Compliance Just Got Easier: Stay ahead of regulatory changes with instant notifications on updates that matter.
['Air Programs']
['Greenhouse Gases']
12/18/2024

Copyright 2026 J. J. Keller & Associate, Inc. For re-use options please contact copyright@jjkeller.com or call 800-558-5011.
If you are measuring destruction or removal efficiency of a point-of-use abatement device according to EPA 430-R-10-003 (incorporated by reference, see §98.7) as specified in §98.94(f)(4), you may follow the alternative procedures specified in paragraphs (a) through (c) of this appendix.
(a) In place of the Quadrupole Mass Spectrometry protocol requirements specified in section 2.2.4 of EPA 430-R-10-003 (incorporated by reference, see §98.7), you must conduct mass spectrometry testing in accordance with the provisions in paragraph (a)(1) through (a)(15) of this appendix.
(1) Detection limits. The mass spectrometer chosen for this application must have the necessary sensitivity to detect the selected effluent species at or below the maximum field detection limits specified in Table 3 of section 2.2.7 of EPA 430-R-10-003 (incorporated by reference, see §98.7).
(2) Sampling location. The sample at the inlet of the point-of-use abatement device must be taken downstream of the process tool and pump package. The sample exhaust must be vented back into the corrosive house ventilation system at a point downstream of the sample inlet location.
(3) Sampling conditions. For etch processes, destruction or removal efficiencies must be determined while etching a substrate (product, dummy, or test). For chemical vapor deposition processes, destruction or removal efficiencies must be determined during a chamber clean after deposition (destruction or removal efficiencies must not be determined in a clean chamber). All sampling must be performed non-intrusively during wafer processing. Samples must be drawn through the mass spectrometer source by an external sample pump. Because of the volatility, vapor pressure, stability and inertness of CF4, C2F6, C3F8, CHF3, NF3, and SF6, the sample lines do not need to be heated.
(4) Mass spectrometer parameters. The specific mass spectrometer operating conditions such as electron energy, secondary electron multiplier voltage, emission current, and ion focusing voltage must be selected according to the specifications provided by the mass spectrometer manufacturer, the mass spectrometer system manual, basic mass spectrometer textbook, or other such sources. The mass spectrometer responses to each of the target analytes must all be calibrated under the same mass spectrometer operating conditions.
(5) Flow rates. A sample flow rate of 0.5-1.5 standard liters per minute (slm) must be drawn from the process tool exhaust stream under study.
(6) Sample frequency. The mass spectrometer sampling frequency for etch processes must be in the range of 0.5 to 1 cycles per second, and for chemical vapor deposition processes must be in the range of 0.25 to 0.5 cycles per second. As an alternative you may use the sampling frequencies specified in section 2.2.4 of EPA 430-R-10-003 (incorporated by reference, see §98.7).
(7) Dynamic dilution calibration parameters. The quadrupole mass spectrometer must be calibrated for both mass location and response to analytes. A dynamic dilution calibration system may be used to perform both types of mass spectrometer system calibrations using two mass flow controllers. Use one mass flow controller to regulate the flow rate of the standard component used to calibrate the system and the second mass flow controller to regulate the amount of diluent gas used to mix with the standard to generate the calibration curve for each compound of interest. The mass flow controller must be calibrated using the single component gas being used with them, for example, nitrogen (N2) for the diluent. A mass flow controller used with calibration mixtures must be calibrated with the calibration mixture balance gas (for example, N2 or He) if the analyte components are 2 percent or less of the volume of the sample. All calibration mixtures must be National Institute of Standards and Technology Traceable gases or equivalent. They must be calibrated over their range of use and must be operated in their experimentally determined dynamic linear range. If compressed gas standards cannot be brought into the fab, metered gas flows of target compounds into the process chamber, under no thermal or plasma conditions and with no wafer(s) present, and with no process emissions from other tools contributing to the sample location, must then be performed throughout the appropriate concentration ranges to derive calibration curves for the subsequent destruction or removal efficiency tests.
(8) Mass location calibration. A mixture containing 1 percent He, Ar, Kr, and Xe in a balance gas of nitrogen must be used to assure the alignment of the quadrupole mass filter (see EPA Method 205 at 40 CFR part 51, appendix M as reference). The mass spectrometer must be chosen so that the mass range is sufficient to detect the predominant peaks of the components under study.
(9) Quadrupole mass spectrometer response calibration. A calibration curve must be generated for each compound of interest.
(10) Calibration frequency. The mass spectrometer must be calibrated at the start of testing a given process. The calibration must be checked at the end of testing.
(11) Calibration range. The mass spectrometer must be calibrated over the expected concentration range of analytes using a minimum of five concentrations including a zero. The zero point is defined as diluent containing no added analyte.
(12) Operating procedures. You must follow the operating procedures specified in paragraphs (a)(12)(i) through (v) of this appendix.
(i) You must perform a qualitative mass calibration by running a standard (or by flowing chamber gases under non-process conditions) containing stable components such as Ar, Kr, and Xe that provide predominant signals at m/e values distributed throughout the mass range to be used. You must adjust the quadrupole mass filter as needed to align with the inert gas fragments.
(ii) You must quantitatively calibrate the quadrupole mass spectrometer for each analyte of interest. The analyte concentrations during calibration must include the expected concentrations in the process effluent. The calibration must be performed under the same operating conditions, such as inlet pressure, as when sampling process exhaust. If the calibration inlet pressure differs from the sampling inlet pressure then the relationship between inlet pressure and quadrupole mass spectrometer signal response must be empirically determined and applied to correct for any differences between calibration and process emissions monitoring data.
(iii) To determine the response time of the instrument to changes in a process, a process gas such as C2F6 must be turned on at the process tool for a fixed period of time (for example, 20 seconds), after which the gas is shut off. The sample flow rate through the system must be adjusted so that the signal increases to a constant concentration within a few seconds and decreases to background levels also within a few seconds.
(iv) You must sample the process effluent through the quadrupole mass spectrometer and acquire data for the required amount of time to track the process, as determined in paragraph (a)(12)(iii) of this appendix. You must set the sample frequency to monitor the changes in the process as specified in paragraph (a)(6) of this appendix. You must repeat this for at least five substrates on the same process and calculate the average and standard deviation of the analyte concentration.
(v) You must repeat the quantitative calibration at the conclusion of sampling to identify any drifts in quadrupole mass spectrometer sensitivity. If drift is observed, you must use an internal standard to correct for changes in sensitivity.
(13) Sample analysis. To determine the concentration of a specific component in the sample, you must divide the ion intensity of the sample response by the calibrated response factor for each component.
(14) Deconvolution of interfering peaks. The effects of interfering peaks must be deconvoluted from the mass spectra for each target analyte.
(15) Calculations. Plot ion intensity versus analyte concentration for a given compound obtained when calibrating the analytical system. Determine the slope and intercept for each calibrated species to obtain response factors with which to calculate concentrations in the sample. For an acceptable calibration, the R2 value of the calibration curve must be at least 0.98.
(b) In place of the Fourier Transform Infrared Spectroscopy protocol requirements specified in section 2.2.4 of EPA 430-R-10-003 (incorporated by reference, see §98.7), you may conduct Fourier Transform Infrared Spectroscopy testing in accordance with the provisions in paragraph (b)(1) through (17) of this appendix, including the laboratory study phase described in paragraphs (b)(1) through (7), and the field study phase described in paragraphs (b)(8) through (17) of this appendix.
(1) Conformance with provisions associated with the Calibration Transfer Standard. This procedure calls for the use of a calibration transfer standard in a number of instances. The use of a calibration transfer standard is necessary to validate optical pathlength and detector response for spectrometers where cell temperature, cell pressure, and cell optical pathlength are potentially variable. For fixed pathlength spectrometers capable of controlling cell temperature and pressure to within +/− 10 percent of a desired set point, the use of a calibration transfer standard, as described in paragraphs (b)(2) to (17) this appendix is not required.
(2) Defining spectroscopic conditions. Define a set of spectroscopic conditions under which the field studies and subsequent field applications are to be carried out. These include the minimum instrumental line-width, spectrometer wave number range, sample gas temperature, sample gas pressure, absorption pathlength, maximum sampling system volume (including the absorption cell), minimum sample flow rate, and maximum allowable time between consecutive infrared analyses of the effluent.
(3) Criteria for reference spectral libraries. On the basis of previous emissions test results and/or process knowledge (including the documentation of results of any initial and subsequent tests, and the final reports required in §98.97(d)(4)(i)), estimate the maximum concentrations of all of the analytes in the effluent and their minimum concentrations of interest (those concentrations below which the measurement of the compounds is of no importance to the analysis). Values between the maximum expected concentration and the minimum concentration of interest are referred to below as the “expected concentration range.” A minimum of three reference spectra is sufficient for a small expected concentration range (e.g., a difference of 30 percent of the range between the low and high ends of the range), but a minimum of four spectra are needed where the range is greater, especially for concentration ranges that may differ by orders of magnitude. If the measurement method is not linear then multiple linear ranges may be necessary. If this approach is adopted, then linear range must be demonstrated to pass the required quality control. When the set of spectra is ordered according to absorbance, the absorbance levels of adjacent reference spectra should not differ by more than a factor of six. Reference spectra for each analyte should be available at absorbance levels that bracket the analyte's expected concentration range; minimally, the spectrum whose absorbance exceeds each analyte's expected maximum concentration or is within 30 percent of it must be available. The reference spectra must be collected at or near the same temperature and pressure at which the sample is to be analyzed under. The gas sample pressure and temperature must be continuously monitored during field testing and you must correct for differences in temperature and pressure between the sample and reference spectra. Differences between the sample and reference spectra conditions must not exceed 50 percent for pressure and 40°C for temperature.
(4) Spectra without reference libraries. If reference spectral libraries meeting the criteria in paragraph (b)(3) of this appendix do not exist for all the analytes and interferants or cannot be accurately generated from existing libraries exhibiting lower minimum instrumental line-width values than those proposed for the testing, prepare the required spectra according to the procedures specified in paragraphs (b)(4)(i) and (ii) of this appendix.
(i) Reference spectra at the same absorbance level (to within 10 percent) of independently prepared samples must be recorded. The reference samples must be prepared from neat forms of the analyte or from gas standards of the highest quality commonly available from commercial sources. Either barometric or volumetric methods may be used to dilute the reference samples to the required concentrations, and the equipment used must be independently calibrated to ensure suitable accuracy. Dynamic and static reference sample preparation methods are acceptable, but dynamic preparations must be used for reactive analytes. Any well characterized absorption pathlength may be employed in recording reference spectra, but the temperature and pressure of the reference samples should match as closely as possible those of the proposed spectroscopic conditions.
(ii) If a mercury cadmium telluride or other potentially non-linear detector (i.e., a detector whose response vs. total infrared power is not a linear function over the range of responses employed) is used for recording the reference spectra, you must correct for the effects of this type of response on the resulting concentration values. As needed, spectra of a calibration transfer standard must be recorded with the laboratory spectrometer system to verify the absorption pathlength and other aspects of the system performance. All reference spectral data must be recorded in interferometric form and stored digitally.
(5) Sampling system preparation. Construct a sampling system suitable for delivering the proposed sample flow rate from the effluent source to the infrared absorption cell. For the compounds of interest, the surfaces of the system exposed to the effluent stream may need to be stainless steel or Teflon; because of the potential for generation of inorganic automated gases, glass surfaces within the sampling system and absorption cell may need to be Teflon-coated. The sampling system should be able to deliver a volume of sample that results in a necessary response time.
(6) Preliminary analytical routines. For the proposed absorption pathlength to be used in actual emissions testing, you must prepare an analysis method containing of all the effluent compounds at their expected maximum concentrations plus the field calibration transfer standard compound at 20 percent of its full concentration as needed.
(7) Documentation. The laboratory techniques used to generate reference spectra and to convert sample spectral information to compound concentrations must be documented. The required level of detail for the documentation is that which allows an independent analyst to reproduce the results from the documentation and the stored interferometric data.
(8) Spectroscopic system performance. The performance of the proposed spectroscopic system, sampling system, and analytical method must be rigorously examined during and after a field study. Several iterations of the analysis method may need to be applied depending on observed concentrations, absorbance intensities, and interferences. During the field study, all the sampling and analytical procedures envisioned for future field applications must be documented. Additional procedures not required during routine field applications, notably dynamic spiking studies of the analyte gases, may be performed during the field study. These additional procedures need to be performed only once if the results are acceptable and if the effluent sources in future field applications prove suitably similar to those chosen for the field study. If changes in the effluent sources in future applications are noted and require substantial changes to the analytical equipment and/or conditions, a separate field study must be performed for the new set of effluent source conditions. All data recorded during the study must be retained and documented, and all spectral information must be permanently stored in interferometric form.
(9) System installation. The spectroscopic and sampling sub-systems must be assembled and installed according to the manufacturers' recommendations. For the field study, the length of the sample lines used must not be less than the maximum length envisioned for future field applications. The system must be given sufficient time to stabilize before testing begins.
(10) Pre-Test calibration. Record a suitable background spectrum using pure nitrogen gas; alternatively, if the analytes of interest are in a sample matrix consistent with ambient air, it is beneficial to use an ambient air background to control interferences from water and carbon dioxide. For variable pathlength Fourier Transform Infrared Spectrometers, introduce a sample of the calibration transfer standard gas directly into the absorption cell at the expected sample pressure and record its absorbance spectrum (the “initial field calibration transfer standard spectrum”). Compare it to the laboratory calibration transfer standard spectra to determine the effective absorption pathlength. If possible, record spectra of field calibration gas standards (single component standards of the analyte compounds) and determine their concentrations using the reference spectra and analytical routines developed in paragraphs (b)(2) through (7) of this appendix; these spectra may be used instead of the reference spectra in actual concentration and uncertainty calculations.
(11) Deriving the calibration transfer standard gas from tool chamber gases. The calibration transfer standard gas may be derived by flowing appropriate semiconductor tool chamber gases under non-process conditions (no thermal or plasma conditions and with no wafer(s) present) if compressed gas standards cannot be brought on-site.
(12) Reactivity and response time checks. While sampling ambient air and continuously recording absorbance spectra, suddenly replace the ambient air flow with calibration transfer standard gas introduced as close as possible to the probe tip. Examine the subsequent spectra to determine whether the flow rate and sample volume allow the system to respond quickly enough to changes in the sampled gas. Should a corrosive or reactive gas be of interest in the sample matrix it would be beneficial to determine the reactivity in a similar fashion, if practical. Examine the subsequent spectra to ensure that the reactivities of the analytes with the exposed surfaces of the sampling system do not limit the time response of the analytical system. If a pressure correction routine is not automated, monitor the absorption cell temperature and pressure; verify that the (absolute) pressure remains within 2 percent of the pressure specified in the proposed system conditions.
(13) Analyte spiking. Analyte spiking must be performed. While sampling actual source effluent, introduce a known flow rate of calibration transfer standard gas into the sample stream as close as possible to the probe tip or between the probe and extraction line. Measure and monitor the total sample flow rate, and adjust the spike flow rate until it represents 10 percent to 20 percent of the total flow rate. After waiting until at least four absorption cell volumes have been sampled, record four spectra of the spiked effluent, terminate the calibration transfer standard spike flow, pause until at least four cell volumes are sampled, and then record four (unspiked) spectra. Repeat this process until 12 spiked and 12 unspiked spectra have been obtained. If a pressure correction routine is not automated, monitor the absorption cell temperature and pressure; verify that the pressure remains within 2 percent of the pressure specified in the proposed system conditions. Calculate the expected calibration transfer standard compound concentrations in the spectra and compare them to the values observed in the spectrum. This procedure is best performed using a spectroscopic tracer to calculate dilution (as opposed to measured flow rates) of the injected calibration transfer standard (or analyte). The spectroscopic tracer should be a component not in the gas matrix that is easily detectable and maintains a linear absorbance over a large concentration range. Repeat this spiking process with all effluent compounds that are potentially reactive with either the sampling system components or with other effluent compounds. The gas spike is delivered by a mass flow controller, and the expected concentration of analyte of interest (AOITheoretical) is calculated as follows:
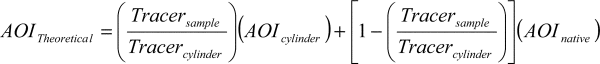
Where:
AOITheoretical = Theoretical analyte of interest concentration (parts per million (ppm)).
Tracersample = Tracer concentration (ppm) as seen by the Fourier Transform Infrared Spectrometer during spiking.
Tracercylinder = The concentration (ppm) of tracer recorded during direct injection of the cylinder to the Fourier Transform Infrared Spectrometer cell.
AOIcylinder = The supplier-certified concentration (ppm) of the analyte of interest gas standard.
AOInative = The native AOI concentration (ppm) of the effluent during stable conditions.
(14) Post-test calibration. At the end of a sampling run and at the end of the field study, record the spectrum of the calibration transfer standard gas. The resulting “final field calibration transfer standard spectrum” must be compared to the initial field calibration transfer standard spectrum to verify suitable stability of the spectroscopic system throughout the course of the field study.
(15) Amendment of analytical routines. The presence of unanticipated interferant compounds and/or the observation of compounds at concentrations outside their expected concentration ranges may necessitate the repetition of portions of the procedures in paragraphs (b)(2) through (14) of this appendix. Such amendments are allowable before final analysis of the data, but must be represented in the documentation required in paragraph (b)(16) of this appendix.
(16) Documentation. The sampling and spiking techniques used to generate the field study spectra and to convert sample spectral information to concentrations must be documented at a level of detail that allows an independent analyst to reproduce the results from the documentation and the stored interferometric data.
(17) Method application. When the required laboratory and field studies have been completed and if the results indicate a suitable degree of accuracy, the methods developed may be applied to practical field measurement tasks. During field applications, the procedures demonstrated in the field study specified in paragraphs (b)(8) through (16) of this appendix must be adhered to as closely as possible, with the following exceptions specified in paragraphs (b)(17)(i) through (iii) of this appendix:
(i) The sampling lines employed should be as short as practically possible and not longer than those used in the field study.
(ii) Analyte spiking and reactivity checks are required after the installation of or major repair to the sampling system or major change in sample matrix. In these cases, perform three spiked/unspiked samples with calibration transfer standard or a surrogate analyte on a daily basis if time permits and gas standards are easy to obtain and get on-site.
(iii) Sampling and other operational data must be recorded and documented as during the field study, but only the interferometric data needed to sufficiently reproduce actual test and spiking data must be stored permanently. The format of this data does not need to be interferograms but may be absorbance spectra or single beams.
(c) When using the flow and dilution measurement protocol specified in section 2.2.6 of EPA 430-R-10-003 (incorporated by reference, see §98.7), you may determine point-of-use abatement device total volume flow with the modifications specified in paragraphs (c)(1) through (3) of this appendix.
(1) You may introduce the non-reactive, non-native gas used for determining total volume flow and dilution across the point-of-use abatement device at a location in the exhaust of the point-of-use abatement device. For abatement systems operating in a mode where specific F-GHG are not readily abated, you may introduce the non-reactive, non-native gas used for determining total volume flow and dilution across the point-of-use abatement device prior to the point-of-use abatement system; in this case, the tracer must be more difficult to destroy than the target compounds being measured based on the thermal stability of the tracer and target.
(2) You may select a location for downstream non-reactive, non-native gas analysis that complies with the requirements in this paragraph (c)(2) of this appendix. The sampling location should be traversed with the sampling probe measuring the non-reactive, non-native gas concentrations to ensure homogeneity of the non-reactive gas and point-of-use abatement device effluent (i.e., stratification test). To test for stratification, measure the non-reactive, non-native gas concentrations at three points on a line passing through the centroidal area. Space the three points at 16.7, 50.0, and 83.3 percent of the measurement line. Sample for a minimum of twice the system response time, determined according to paragraph (c)(3) of this appendix, at each traverse point. Calculate the individual point and mean non-reactive, non-native gas concentrations. If the non-reactive, non-native gas concentration at each traverse point differs from the mean concentration for all traverse points by no more than ±5.0 percent of the mean concentration, the gas stream is considered unstratified and you may collect samples from a single point that most closely matches the mean. If the 5.0 percent criterion is not met, but the concentration at each traverse point differs from the mean concentration for all traverse points by no more than ±10.0 percent of the mean, you may take samples from two points and use the average of the two measurements. Space the two points at 16.7, 50.0, or 83.3 percent of the measurement line. If the concentration at each traverse point differs from the mean concentration for all traverse points by more than ±10.0 percent of the mean but less than 20.0 percent, take samples from three points at 16.7, 50.0, and 83.3 percent of the measurement line and use the average of the three measurements. If the gas stream is found to be stratified because the 20.0 percent criterion for a 3-point test is not met, locate and sample the non-reactive, non- native gas from traverse points for the test in accordance with Sections 11.2 and 11.3 of EPA Method 1 in 40 CFR part 60, Appendix A-1. A minimum of 40 non-reactive gas concentration measurements will be collected at three to five different injected non-reactive gas flow rates for determination of point-of-use abatement device effluent flow. The total volume flow of the point-of-use abatement device exhaust will be calculated consistent with the EPA 430-R-10-003 (incorporated by reference, see §98.7) Equations 1 through 7.
(3) You must determine the measurement system response time according to paragraphs (c)(3)(i) through (iii) of this appendix.
(i) Before sampling begins, introduce ambient air at the probe upstream of all sample condition components in system calibration mode. Record the time it takes for the measured concentration of a selected compound (for example, carbon dioxide) to reach steady state.
(ii) Introduce nitrogen in the system calibration mode and record the time required for the concentration of the selected compound to reach steady state.
(iii) Observe the time required to achieve 95 percent of a stable response for both nitrogen and ambient air. The longer interval is the measurement system response time.
[78 FR 68234, Nov. 13, 2013]
['Air Programs']
['Greenhouse Gases']
UPGRADE TO CONTINUE READING