Compliance Just Got Easier: Stay ahead of regulatory changes with instant notifications on updates that matter.
['Toxic and Hazardous Substances - OSHA']
['Acrylonitrile', 'Toxic and Hazardous Substances - OSHA']
12/16/2024

Copyright 2026 J. J. Keller & Associate, Inc. For re-use options please contact copyright@jjkeller.com or call 800-558-5011.
There are many methods available for monitoring employee exposures to acrylonitrile. Most of these involve the use of charcoal tubes and sampling pumps, with analysis by gas chromatograph. The essential differences between the charcoal tube methods include, among others, the use of different desorbing solvents, the use of different lots of charcoal, and the use of different equipment for analysis of the samples.
Besides charcoal, considerable work has been performed on methods using porous polymer sampling tubes and passive dosimeters. In addition, there are several portable gas analyzers and monitoring units available on the open market.
This appendix contains details for the methods which have been tested at OSHA Analytical Laboratory in Salt Lake City, and NIOSH in Cincinnati. Each is a variation on NIOSH Method S-156, which is also included for reference. This does not indicate that these methods are the only ones which will be satisfactory. There also may be workplace situations in which these methods are not adequate, due to such factors as high humidity. Copies of the other methods available to OSHA are available in the rulemaking record, and may be obtained from the OSHA Docket Office. These include, the Union Carbide, Monsanto, Dow Chemical and Dow Badische methods, as well as NISOH Method P & CAM 127.
Employers who note problems with sample breakthrough should try larger charcoal tubes. Tubes of larger capacity are available, and are often used for sampling vinyl chloride. In addition, lower flow rates and shorter sampling times should be beneficial in minimizing breakthrough problems.
Whatever method the employer chooses, he must assure himself of the method’s accuracy and precision under the unique conditions present in his workplace.
NIOSH METHOD S-156 (UNMODIFIED)
Analyte: Acrylonitrile.
Matrix: Air.
Procedure: Absorption on charcoal, desorption with methanol, GC.
1. Principle of the method (Reference 11.1).
1.1 A known volume of air is drawn through a charcoal tube to trap the organic vapors present.
1.2 The charcoal in the tube is transferred to a small, stoppered sample container, and the analyte is desorbed with methanol.
1.3 An aliquot of the desorbed sample is injected into a gas chromatograph.
1.4 The area of the resulting peak is determined and compared with areas obtained for standards.
2. Range and sensitivity.
2.1 This method was validated over the range of 17.5-70.0 mg/cu m at an atmospheric temperature and pressure of 22°C and 760 MM Hg, using a 20-liter sample. Under the conditions of sample size (20-liters) the probable useful range of this method is 4.5-135 mg/cu m. The method is capable of measuring much smaller amounts if the desorption efficiency is adequate. Desorption efficiency must be determined over the range used.
2.2 The upper limit of the range of the method is dependent on the adsorptive capacity of the charcoal tube. This capacity varies with the concentrations of acrylonitrile and other substances in the air. The first section of the charcoal tube was found to hold at least 3.97 mg of acrylonitrile when a test atmosphere containing 92.0 mg/cu m of acrylonitrile in air was sampled 0.18 liter per minute for 240 minutes; at that time the concentration of acrylonitrile in the effluent was less than 5 percent of that in the influent. (The charcoal tube consists of two sections of activated charcoal separated by a section of urethane foam. See section 6.2.) If a particular atmosphere is suspected of containing a large amount of contaminant, a smaller sampling volume should be taken.
3. Interference.
3.1 When the amount of water in the air is so great that condensation actually occurs in the tube, organic vapors will not be trapped efficiently. Preliminary experiments using toluene indicate that high humidity severely decreases the breakthrough volume.
3.2 When interfering compounds are known or suspected to be present in the air, such information, including their suspected identities, should be transmitted with the sample.
3.3 It must be emphasized that any compound which has the same retention time as the analyte at the operating conditions described in this method is an interference. Retention time data on a single column cannot be considered proof of chemical identity.
3.4 If the possibility of interference exists, separation conditions (column packing, temperature, etc.) must be changed to circumvent the problem.
4. Precision and accuracy.
4.1 The Coefficient of Variation (CV T ) for the total analytical and sampling method in the range of 17.5-70.0 mg/cu m was 0.073. This value corresponds to a 3.3 mg/cu m standard deviation at the (previous) OSHA standard level (20 ppm). Statistical information and details of the validation and experimental test procedures can be found in Reference 11.2.
4.2 On the average the concentrations obtained at the 20 ppm level using the overall sampling and analytical method were 6.0 percent lower than the “true” concentrations for a limited number of laboratory experiments. Any difference between the “found” and “true” concentrations may not represent a bias in the sampling and analytical method, but rather a random variation from the experimentally determined “true” concentration. Therefore, no recovery correction should be applied to the final result in section 10.5.
5. Advantages and disadvantages of the method.
5.1 The sampling device is small, portable, and involves no liquids. Interferences are minimal, and most of those which do occur can be eliminated by altering chromatographic conditions. The tubes are analyzed by means of a quick, instrumental method.
The method can also be used for the simultaneous analysis of two or more substances suspected to be present in the same sample by simply changing gas chromatographic conditions.
5.2 One disadvantage of the method is that the amount of sample which can be taken is limited by the number of milligrams that the tube will hold before overloading. When the sample value obtained for the backup section of the charcoal tube exceeds 25 percent of that found on the front section, the possibility of sample loss exists.
5.3 Furthermore, the precision of the method is limited by the reproducibility of the pressure drop across the tubes. This drop will affect the flow rate and cause the volume to be imprecise, because the pump is usually calibrated for one tube only.
6. Apparatus.
6.1 A calibrated personal sampling pump whose flow can be determined within ±5 percent at the recommended flow rate. (Reference 11.3).
6.2 Charcoal tubes: Glass tubes with both ends flame sealed, 7 cm long with a 6-mm O.D. and a 4-mm I.D., containing 2 sections of 20/40 mesh activated charcoal separated by a 2-mm portion of urethane foam. The activated charcoals prepared from coconut shells and is fired at 600°C prior to packing. The adsorbing section contains 100 mg of charcoal, the backup section 50 mg. A 3-mm portion of urethane foam is placed between the outlet end of the tube and the backup section. A plug of silicated glass wool is placed in front of the adsorbing section. The pressure drop across the tube must be less than 1 inch of mercury at a flow rate of 1 liter per minute.
6.3 Gas chromatograph equipped with a flame ionization detector.
6.4 Column (4-ft × 1/4 -in stainless steel) packed with 50/80 mesh Poropak, type Q.
6.5 An electronic integrator or some other suitable method for measuring peak areas.
6.6 Two-milliliter sample containers with glass stoppers or Teflon-lined caps. If an automatic sample injector is used, the associated vials may be used.
6.7 Microliter syringes: 10-microliter and other convenient sizes for making standards.
6.8 Pipets: 1.0-ml delivery pipets.
6.9 Volumetric flask: 10-ml or convenient sizes for making standard solutions.
7. Reagents.
7.1 Chromatographic quality methanol.
7.2 Acrylonitrile, reagent grade.
7.3 Hexane, reagent grade.
7.4 Purified nitrogen.
7.5 Prepurified hydrogen.
7.6 Filtered compressed air.
8. Procedure.
8.1 Cleaning of equipment. All glassware used for the laboratory analysis should be detergent washed and thoroughly rinsed with tap water and distilled water.
8.2 Calibration of personal pumps. Each personal pump must be calibrated with a representative charcoal tube in the line. This will minimize errors associated with uncertainties in the sample volume collected.
8.3 Collection and shipping of samples.
8.3.1 Immediately before sampling, break the ends of the tube to provide an opening at least one-half the internal diameter of the tube (2 mm).
8.3.2 The smaller section of charcoal is used as a backup and should be positioned nearest the sampling pump.
8.3.3 The charcoal tube should be placed in a vertical direction during sampling to minimize channeling through the charcoal.
8.3.4 Air being sampled should not be passed through any hose or tubing before entering the charcoal tube.
8.3.5 A maximum sample size of 20 liters is recommended. Sample at a flow of 0.20 liter per minute or less. The flow rate should be known with an accuracy of at least ±5 percent.
8.3.6 The temperature and pressure of the atmosphere being sampled should be recorded. If pressure reading is not available, record the elevation.
8.3.7 The charcoal tubes should be capped with the supplied plastic caps immediately after sampling. Under no circumstances should rubber caps be used.
8.3.8 With each batch of 10 samples submit one tube from the same lot of tubes which was used for sample collection and which is subjected to exactly the same handling as the samples except that no air is drawn through it. Label this as a blank.
8.3.9 Capped tubes should be packed tightly and padded before they are shipped to minimize tube breakage during shipping.
8.3.10 A sample of the bulk material should be submitted to the laboratory in a glass container with a Teflon-lined cap. This sample should not be transported in the same container as the charcoal tubes.
8.4 Analysis of samples.
8.4.1 Preparation of samples. In preparation for analysis, each charcoal tube is scored with a file in front of the first section of charcoal and broken open. The glass wool is removed and discarded. The charcoal in the first (larger) section is transferred to a 2-ml stoppered sample container. The separating section of foam is removed and discarded; the second section is transferred to another stoppered container. These two sections are analyzed separately.
8.4.2 Desorption of samples. Prior to analysis, 1.0 ml of methanol is pipetted into each sample container. Desorption should be done for 30 minutes. Tests indicate that this is adequate if the sample is agitated occasionally during this period. If an automatic sample injector is used, the sample vials should be capped as soon as the solvent is added to minimize volatilization.
8.4.3 GC conditions. The typical operating conditions for the gas chromatograph are:
1. 50 ml/min (60 psig) nitrogen carrier gas flow.
2. 65 ml/min (24 psig) hydrogen gas flow to detector.
3. 500 ml/min (50 psig) air flow to detector.
4. 235°C injector temperature.
5. 255°C manifold temperature (detector).
6. 155°C column temperature.
8.4.4 Injection. The first step in the analysis is the injection of the sample into the gas chromatograph. To eliminate difficulties arising from blow-back or distillation within the syringe needle, one should employ the solvent flush injection technique. The 10-microliter syringe is first flushed with solvent several times to wet the barrel and plunger. Three microliters of solvent are drawn into the syringe to increase the accuracy and reproducibility of the injected sample volume. The needle is removed from the solvent, and the plunger is pulled back about 0.2 microliter to separate the solvent flush from the sample with a pocket of air to be used as a marker. The needle is then immersed in the sample, and a 5-microliter aliquot is withdrawn, taking into consideration the volume of the needle, since the sample in the needle will be completely injected. After the needle is removed from the sample and prior to injection, the plunger is pulled back 1.2 microliters to minimize evaporation of the sample from the tip of the needle. Observe that the sample occupies 4.9-5.0 microliters in the barrel of the syringe. Duplicate injections of each sample and standard should be made. No more than a 3 percent difference in area is to be expected. An automatic sample injector can be used if it is shown to give reproducibility at least as good as the solvent flush method.
8.4.5 Measurement of area. The area of the sample peak is measured by an electronic integrator or some other suitable form of area measurement, and preliminary results are read from a standard curve prepared as discussed below.
8.5 Determination of desorption efficiency.
8.5.1 Importance of determination. The desorption efficiency of a particular compound can vary from one laboratory to another and also from one batch of charcoal to another. Thus, it is necessary to determine at least once the percentage of the specific compound that is removed in the desorption process, provided the same batch of charcoal is used.
8.5.2 Procedure for determining desorption efficiency. Activated charcoal equivalent to the amount in the first section of the sampling tube (100 mg) is measured into a 2.5 in, 4-mm I.D. glass tube, flame sealed at one end. This charcoal must be from the same batch as that used in obtaining the samples and can be obtained from unused charcoal tubes. The open end is capped with Parafilm. A known amount of hexane solution of acrylonitrile containing 0.239 g/ml is injected directly into the activated charcoal with a microliter syringe, and tube is capped with more Parafilm. When using an automatic sample injector, the sample injector vials, capped with Teflon-faced septa, may be used in place of the glass tube.
The amount injected is equivalent to that present in a 20-liter air sample at the selected level.
Six tubes at each of three levels (0.5X, 1X, and 2X of the standard) are prepared in this manner and allowed to stand for at least overnight to assure complete adsorption of the analyte onto the charcoal. These tubes are referred to as the sample. A parallel blank tube should be treated in the same manner except that no sample is added to it. The sample and blank tubes are desorbed and analyzed in exactly the same manner as the sampling tube described in section 8.4.
Two or three standards are prepared by injecting the same volume of compound into 1.0 ml of methanol with the same syringe used in the preparation of the samples. These are analyzed with the samples.
The desorption efficiency (D.E.) equals the average weight in mg recovered from the tube divided by the weight in mg added to the tube, or
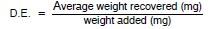 |
The desorption efficiency is dependent on the amount of analyte collected on the charcoal. Plot the desorption efficiency versus weight of analyte found. This curve is used in section 10.4 to correct for adsorption losses.
9. Calibration and standards.
It is convenient to express concentration of standards in terms of mg/1.0 ml methanol, because samples are desorbed in this amount of methanol. The density of the analyte is used to convert mg into microliters for easy measurement with a microliter syringe. A series of standards, varying in concentration over the range of interest, is prepared and analyzed under the same GC conditions and during the same time period as the unknown samples. Curves are established by plotting concentration in mg/1.0 ml versus peak area.
NOTE: Since no internal standard is used in the method, standard solutions must be analyzed at the same time that the sample analysis is done. This will minimize the effect of known day-to-day variations and variations during the same day of the FID response.
10. Calculations.
10.1 Read the weight, in mg, corresponding to each peak area from the standard curve. No volume corrections are needed, because the standard curve is based on mg/1.0 ml methanol and the volume of sample injected is identical to the volume of the standards injected.
10.2 Corrections for the bank must be made for each sample.
| Where:mg sample = mg found in front section of sample tube.mg sample = mg found in front section of blank tube. |
A similar procedure is followed for the backup sections.
10.3 Add the weights found in the front and backup sections to get the total weight in the sample.
10.4 Read the desorption efficiency from the curve (see sec. 8.5.2) for the amount found in the front section. Divide the total weight by this desorption efficiency to obtain the corrected mg/sample. 
10.5 The concentration of the analyte in the air sampled can be expressed in mg/cu m. 
10.6 Another method of expressing concentration is ppm.
| Where:P = Pressure (mm Hg) of air sampled. T = Temperature (°C) of air sampled. 24.45 = Molar volume (liter/mole) at 25°C and 760 mm Hg.M.W. = Molecular weight (g/mole) of analyte. 760 = Standard pressure (mm Hg).298 = Standard temperature (°K). |
11. References.
11.1 White, L. D. et al., “A Convenient Optimized Method for the Analysis of Selected Solvent Vapors in the Industrial Atmosphere,” Amer. Ind. Hyg. Assoc. J., 31:225 (1970).
11.2 Documentation of NIOSH Validation Tests, NIOSH Contract No. CDC-99-74-45.
11.3 Final Report, NIOSH Contract HSM-99-71-31, “Personal Sampler Pump for Charcoal Tubes,” September 15, 1972.
NIOSH Modification of NIOSH Method S-156
The NIOSH recommended method for low levels for acrylonitrile is a modification of method S-156. It differs in the following respects:
(1) Samples are desorbed using 1 ml of 1 percent acetone in CS 2 rather than methanol.
(2) The analytical column and conditions are:
Column: 20 percent SP-1000 on 80/100 Supelcoport 10 feet x 1/8 inch S.S.
Conditions:
- Injector temperature: 200°C.
- Detector temperature: 100°C.
- Column temperature: 85°C.
- Helium flow: 25 ml/min.
- Air flow: 450 ml/min.
- Hydrogen flow: 55 ml/min.
(3) A 2 μl injection of the desorbed analyte is used.
(4) A sampling rate of 100 ml/min is recommended.
OSHA Laboratory Modification of NIOSH Method S-156
Analyte: Acrylonitrile.
Matrix: Air.
Procedure: Adsorption on charcoal, desorption with methanol, GC.
1. Principle of the Method (Reference 1).
1.1 A known volume of air is drawn through a charcoal tube to trap the organic vapors present.
1.2 The charcoal in the tube is transferred to a small, stoppered sample vial, and the analyte is desorbed with methanol.
1.3 An aliquot of the desorbed sample is injected into a gas chromatograph.
1.4 The area of the resulting peak is determined and compared with areas obtained for standards.
2. Advantages and disadvantages of the method.
2.1 The sampling device is small, portable, and involves no liquids. Interferences are minimal, and most of those which do occur can be eliminated by altering chromatographic conditions. The tubes are analyzed by means of a quick, instrumental method.
2.2 This method may not be adequate for the simultaneous analysis of two or more substances.
2.3 The amount of sample which can be taken is limited by the number of milligrams that the tube will hold before overloading. When the sample value obtained for the backup section of the charcoal tube exceeds 25 percent of that found on the front section, the possibility of sample loss exists.
2.4 The precision of the method is limited by the reproducibility of the pressure drop across the tubes. This drop will affect the flow rate and cause the volume to be imprecise, because the pump is usually calibrated for one tube only.
3. Apparatus.
3.1 A calibrated personal sampling pump whose flow can be determined within ±5 percent at the recommended flow rate.
3.2 Charcoal tubes: Glass tube with both ends flame sealed, 7 cm long with a 6-mm O.D. and a 4-mm I.D., containing 2 sections of 20/40 mesh activated charcoal separated by a 2-mm portion of urethane foam. The activated charcoal is prepared from coconut shells and is fired at 600°C prior to packing. The adsorbing section contains 100 mg of charcoal, the back-up section 50 mg. A 3-mm portion of urethane foam is placed between the outlet end of the tube and the back-up section. A plug of sililated glass wool is placed in front of the adsorbing section. The pressure drop across the tube must be less than one inch of mercury at a flow rate of 1 liter per minute.
3.3 Gas chromatograph equipped with a nitrogen phosphorus detector.
3.4 Column (10-ft x 1/8 "-in stainless steel) packed with 100/120 Supelcoport coated with 10 percent SP 1000.
3.5 An electronic integrator or some other suitable method for measuring peak area.
3.6 Two-milliliter sample vials with Teflon-lined caps
3.7 Microliter syringes: 10-microliter, and other convenient sizes for making standards.
3.8 Pipets: 1.0-ml delivery pipets.
3.9 Volumetric flasks: convenient sizes for making standard solutions.
4. Reagents.
4.1 Chromatographic quality methanol.
4.2 Acrylonitrile, reagent grade.
4.3 Filtered compressed air.
4.4 Purified hydrogen.
4.5 Purified helium.
5. Procedure.
5.1 Cleaning of equipment. All glassware used for the laboratory analysis should be properly cleaned and free of organics which could interfere in the analysis.
5.2 Calibration of personal pumps. Each pump must be calibrated with a representative charcoal tube in the line.
5.3 Collection and shipping of samples.
5.3.1 Immediately before sampling, break the ends of the tube to provide an opening at least one-half the internal diameter of the tube (2 mm).
5.3.2 The smaller section of the charcoal is used as the backup and should be placed nearest the sampling pump.
5.3.3 The charcoal should be placed in a vertical position during sampling to minimize channeling through the charcoal.
5.3.4 Air being sampled should not be passed through any hose or tubing before entering the charcoal tube.
5.3.5 A sample size of 20 liters is recommended. Sample at a flow rate of approximately 0.2 liters per minute. The flow rate should be known with an accuracy of at least ± 5 percent.
5.3.6 The temperature and pressure of the atmosphere being sampled should be recorded.
5.3.7 The charcoal tubes should be capped with the supplied plastic caps immediately after sampling. Rubber caps should not be used.
5.3.8 Submit at least one blank tube (a charcoal tube subjected to the same handling procedures, without having any air drawn through it) with each set of samples.
5.3.9. Take necessary shipping and packing precautions to minimize breakage of samples.
5.4 Analysis of samples.
5.4.1 Preparation of samples. In preparation for analysis, each charcoal tube is scored with a file in front of the first section of charcoal and broken open. The glass wool is removed and discarded. The charcoal in the first (larger) section is transferred to a 2-ml vial. The separating section of foam is removed and discarded; the section is transferred to another capped vial. These two sections are analyzed separately.
5.4.2 Desorption of samples. Prior to analysis, 1.0 ml of methanol is pipetted into each sample container. Desorption should be done for 30 minutes in an ultrasonic bath. The sample vials are recapped as soon as the solvent is added.
5.4.3 GC conditions. The typical operating conditions for the gas chromatograph are:
1. 30 ml/min (60 psig) helium carrier gas flow.
2. 3.0 ml/min (30 psig) hydrogen gas flow to detector.
3. 50 ml/min (60 psig) air flow to detector.
4. 200°C injector temperature.
5. 200°C dejector temperature.
6. 100°C column temperature.
5.4.4 Injection. Solvent flush technique or equivalent.
5.4.5 Measurement of area. The area of the sample peak is measured by an electronic integator or some other suitable form of area measurement, and preliminary results are read from a standard curve prepared as discussed below.
5.5 Determination of desorption efficiency.
5.5.1 Importance of determination. The desorption efficiency of a particular compound can vary from one laboratory to another and also from one batch of charcoal to another. Thus, it is necessary to determine, at least once, the percentage of the specific compound that is removed in the desorption process, provided the same batch of charcoal is used.
5.5.2 Procedure for determining desorption efficiency. The reference portion of the charcoal tube is removed. To the remaining portion, amounts representing 0.5X, 1X, and 2X (X represents TLV) based on a 20 l air sample are injected onto several tubes at each level. Dilutions of acrylonitrile with methanol are made to allow injection of measurable quantities. These tubes are then allowed to equilibrate at least overnight. Following equilibration they are analyzed following the same procedure as the samples. A curve of the desorption efficiency amt recovered/amt added is plotted versus amount of analyte found. This curve is used to correct for adsorption losses.
6. Calibration and standards.
A series of standards, varying in concentration over the range of interest, is prepared and analyzed under the same GC conditions and during the same time period as the unknown samples. Curves are prepared by plotting concentration versus peak area.
NOTE: Since no internal standard is used in the method, standard solutions must be analyzed at the same time that the sample analysis is done. This will minimize the effect of known day-to-day variations and variations during the same day of the NPD response. Multiple injections are necessary.
7. Calculations.
Read the weight, corresponding to each peak area from the standard curve, correct for the blank, correct for the desorption efficiency, and make necessary air volume corrections.
8. Reference. NIOSH Method S-156.
['Toxic and Hazardous Substances - OSHA']
['Acrylonitrile', 'Toxic and Hazardous Substances - OSHA']
UPGRADE TO CONTINUE READING