | 3.0 |
| Sr | 0.129 | 0.1 | 105 | 0.4 | 0.0 | 0.4 | 99 | 0.1 | 0.2 |
| Tl | <0.02 | 0.1 | 103 | 1.1 | 2.9 | 0.4 | 97 | 1.3 | 3.9 |
| V | 0.003 | 0.05 | 94 | 0.4 | 0.0 | 0.2 | 98 | 0.1 | 0.0 |
| Zn | 0.006 | 0.05 | 97 | 1.6 | 1.8 | 0.2 | 94 | 0.4 | 0.0 |
| Well Water | |||||||||
| Ag | <0.002 | 0.05 | 97 | 0.7 | 2.1 | 0.2 | 96 | 0.2 | 0.5 |
| Al | 0.036 | 0.05 | 107 | 7.6 | 10.1 | 0.2 | 101 | 1.1 | 0.8 |
| As | <0.008 | 0.05 | 107 | 0.7 | 1.9 | 0.2 | 104 | 0.4 | 1.0 |
| B | 0.063 | 0.1 | 97 | 0.6 | 0.7 | 0.4 | 98 | 0.8 | 2.1 |
| Ba | 0.102 | 0.05 | 102 | 3.0 | 0.0 | 0.2 | 99 | 0.9 | 1.0 |
| Be | <0.0003 | 0.01 | 100 | 0.0 | 0.0 | 0.1 | 100 | 0.0 | 0.0 |
| Ca | 93.8 | 5.0 | * | * | 2.1 | 20.0 | 100 | 4.1 | 0.1 |
| Cd | 0.002 | 0.01 | 90 | 0.0 | 0.0 | 0.1 | 96 | 0.0 | 0.0 |
| Co | <0.002 | 0.02 | 94 | 0.4 | 1.1 | 0.2 | 94 | 0.4 | 1.1 |
| Cr | <0.004 | 0.01 | 100 | 7.1 | 20.0 | 0.1 | 100 | 0.4 | 1.0 |
| Cu | <0.005 | 0.02 | 100 | 1.1 | 0.4 | 0.2 | 96 | 0.5 | 1.5 |
| Fe | 0.042 | 0.1 | 99 | 2.3 | 1.4 | 0.4 | 97 | 1.4 | 3.3 |
| Hg | <0.007 | 0.05 | 94 | 2.8 | 8.5 | 0.2 | 93 | 1.2 | 3.8 |
| K | 6.21 | 5.0 | 96 | 3.4 | 3.6 | 20.0 | 101 | 1.2 | 2.3 |
| Li | 0.001 | 0.02 | 100 | 7.6 | 9.5 | 0.2 | 104 | 1.0 | 1.9 |
| Mg | 24.5 | 5.0 | 95 | 5.6 | 0.3 | 20.0 | 93 | 1.6 | 1.2 |
| Mn | 2.76 | 0.01 | * | * | 0.4 | 0.1 | * | * | 0.7 |
| Mo | <0.004 | 0.02 | 108 | 1.8 | 4.7 | 0.2 | 101 | 0.2 | 0.5 |
| Na | 35.0 | 5.0 | 101 | 11.4 | 0.8 | 20.0 | 100 | 3.1 | 1.5 |
| Ni | <0.005 | 0.02 | 112 | 1.8 | 4.4 | 0.2 | 96 | 0.2 | 0.5 |
| P | 0.197 | 0.1 | 95 | 12.7 | 1.9 | 0.4 | 98 | 3.4 | 0.9 |
| Pb | <0.01 | 0.05 | 87 | 4.9 | 16.1 | 0.2 | 95 | 0.2 | 0.5 |
| Sb | <0.008 | 0.05 | 98 | 2.8 | 8.2 | 0.2 | 99 | 1.4 | 4.0 |
| Se | <0.02 | 0.1 | 102 | 0.4 | 1.0 | 0.4 | 94 | 1.1 | 3.4 |
| SiO2 | 13.1 | 5.0 | 93 | 4.8 | 2.8 | 20.0 | 99 | 0.8 | 0.0 |
| Sn | <0.007 | 0.05 | 98 | 2.8 | 8.2 | 0.2 | 94 | 0.2 | 0.5 |
| Sr | 0.274 | 0.1 | 94 | 5.7 | 2.7 | 0.4 | 95 | 1.7 | 2.2 |
| Tl | <0.02 | 0.1 | 92 | 0.4 | 1.1 | 0.4 | 95 | 1.1 | 3.2 |
| V | <0.003 | 0.05 | 98 | 0.0 | 0.0 | 0.2 | 99 | 0.4 | 1.0 |
| Zn | 0.538 | 0.05 | * | * | 0.7 | 0.2 | 99 | 2.5 | 1.1 |
| Sewage Treatment Effluent | |||||||||
| Ag | 0.009 | 0.05 | 92 | 1.5 | 3.6 | 0.2 | 95 | 0.1 | 0.0 |
| Al | 1.19 | 0.05 | * | * | 0.9 | 0.2 | 113 | 12.4 | 2.1 |
| As | <0.008 | 0.05 | 99 | 2.1 | 6.1 | 0.2 | 93 | 2.1 | 6.5 |
| B | 0.226 | 0.1 | 217 | 16.3 | 9.5 | 0.4 | 119 | 13.1 | 20.9 |
| Ba | 0.189 | 0.05 | 90 | 6.8 | 1.7 | 0.2 | 99 | 1.6 | 0.5 |
| Be | <0.0003 | 0.01 | 94 | 0.4 | 1.1 | 0.1 | 100 | 0.4 | 1.0 |
| Ca | 87.9 | 5.0 | * | * | 0.6 | 20.0 | 101 | 3.7 | 0.0 |
| Cd | 0.009 | 0.01 | 89 | 2.6 | 2.3 | 0.1 | 97 | 0.4 | 1.0 |
| Co | 0.016 | 0.02 | 95 | 3.1 | 0.0 | 0.2 | 93 | 0.4 | 0.5 |
| Cr | 0.128 | 0.01 | * | * | 1.5 | 0.1 | 97 | 2.4 | 2.7 |
| Cu | 0.174 | 0.02 | 98 | 33.1 | 4.7 | 0.2 | 98 | 3.0 | 1.4 |
| Fe | 1.28 | 0.1 | * | * | 2.8 | 0.4 | 111 | 7.0 | 0.6 |
| Hg | <0.007 | 0.05 | 102 | 1.4 | 3.9 | 0.2 | 98 | 0.5 | 1.5 |
| K | 10.6 | 5.0 | 104 | 2.8 | 1.3 | 20.0 | 101 | 0.6 | 0.0 |
| Li | 0.011 | 0.02 | 103 | 8.5 | 3.2 | 0.2 | 105 | 0.8 | 0.5 |
| Mg | 22.7 | 5.0 | 100 | 4.4 | 0.0 | 20.0 | 92 | 1.1 | 0.2 |
| Mn | 0.199 | 0.01 | * | * | 2.0 | 0.1 | 104 | 1.9 | 0.3 |
| Mo | 0.125 | 0.02 | 110 | 21.2 | 6.8 | 0.2 | 102 | 1.3 | 0.9 |
| Na | 0.236 | 5.0 | * | * | 0.0 | 20.0 | * | * | 0.4 |
| Ni | 0.087 | 0.02 | 122 | 10.7 | 4.5 | 0.2 | 98 | 0.8 | 1.1 |
| P | 4.71 | 0.1 | * | * | 2.6 | 0.4 | * | * | 1.4 |
| Pb | 0.015 | 0.05 | 91 | 3.5 | 5.0 | 0.2 | 96 | 1.3 | 2.9 |
| Sb | <0.008 | 0.05 | 97 | 0.7 | 2.1 | 0.2 | 103 | 1.1 | 2.9 |
| Se | <0.02 | 0.1 | 108 | 3.9 | 10.0 | 0.4 | 101 | 2.6 | 7.2 |
| SiO2 | 16.7 | 5.0 | 124 | 4.0 | 0.9 | 20.0 | 108 | 1.1 | 0.8 |
| Sn | 0.016 | 0.05 | 90 | 3.8 | 0.0 | 0.2 | 95 | 1.0 | 0.0 |
| Sr | 0.515 | 0.1 | 103 | 6.4 | 0.5 | 0.4 | 96 | 1.6 | 0.2 |
| Tl | <0.02 | 0.1 | 105 | 0.4 | 1.0 | 0.4 | 95 | 0.0 | 0.0 |
| V | 0.003 | 0.05 | 93 | 0.9 | 2.0 | 0.2 | 97 | 0.2 | 0.5 |
| Zn | 0.160 | 0.05 | 98 | 3.3 | 1.9 | 0.2 | 101 | 1.0 | 1.4 |
| Industrial Effluent | |||||||||
| Ag | <0.0003 | 0.05 | 88 | 0.0 | 0.0 | 0.2 | 84 | 0.9 | 3.0 |
| Al | 0.054 | 0.05 | 88 | 11.7 | 12.2 | 0.2 | 90 | 3.9 | 8.1 |
| As | <0.02 | 0.05 | 82 | 2.8 | 9.8 | 0.2 | 88 | 0.5 | 1.7 |
| B | 0.17 | 0.1 | 162 | 17.6 | 13.9 | 0.4 | 92 | 4.7 | 9.3 |
| Ba | 0.083 | 0.05 | 86 | 8.2 | 1.6 | 0.2 | 85 | 2.3 | 2.4 |
| Be | <0.0006 | 0.01 | 94 | 0.4 | 1.1 | 0.1 | 82 | 1.4 | 4.9 |
| Ca | 500 | 5.0 | * | * | 2.8 | 20.0 | * | * | 2.3 |
| Cd | 0.008 | 0.01 | 85 | 4.7 | 6.1 | 0.1 | 82 | 1.4 | 4.4C |
| Co | <0.004 | 0.02 | 93 | 1.8 | 5.4 | 0.2 | 83 | 0.4 | 1.2 |
| Cr | 0.165 | 0.01 | * | * | 4.5 | 0.1 | 106 | 6.6 | 5.6 |
| Cu | 0.095 | 0.02 | 93 | 23.3 | 0.9 | 0.2 | 95 | 2.7 | 2.8 |
| Fe | 0.315 | 0.1 | 88 | 16.4 | 1.0 | 0.4 | 99 | 6.5 | 8.0 |
| Hg | <0.01 | 0.05 | 87 | 0.7 | 2.3 | 0.2 | 86 | 0.4 | 1.2 |
| K | 2.87 | 5.0 | 101 | 3.4 | 2.4 | 20.0 | 100 | 0.8 | 0.4 |
| Li | 0.069 | 0.02 | 103 | 24.7 | 5.6 | 0.2 | 104 | 2.5 | 2.2 |
| Mg | 6.84 | 5.0 | 87 | 3.1 | 0.0 | 20.0 | 87 | 0.9 | 1.2 |
| Mn | 0.141 | 0.01 | * | * | 1.2 | 0.1 | 89 | 6.6 | 4.8 |
| Mo | 1.27 | 0.02 | * | * | 0.0 | 0.2 | 100 | 15.0 | 2.7 |
| Na | 1500 | 5.0 | * | * | 2.7 | 20.0 | * | * | 2.0 |
| Ni | 0.014 | 0.02 | 98 | 4.4 | 3.0 | 0.2 | 87 | 0.5 | 1.1 |
| P | 0.326 | 0.1 | 105 | 16.0 | 4.7 | 0.4 | 97 | 3.9 | 1.4 |
| Pb | 0.251 | 0.05 | 80 | 19.9 | 1.4 | 0.2 | 88 | 5.0 | 0.9 |
| Sb | 2.81 | 0.05 | * | * | 0.4 | 0.2 | * | * | 2.0 |
| Se | 0.021 | 0.1 | 106 | 2.6 | 3.2 | 0.4 | 105 | 1.9 | 4.6 |
| SiO2 | 6.83 | 5.0 | 99 | 6.8 | 1.7 | 20.0 | 100 | 2.2 | 3.0 |
| Sn | <0.01 | 0.05 | 87 | 0.7 | 2.3 | 0.2 | 86 | 0.4 | 1.2 |
| Sr | 6.54 | 0.1 | * | * | 2.0 | 0.4 | * | * | 2.7 |
| Tl | <0.03 | 0.1 | 87 | 1.8 | 5.8 | 0.4 | 84 | 1.1 | 3.6 |
| V | <0.005 | 0.05 | 90 | 1.4 | 4.4 | 0.2 | 84 | 1.1 | 3.6 |
| Zn | 0.024 | 0.05 | 89 | 6.0 | 4.4 | 0.2 | 91 | 3.5 | 8.9 |
| S (R) Standard deviation of percent recovery. | |||||||||
| RPD Relative percent difference between duplicate spike determinations. | |||||||||
| < Sample concentration below established method detection limit. | |||||||||
| * Spike concentration <10% of sample background concentration. | |||||||||
| Analyte | Sample conc. mg/kg | Low spike mg/kg | Average recovery R (%) | S (R) | RPD | High spike mg/kg | Average recovery R (%) | S (R) | RPD |
| EPA Hazardous Soil #884 | |||||||||
| Ag | 1.1 | 20 | 98 | 0.7 | 1.0 | 100 | 96 | 0.2 | 0.6 |
| Al | 5080 | 20 | * | * | 7.2 | 100 | * | * | 5.4 |
| As | 5.7 | 20 | 95 | 5.4 | 10.6 | 100 | 96 | 1.4 | 3.6 |
| B | 20.4 | 100 | 93 | 2.7 | 5.3 | 400 | 100 | 2.1 | 5.5B |
| Ba | 111 | 20 | 98 | 71.4 | 22.2 | 100 | 97 | 10.0 | 1.0 |
| Be | 0.66 | 20 | 97 | 0.7 | 2.3 | 100 | 99 | 0.1 | 0.2 |
| Ca | 85200 | − | − | − | − | − | − | − | − |
| Cd | 2 | 20 | 93 | 0.7 | 1.0 | 100 | 94 | 0.2 | 0.4 |
| Co | 5.5 | 20 | 96 | 3.5 | 7.7 | 100 | 93 | 0.8 | 2.1 |
| Cr | 79.7 | 20 | 87 | 28.8 | 16.5 | 100 | 104 | 1.3 | 1.1 |
| Cu | 113 | 20 | 110 | 16.2 | 4.4 | 100 | 104 | 4.0 | 4.2 |
| Fe | 16500 | − | − | − | − | − | − | − | − |
| Hg | <1.4 | 10 | 92 | 2.5 | 7.7 | 40 | 98 | 0.0 | 0.0 |
| K | 621 | 500 | 121 | 1.3 | 0.0 | 2000 | 107 | 0.9 | 1.8 |
| Li | 6.7 | 10 | 113 | 3.5 | 4.4 | 40 | 106 | 0.6 | 0.6 |
| Mg | 24400 | 500 | * | * | 8.4 | 2000 | * | * | 10.1 |
| Mn | 343 | 20 | * | * | 8.5 | 100 | 95 | 11.0 | 1.6 |
| Mo | 5.3 | 20 | 88 | 5.3 | 13.2 | 100 | 91 | 1.4 | 4.1 |
| Na | 195 | 500 | 102 | 2.2 | 2.4 | 2000 | 100 | 1.5 | 3.7 |
| Ni | 15.6 | 20 | 100 | 1.8 | 0.0 | 100 | 94 | 1.5 | 3.6 |
| P | 595 | 500 | 106 | 13.4 | 8.0 | 2000 | 103 | 3.2 | 2.7 |
| Pb | 145 | 20 | 88 | 51.8 | 17.9 | 100 | 108 | 15.6 | 17.4 |
| Sb | 6.1 | 20 | 83 | 3.9 | 7.5 | 100 | 81 | 1.9 | 5.9 |
| Se | <5 | 20 | 79 | 14.7 | 52.4 | 100 | 99 | 0.7 | 2.1 |
| Sn | 16.6 | 20 | 91 | 34.6 | 5.8 | 80 | 112 | 8.7 | 2.8 |
| Sr | 102 | 100 | 84 | 9.6 | 10.8 | 400 | 94 | 2.5 | 4.6 |
| Tl | <4 | 20 | 92 | 4.8 | 14.6 | 100 | 91 | 1.5 | 4.6 |
| V | 16.7 | 20 | 104 | 4.2 | 5.4 | 100 | 99 | 0.8 | 1.7 |
| Zn | 131 | 20 | 103 | 31.2 | 7.3 | 100 | 104 | 7.2 | 6.4 |
| EPA Electroplating Sludge #286 | |||||||||
| Ag | 6 | 20 | 96 | 0.2 | 0.4 | 100 | 93 | 0.1 | 0.4 |
| Al | 4980 | 20 | * | * | 4.4 | 100 | * | * | 5.6 |
| As | 32 | 20 | 94 | 1.3 | 0.8 | 100 | 97 | 0.7 | 1.6 |
| B | 210 | 100 | 113 | 2.0 | 1.6 | 400 | 98 | 1.9 | 3.5 |
| Ba | 39.8 | 20 | 0 | 6.8 | 0.3 | 100 | 0 | 1.6 | 5.7 |
| Be | 0.32 | 20 | 96 | 0.2 | 0.5 | 100 | 101 | 0.7 | 2.0 |
| Ca | 48500 | − | − | − | − | − | − | − | − |
| Cd | 108 | 20 | 98 | 2.5 | 0.8 | 100 | 96 | 0.5 | 0.5 |
| Co | 5.9 | 20 | 93 | 2.9 | 5.7 | 100 | 93 | 0.6 | 1.5 |
| Cr | 7580 | 20 | * | * | 0.7 | 100 | * | * | 1.3 |
| Cu | 806 | 20 | * | * | 1.5 | 100 | 94 | 8.3 | 0.7 |
| Fe | 31100 | − | − | − | − | − | − | − | − |
| Hg | 6.1 | 10 | 90 | 2.5 | 4.0 | 40 | 97 | 1.7 | 4.3 |
| K | 2390 | 500 | 75 | 8.3 | 4.0 | 2000 | 94 | 2.9 | 3.8 |
| Li | 9.1 | 10 | 101 | 2.8 | 0.5 | 40 | 106 | 1.6 | 3.1 |
| Mg | 1950 | 500 | 110 | 2.0 | 0.8 | 2000 | 108 | 2.3 | 3.2 |
| Mn | 262 | 20 | * | * | 1.8 | 100 | 91 | 1.2 | 0.9 |
| Mo | 13.2 | 20 | 92 | 2.1 | 2.9 | 100 | 92 | 0.3 | 0.0 |
| Na | 73400 | 500 | * | * | 1.7 | 2000 | * | * | 1.4 |
| Ni | 456 | 20 | * | * | 0.4 | 100 | 88 | 2.7 | 0.9 |
| P | 9610 | 500 | * | * | 2.9 | 2000 | 114 | 7.4 | 3.4 |
| Pb | 1420 | 20 | * | * | 2.1 | 100 | * | * | 1.3 |
| Sb | <2 | 20 | 76 | 0.9 | 3.3 | 100 | 75 | 2.8 | 10.7 |
| Se | 6.3 | 20 | 86 | 9.0 | 16.6 | 100 | 103 | 1.6 | 2.7 |
| Sn | 24.0 | 20 | 87 | 4.0 | 2.7 | 80 | 92 | 0.7 | 0.0 |
| Sr | 145 | 100 | 90 | 8.1 | 8.1 | 400 | 93 | 2.4 | 4.6 |
| Tl | 16 | 20 | 89 | 4.6 | 5.3 | 100 | 92 | 0.8 | 0.9 |
| V | 21.7 | 20 | 95 | 1.2 | 1.0 | 100 | 96 | 0.4 | 0.9 |
| Zn | 12500 | 20 | * | * | 0.8 | 100 | * | * | 0.8 |
| NBS 1645 River Sediment | |||||||||
| Ag | 1.6 | 20 | 92 | 0.4 | 1.0 | 100 | 96 | 0.3 | 0.9 |
| Al | 5160 | 20 | * | * | 8.4 | 100 | * | * | 2.4 |
| As | 62.8 | 20 | 89 | 14.4 | 9.7 | 100 | 97 | 2.9 | 5.0 |
| B | 31.9 | 100 | 116 | 7.1 | 13.5 | 400 | 95 | 0.6 | 1.5 |
| Ba | 54.8 | 20 | 95 | 6.1 | 2.8 | 100 | 98 | 1.2 | 1.3 |
| Be | 0.72 | 20 | 101 | 0.4 | 1.0 | 100 | 103 | 1.4 | 3.9 |
| Ca | 28000 | − | − | − | − | − | − | − | − |
| Cd | 9.7 | 20 | 100 | 1.1 | 0.0 | 100 | 101 | 0.7 | 1.8 |
| Co | 9.4 | 20 | 98 | 3.8 | 4.8 | 100 | 98 | 0.9 | 1.8 |
| Cr | 28500 | 20 | * | * | 0.4 | 100 | * | * | 0.7 |
| Cu | 109 | 20 | 115 | 8.5 | 0.0 | 100 | 102 | 1.8 | 1.0 |
| Fe | 84800 | − | − | − | − | − | − | − | − |
| Hg | 3.1 | 10 | 99 | 4.3 | 7.7 | 40 | 96 | 0.7 | 1.0 |
| K | 452 | 500 | 98 | 4.1 | 2.0 | 2000 | 106 | 1.4 | 2.3 |
| Li | 3.7 | 10 | 101 | 2.0 | 0.7 | 40 | 108 | 1.3 | 3.0 |
| Mg | 6360 | 500 | * | * | 1.8 | 2000 | 93 | 2.7 | 1.0 |
| Mn | 728 | 20 | * | * | 3.5 | 100 | 97 | 12.4 | 2.2 |
| Mo | 17.9 | 20 | 97 | 12.5 | 18.5 | 100 | 98 | 0.6 | 0.0 |
| Na | 1020 | 500 | 92 | 2.6 | 0.0 | 2000 | 97 | 1.1 | 1.7 |
| Ni | 36.2 | 20 | 94 | 5.9 | 4.0 | 100 | 100 | 1.1 | 1.5 |
| P | 553 | 500 | 102 | 1.4 | 0.9 | 2000 | 100 | 0.8 | 1.6 |
| Pb | 707 | 20 | * | * | 0.8 | 100 | 103 | 5.9 | 0.4 |
| Sb | 22.8 | 20 | 86 | 2.3 | 0.0 | 100 | 88 | 0.6 | 0.9 |
| Se | 6.7 | 20 | 103 | 14.3 | 27.1 | 100 | 98 | 3.1 | 7.6 |
| Sn | 309 | 20 | * | * | 1.0 | 80 | 101 | 7.9 | 2.7 |
| Sr | 782 | 100 | 91 | 12.3 | 3.0 | 400 | 96 | 3.3 | 2.6 |
| Tl | <4 | 20 | 90 | 0.0 | 0.0 | 100 | 95 | 1.3 | 4.0 |
| V | 20.1 | 20 | 89 | 5.4 | 5.8 | 100 | 98 | 0.7 | 0.0 |
| Zn | 1640 | 20 | * | * | 1.8 | 100 | * | * | 1.1 |
| S (R) Standard deviation of percent recovery. | |||||||||
| RPD Relative percent difference between duplicate spike determinations. | |||||||||
| < Sample concentration below established method detection limit. | |||||||||
| * Spike concentration <10% of sample background concentration. | |||||||||
| − Not spiked. | |||||||||
| Equivalent. | |||||||||
| Element | Mean conc. (mg/L) | Nb | RSD (%) | Accuracec (% of Nominal) |
| Al | 14.8 | 8 | 6.3 | 100 |
| Sb | 15.1 | 8 | 7.7 | 102 |
| As | 14.7 | 7 | 6.4 | 99 |
| Ba | 3.66 | 7 | 3.1 | 99 |
| Be | 3.78 | 8 | 5.8 | 102 |
| Cd | 3.61 | 8 | 7.0 | 97 |
| Ca | 15.0 | 8 | 7.4 | 101 |
| Cr | 3.75 | 8 | 8.2 | 101 |
| Co | 3.52 | 8 | 5.9 | 95 |
| Cu | 3.58 | 8 | 5.6 | 97 |
| Fe | 14.8 | 8 | 5.9 | 100 |
| Pb | 14.4 | 7 | 5.9 | 97 |
| Mg | 14.1 | 8 | 6.5 | 96 |
| Mn | 3.70 | 8 | 4.3 | 100 |
| Mo | 3.70 | 8 | 6.9 | 100 |
| Ni | 3.70 | 7 | 5.7 | 100 |
| K | 14.1 | 8 | 6.6 | 95 |
| Se | 15.3 | 8 | 7.5 | 10 |
| Na | 14.0 | 8 | 4.2 | 95 |
| Tl | 15.1 | 7 | 8.5 | 102 |
| V | 3.51 | 8 | 6.6 | 95 |
| Zn | 3.57 | 8 | 8.3 | 96 |
| aThese performance values are independent of sample preparation because the labs analyzed portions of the same solutions using sequential or simultaneous instruments. | ||||
| bN = Number of measurements for mean and relative standard deviation (RSD). | ||||
| cAccuracy is expressed as a percentage of the nominal value for each analyte in the acidified, multi-element solutions. | ||||
| Analyte | Concentrationμg/L | Total recoverable digestionμ/L |
| Aluminum | 69-4792 | X = 0.9380 (C) 22.1 |
| SR = 0.0481 (X) 18.8 | ||
| Antimony | 77-1406 | 0.8908 (C) 0.9 |
| SR = 0.0682 (X) 2.5 | ||
| Arsenic | 69-1887 | X = 1.0175 (C) 3.9 |
| SR = 0.0643 (X) 10.3 | ||
| Barium | 9-377 | X = 0.8.80 (C) 1.68 |
| SR = 0.0826 (X) 3.54 | ||
| Beryllium | 3-1906 | X = 1.0177 (C) − 0.55 |
| SR = 0.0445 (X) − 0.10 | ||
| Boron | 19-5189 | X = 0.9676 (C) 18.7 |
| SR = 0.0743 (X) 21.1 | ||
| Cadmium | 9-1943 | X = 1.0137 (C) − 0.65 |
| SR = 0.0332 (X) 0.90 | ||
| Calcium | 17-47170 | X = 0.9658 (C) 0.8 |
| SR = 0.0327 (X) 10.1 | ||
| Chromium | 13-1406 | X = 1.0049 (C) − 1.2 |
| SR = 0.0571 (X) 1.0 | ||
| Cobalt | 17-2340 | X = 0.9278 (C) 1.5 |
| SR = 0.0407 (X) 0.4 | ||
| Copper | 8-1887 | X = 0.9647 (C) − 3.64 |
| SR = 0.0406 (X) 0.96 | ||
| Iron | 13-9359 | X = 0.9830 (C) 5.7 |
| SR = 0.0790 (X) 11.5 | ||
| Lead | 42-4717 | X = 1.0056 (C) 4.1 |
| SR = 0.0448 (X) 3.5 | ||
| Magnesium | 34-13868 | X = 0.9879 (C) 2.2 |
| SR = 0.0268 (X) 8.1 | ||
| Manganese | 4-1887 | X = 0.9725 (C) 0.07 |
| SR = 0.0400 (X) 0.82 | ||
| Molybdenum | 17-1830 | X = 0.9707 (C) − 2.3 |
| SR = 0.0529 (X) 2.1 | ||
| Nickel | 17-47170 | X = 0.9869 (C) 1.5 |
| SR = 0.0393 (X) 2.2 | ||
| Potassium | 347-14151 | X = 0.9355 (C) − 183.1 |
| SR = 0.0329 (X) 60.9 | ||
| Selenium | 69-1415 | X = 0.9737 (C) − 1.0 |
| SR = 0.0443 (X) 6.6 | ||
| Silicon | 189-9434 | X = 0.9737 (C) − 22.6 |
| SR = 0.2133 (X) 22.6 | ||
| Silver | 8-189 | X = 0.3987 (C) 8.25 |
| SR = 0.1836 (X) − 0.27 | ||
| Sodium | 35-47170 | X = 1.0526 (C) 26.7 |
| SR = 0.0884 (X) 50.5 | ||
| Thallium | 79-1434 | X = 0.9238 (C) 5.5 |
| SR = 0.0106 (X) 48.0 | ||
| Vanadium | 13-4698 | X = 0.9551 (C) 0.4 |
| SR = 0.0472 (X) 0.5 | ||
| Zinc | 7-7076 | X = 0.9500 (C) 1.82 |
| SR = 0.0153 (X) 7.78 | ||
| *-Regression equations abstracted from Reference 16. | ||
| X = Mean Recovery, μg/L. | ||
| C = True Value for the Concentration, μg/L. | ||
| SR = Single-analyst Standard Deviation, μg/L. | ||
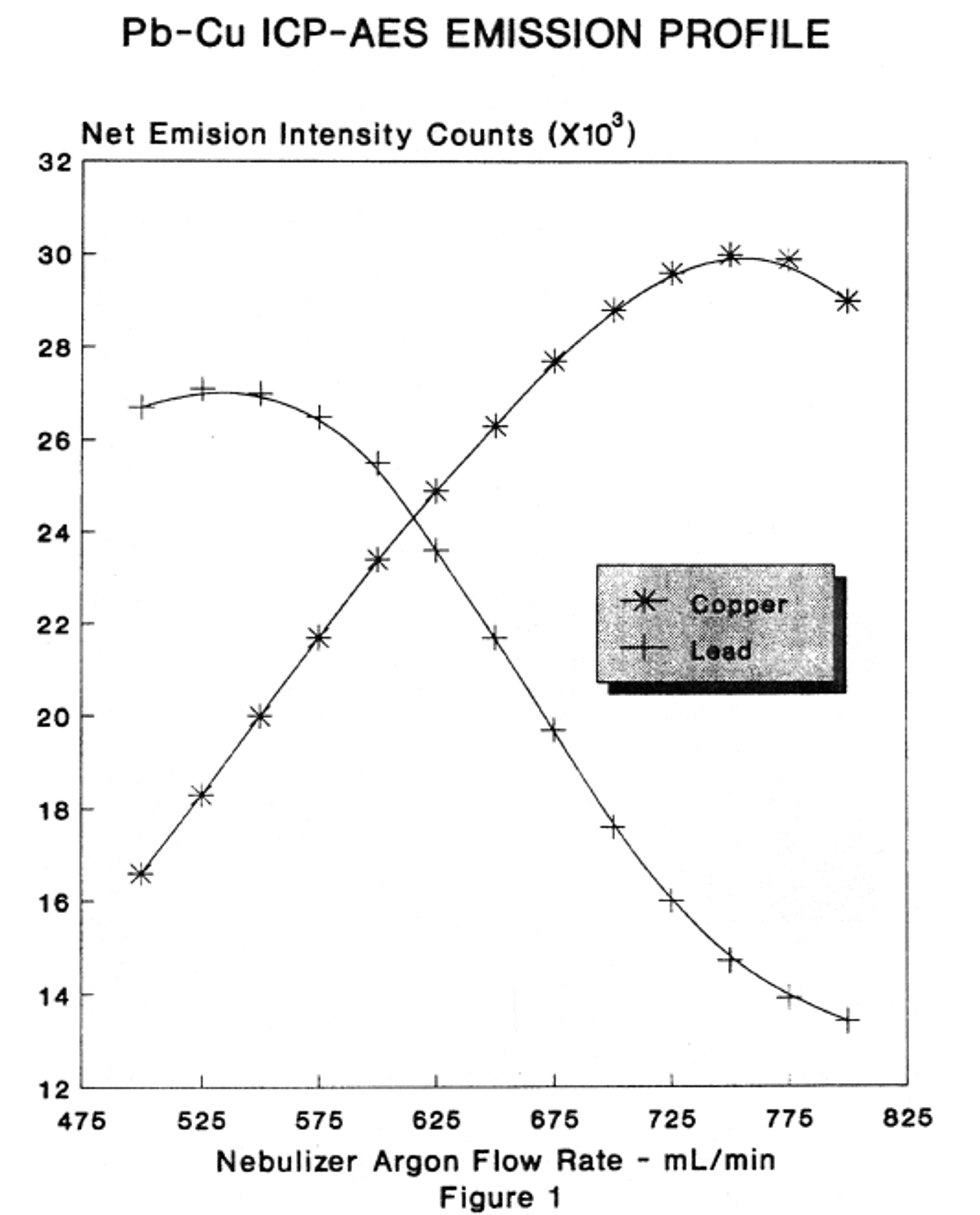
[77 FR 29813, May 18, 2012]
['Water Programs']
['Water Analysis', 'Water Quality']
UPGRADE TO CONTINUE READING